

Abstract
Medulloblastoma is an invasive embryonal tumor of the cerebellum with predominant neuronal differentiation. Although several genes have been implicated in medulloblasoma formation, such as Patched (Ptc1) and the adenomatous polyposis coli gene (Apc), the majority of these tumors cannot be explained by mutations in these genes. The cellular origin as well as the genetic and molecular changes involved in the genesis and progression of human medulloblastomas remain largely unknown. Here we show that disruption of poly(ADP-ribose) polymerase (PARP-1) causes a high incidence (49%) of aggressive brain tumors in p53 null mice, with typical features of human cerebellar medulloblastomas. At as early as 8 weeks of age, lesions started on the outer surface of the cerebellum from remnant granule cell precursors of the developmental external germinal layer. Progression of these tumors is associated with the re-activation of the neuronal specific transcription factor Math1, dysregulation of Shh/Ptc1 signaling pathway, and chromosomal aberrations, including triradial and quadriradial chromosomes. The present study indicates that the loss of function of DNA double-strand break-sensing and repair molecules is an etiological factor in the evolution of the cerebellar medulloblastomas. These PARP-1/p53 double null mice represent a novel model for the pathogenesis of human medulloblastomas.
Primitive neuroectodermal tumors (PNETs) are highly malignant embryonal neoplasms of the central nervous system (CNS) with the cerebellar medulloblastoma as the most common and best characterized lesion. These tumors manifest predominantly in children and adolescents, and comprise approximately 20% of all pediatric CNS neoplasms. 1,2 Significant progress has been made in the treatment of children with medulloblastoma, but large gaps still exist in our understanding of the origin and pathogenesis of this malignancy.
A minority of medulloblastoma occur in the setting of hereditary cancer syndromes, including Li-Fraumeni syndrome caused by germline mutations of the tumor suppressor gene p53, 3,4 Gorlin syndrome with mutations in human homologue of the Drosophila gene Patched (Ptc1), 5,6 and Turcot’s syndrome carrying mutations in the adenomatous polyposis coli gene (APC). 7 However, mutations of p53, 3,8 ptc1, 9-12 APC, and genes involved in Wnt signaling 13-15 have only been detected in a small proportion of sporadic tumors, suggesting that additional genetic defects must be involved.
A causal link between gene mutations and the development of medulloblastomas comes from the inactivation of ptc1 in mice. Ptc1 is a receptor for Sonic hedgehog (Shh) and thereby suppresses Shh-mediated mitogen signaling for growth of granule cell precursors (GCPs) of the cerebellum. Ptc1 is primarily expressed in neuronal cells in the internal granule cell layer in the newborn cerebellum and in the granule cell layer of adults derived thereof. Ptc1 mutations release its suppression on Shh leading to uncontrolled proliferation of neuronal precursors and transformation. 16 Therefore, Shh/Ptc1 signaling plays a critical role during cerebellum development and in neuronal tumorigenesis. Inactivation of ptc1 in mice causes the death of homozygotes during embryogenesis with an open and overgrown neural tube. Interestingly, 14% of ptc1 heterozygous mutant mice develop aggressive cerebellar tumors, 17,18 suggesting that Shh signaling is involved in medulloblastoma formation. Whereas mice lacking p53 rarely develop brain tumors, 19-21 and mutations of the Rb gene have not been found in human medulloblastomas, null mutation of p53 induces a high frequency of medulloblastomas in Rb “conditional” knockout mice. 22 In addition, p53 null mutations dramatically accelerate medulloblastoma formation in ptc1 heterozygous animals. 23
DNA damage signaling/repair molecules play an important role in the maintenance of genomic integrity and in suppressing malignant transformation, most likely via their role in either homologous recombination repair or non-homologous end-joining (NHEJ) pathways. 24,25 Poly(ADP-ribose) polymerase (PARP-1) is a DNA strand break-sensing molecule and activation of PARP-1 is one of the early responses to DNA damage, among other DNA break-sensing molecules, such as ATM, DNA-PK, and p53. PARP-1 has been shown to play a role in protecting mice from γ-irradiation and the alkylatin agent (MNU), suppressing recombination, telomere length regulation, and chromosomal integrity. 26-28
Recent studies using PARP-1 knockout mice have implicated PARP-1 in the suppression of tumorigenesis. 28 Although T- and B-cell development are retarded in DNA-PK catalytic domain-mutated mice (SCID), PARP-1 deficiency partially rescued T-cell development, nevertheless, these mice succumbed to thymic lymphomas. 29 In addition, PARP-1-deficient mice show an increase in sarcoma and adenoma formation when treated with nitrosamine. 30 We also found that PARP-1 deficiency provoked carcinoma formation in p53 heterozygous knockout mice and neuronal malignancy (PNET) in p53 null mice. 31 In the present study, we report that PARP-1/p53 double null mice develop a high frequency of cerebellar medulloblastomas at an early age originating from granule precursors in the cerebellum associated with molecular and genetic alterations. These characteristics identify these PARP-1/p53 double null mice as a model for human medulloblastoma.
Materials and Methods
Animal Breeding Scheme
We bred PARP-1−/− mice in a mixed (129/Sv × C57BL/6) background 32 with p53+/− mice (129/Sv) 19 to generate PARP-1+/−p53+− mice (129/Sv × C57BL/6). These mice were then intercrossed for 5 subsequent generations before setting up a large cohort group. All analyses were carried out using animals from generations 5 to 10. Genotyping for PARP-1 and p53 loci was performed as described previously. 31
Histological and Immunohistochemical Analysis
Animals were euthanized by deep anesthesia when extracranial tumors or severe decline in health were evident and a full necropsy was performed on each animal. Whole mouse brains were fixed in 10% buffered formaldehyde for at least 24 hours. Sections of 3-μm thickness were stained with hematoxylin and eosin (H&E). Immunohistochemical staining was performed using antibodies against NeuN (monoclonal, 1:2000, Chemicon International, Temicula, CA) and MAP-2 (monoclonal, 1:10,000, Roche, Indianapolis, IN). Immunostaining for Math1 (antibody kindly provided by Dr. Johnson, University of Texas Southwestern Medical Center, Dallas, TX) was performed on tumor sections following protocol previously described. 33 Biotinylated secondary antibodies (Elite Kit, Vector Laboratory, Burlingame, CA) were used at a dilution of 1:1000. Visualization of antibody staining was achieved using an avidin/biotin peroxidase (Vector Laboratory) and diaminobenzidine as a chromogen. Tissue structures were visualized by counterstaining with Mayer’s hematoxylin.
RNA Isolation and Northern Blot Analysis
Total RNA was isolated from normal cerebella and medulloblastomas using TriReagent (Sigma, Steinheim, Germany) according to the manufacturer’s instruction. 10 to 30 μg of total RNA were electrophoresed on a 0.8% agarose-formaldehyde gel, transferred to a nitro-cellulose filter (Hybond N+, Amersham, Buckinghamshire, UK), and hybridized with 32P-labeled cDNA probes and exposed to PhosphorImager (Molecular Dynamics, Inc., Sunnyvale, CA).
Cytogenetic Analyses
Primary medulloblastoma cells were isolated according to the protocol 34 with modification. Briefly, medulloblastoma cells were mechanically dissociated with 40-μm Cell Strainer (Becton Dickinson Labware, Franklin Lakes, NJ) followed by three-time washing with Dulbecco’s modified Eagle’s medium (DMEM) containing 200 units/ml penicillin, 200 μg/ml streptomycin, and 50 μg/ml gentamicin. All cell culture medium and reagents were from Life Technologies (Paisley, UK). These tumor cells were seeded onto gelatin-coated plates in DMEM containing 10% fetal calf serum, 4.0 mmol/L glutamine, 50 units/ml penicillin, 50 μg/ml streptomycin, and 25 μg/ml gentamicin. Metaphase spreads were prepared and stained for fluorescence in situ hybridization (FISH) analysis using a Cy-3-labeled telomeric probe [(CCCTAA)3 peptidenucleic acid] and counterstained by 4′,6′-diamidino-2-phenylindole (DAPI) according to previously described protocols. 31
Results
Null Mutation of PARP-1 Causes a High Incidence of PNETs/Medulloblastomas in p53−/− Mice
In our previous studies, we noted many neuronal tumors in PARP-1−/−p53−/− mice. 31 To better characterize these tumors and to minimize the influence of genetic modifiers, we generated a large cohort of PARP-1/p53 single or double null mice after intercrossing of PARP-1+/−p53+/− mice for 5 or more generations. In addition to a high frequency of lymphomas and angiosarcomas (data not shown), PARP-1 null mutation dramatically increased the incidence of cerebellar tumors in p53 null mice with a median of tumor onset of 16 weeks (P < 0.0001, Figure 1 ▶ ). Gross and histopathological analyses of the brains of these mice revealed that 31 of 63 (49%) PARP-1−/−p53−/− mice developed brain tumors starting at the age of 8 weeks (Figure 1) ▶ , and there was a predisposition in male versus female mice (2.1:1). Among these brain tumors, 30 (97%) were localized in the cerebellum (Figure 1) ▶ exhibiting an invasive growth pattern with poorly defined margins (Figure 2B ▶ , insert) and one tumor was located in the cerebrum.
Figure 1.

Incidence of brain tumors in PARP-1−/−p53−/− mice. A high frequency of brain tumors was only observed in PARP-1/p53 double mutant mice. All brain tumors were identified by macro- and microscopic examinations. The groups of various genotypes were monitored for 24 weeks and the curves show only brain tumors in various genotypes. The incidence of lymphomas and soft tissue sarcomas were not shown.
Figure 2.
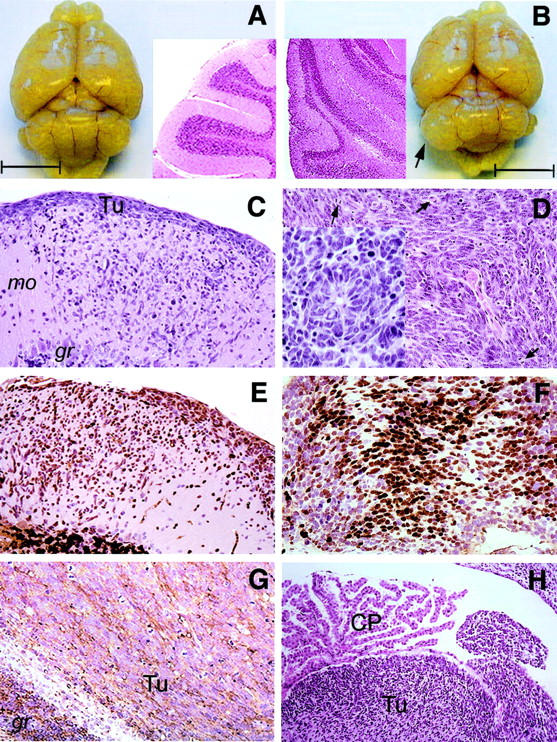
Gross and histological examinations of representative cerebellar tumors in PARP-1/p53 double null mice. A: Gross appearance of normal adult mouse brain at 20 weeks of age and a sagittal section (H&E) of a normal cerebellum is shown in insert. B: Gross appearance of a cerebellar tumor (arrow) in a PARP-1−/−p53−/− mouse at 15 weeks of age. A sagittal section of this tumor stained by H&E and is shown as an inset. Bars in (A) and (B) represent 0.5 cm. C: H&E staining of an early neoplastic lesion, spreading from the cerebellar surface through the molecular layer. D: Advanced medulloblastoma (H&E), consisting of densely packed cells with hyperchromatic nuclei and brisk mitotic activity (arrow). A neuroblastic rosette is shown as insert. (E–G). Early neoplastic lesions are immunoreactive for NeuN (E) and in advanced tumors, most cells express NeuN (F). G: MAP-2 cytoplasmic immunostaining shows a filamentous pattern through out the tumor (Tu). (H) Infiltration by a cerebellar medulloblastoma (Tu) into the fourth ventricle. mo, molecular layer. gr, granule cell layer. CP, choroid plexus. Original magnification of C, D, E, F, and G is ×20 and H is ×10.
In addition to the cerebellar tumors, early neoplastic lesions were observed in eight mice at the outer surface of the cerebellar molecular layer (Figure 2C) ▶ , the region corresponding to the external germinal layer (EGL) of the developing cerebellum. These cells proliferate and migrate inward as indicated by positive NeuN staining (Figure 2E) ▶ . Interestingly, tumor cells at advanced stages infiltrating adjacent cerebellar structure were frequently arranged in sheets and rows with a high mitotic activity (Figure 2D) ▶ . Occasionally, there were neuroblastic rosettes (Figure 2D ▶ , insert). Large tumors often compressed the cerebellar hemispheres, and invaded the fourth ventricle (Figure 2H) ▶ . Infiltration of the subarachnoid space was also occasionally observed (data not shown). These histological features indicate that cerebellar tumors in PARP-1/p53 double null mice tend to spread into neighboring CNS structures.
Cerebellar tumors showed strong immunoreactivity for neuronal nuclei (NeuN, Figure 2, E and F ▶ ) and for the early neuronal differentiation marker nestin (data not shown). In addition, they expressed microtubule-associated protein 2 (MAP-2) (Figure 2G) ▶ , and, weakly, synaptophysin (data not shown). Although the majority of these brain tumors were negative for glial fibrillary acidic protein (GFAP), scattered GFAP-positive cells were observed in a few cases (data not shown). These analyses indicate that cerebellar tumors arising in PARP-1/p53 double null mice exhibit predominantly neuronal differentiation. Together, the pathological and immunohistological characteristics are similar to the phenotypic features of human cerebellar medulloblastomas. 1,2
Math1 Reactivation in Granule Cell Precursors (GCPs) in PARP-1−/− p53 Null Mice
The fact that tumors were mainly localized at the outer surface of the cerebellum and underwent a dramatic proliferation expansion is reminiscent of rapid postnatal development of the cerebellum. Math1 is a neuron-specific basic helix-loop-helix (bHLH) transcription factor and is required for the proliferation of granule cells in the cerebellum. 35 This molecule is specifically expressed in the mitotic precursor cells at the EGL but is shut off in post-mitotic neuronal cells at the EGL and mature granule cells. 33 We performed Northern blot analysis for math1 mRNA expression in developing cerebellum and tumor samples. As expected, the math1 transcript was readily detectable in the cerebellum of wild-type, PARP-1−/−, and PARP-1−/−p53−/− mice at postnatal day 6 (P6) and 10 (P10), but undetectable at day 21 (P21) and week 6 (W6) as well as adjacent non-tumor cerebellum (N, Figure 3A ▶ ). Strikingly, math1 was highly expressed in medulloblastoma samples (Figure 3A) ▶ , suggesting a re-activation of math1 during tumorigenesis.
Figure 3.
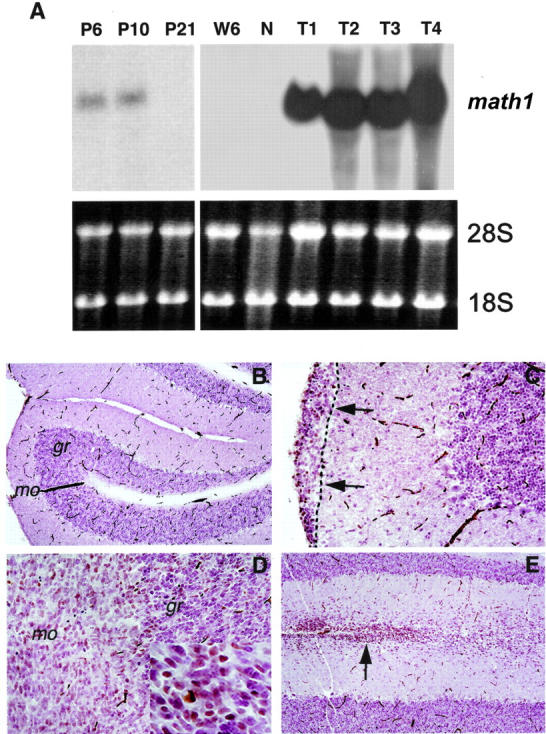
A: Re-activation of neuron-specific transcription factor Math1 in medulloblastomas. Northern blot analysis of math1 expression in a normal cerebellum (N) at postnatal day 6 (P6), P10, P21, and at 6 weeks (W6) in adult mice, and medulloblastomas at the age of 15 to 22 weeks (T1–T4). The math1 transcript (2.5 kb) was detected in the cerebellum at P6 and P10, but was absent at postnatal day 21 and in adult cerebella (W6 and N). However, high expression levels of math1 mRNA were detected in medulloblastomas (T1–T4). Ethidium staining of 18S and 28S rRNA was used to control loading. Immunohistochemistry of Math1 in the normal cerebellum (B) and medulloblastomas (C–E). B: A normal cerebellum from a PARP-1−/−p53−/− mouse (16 weeks old) is devoid of Math1 staining in the molecular (mo) and granule (gr) layers, similar to wild-type animals. C: Tumor cells from an early neoplastic lesion were located in the outer surface of the cerebellar molecular layer (arrows). D: An advanced cerebellar tumor showing a diffused staining for Math1 (see also insert with Math1-positive cells in the granule cell layer). E: Math1-positive cells were also detected in tumor cells infiltrated into the molecular layer at advanced stages. Original magnification of B and E is × 10 and C and D is × 20.
To further localize the expression of Math1 to specific cell types in medulloblastomas, we performed immunohistological staining of the tumors at various stages using an antibody specifically against mouse Math1. In agreement with previous studies, 33 normal granule cells in the adult cerebellum were negative for Math1 expression (Figure 3B) ▶ . Interestingly, in the early lesions of medulloblastoma, tumor cells at the outer surface of the cerebellum were stained positive for Math1 (Figure 3C) ▶ and these cells also co-expressed NeuN (data not shown), indicating that medulloblastomas most likely originated from the granule cell precursors retained in the EGL. In advanced tumors, Math1 was expressed in a diffused manner throughout the tumor mass (Figure 3D) ▶ . Furthermore, tumor cells that infiltrated to the molecular layer of the cerebellum also showed positive staining for Math1 (Figure 3E) ▶ . These data indicate that Math1 is constitutively expressed during medulloblastoma progression, consistent with its role in neuronal cell proliferation.
Characterization of Shh/Ptc1 Signaling in Medulloblastomas
Mutations in ptc1 have been detected in sporadic and hereditary medulloblastomas. 16 We next investigated whether ptc1 is involved in PARP-1 deficiency-induced medulloblastomas by Northern blot analysis. Ptc1 is expressed at normal levels in the cerebellum of PARP-1−/− and PARP-1−/−p53−/− mice at postnatal day P6, P10, P21 (data not shown), and W6 (Figure 4, A and B) ▶ when compared to that in wild-type mice. However, expression of ptc1 was dramatically reduced in the tumors compared to their adjacent non-tumor cerebellar tissues and to that in 6-week-old mice (Figure 4, A and B) ▶ .
Figure 4.

Dysregulation of the Shh/ptc1 signaling pathway in medulloblastomas derived from PARP-1/p53 double null mice. A: Representative Northern blot analysis for ptc1 and gli1 expression in normal adult cerebellum and medulloblastomas, and a quantitation of expression levels of ptc1 (B) and gli1 (C) is shown. Reduced levels of the ptc1 transcript (8 kb) were detected in all tumor samples when compared to non-tumor cerebellar tissue of adult (N4) and 6-week-old (W6) mice. In contrast, weak expression of gli1 (4.0 kb) was seen in the normal cerebellum (W6 and N4), whereas higher expression levels of gli1 were present in medulloblastomas (T1–T4). The β-actin probe is used to control loading.
The transcription factor gli1 is a downstream target of the Shh signaling and is normally repressed by Ptc1. 16 To test whether down-regulation of ptc1 activates the Shh signaling pathway in these medulloblastomas, we analyzed gli1 mRNA expression in normal developing and adult cerebellum as well as tumor samples. While the gli1 transcript was present at low levels in the normal cerebellum of various genotypes at P6, P10 and P21, W6 (Figure 4A ▶ , data not shown), a significant increase of gli1 expression was observed in medulloblastoma samples (Figure 4, A and C) ▶ . Notably, the up-regulation of gli1 expression was negatively correlated with the pattern of ptc1 expression in normal cerebellum and tumors, consistent with the proposed role for Ptc1 in suppressing gli expression. These data suggest that dysregulation of the Shh/Ptc1 pathway is involved in medulloblastoma formation in these PARP-1/p53 double null mice.
Severe Chromosomal Aberrations and Increased Mitotic Recombination in Medulloblastoma Cells Derived from PARP-1−/− p53−/− Mice
To understand the genetic basis of these tumors, we analyzed by FISH more than 33 metaphases from each population of three primary medulloblastomas that we isolated. These tumors were highly aneuploid and harbored various chromosomal aberrations. We noticed a high frequency of fusions (more than one fusion per metaphase) (Figure 5A) ▶ , including Robertsonian translocations (Figure 5, A, C, and D) ▶ and dicentric chromosomes (Figure 5A) ▶ . Chromosome and chromatid breaks (Figure 5A and E–J) ▶ as well as fragmentations (Figure 5, G and H) ▶ were also present in these tumor cells. Intriguingly, a frequent chromosome configuration including triradials and quadriradials has been found in these tumors (Figure 5A and I–K) ▶ . Since the radial chromosomes are usually resulted from interchromatid or intrachromosome exchanges during S phase, this specific chromosome structural change indicates an increased mitotic recombination in medulloblastomas derived from PARP-1/p53 double null mice.
Figure 5.

Cytogenetic analysis of medulloblastoma cells derived from PARP-1/p53 double null mice. Metaphase chromosomes were prepared from primary medulloblastomas and stained with the telomeric FISH probe (D, F, H, J, and L) then counterstained with 4′,6′-diamidino-2-phenylindole (DAPI) (B, C, E, G, I, and K). A: Summary of chromosomal aberrations in primary medulloblastoma cells. RL, Robertsonian-like fusions; Dic, dicentric chromosomes. Radial chromosomes include triradial and quadriradial chromosomes. Representative metaphase spread from medulloblastoma cells is shown in (B) and chromosomal abnormalities include Robertsonian-like fusions (C and D), chromosomal break (E and F), fragmentations (G–J), triradial (I and J), and quadriradial (K and L) chromosomes.
Discussion
The present study shows that loss of function of the DNA strand break-binding molecule PARP-1 results in a high frequency of cerebellar tumors in p53 null mice with onset at an early age. These tumors originate from neuronal cells retained at the outer surface of the cerebellar molecular layer and show a tendency to invade the fourth ventricle and the subarachnoidal space. These features resemble the major pathological characteristics of human medulloblastomas. Although the histogenesis of medulloblastomas has been a controversial issue for more than 70 years, it has been proposed that this tumor type originates from GCPs in the EGL of the cerebellum. 1,2 This view is supported by the observations showing that ptc1 mutations are detected in a subset of medulloblastomas 5,6 and that transcription factor ZIC1 is up-regulated in medulloblastomas, which is predominantly expressed in the cerebellar granule cell lineage of fetal brain. 36,37 In addition, medulloblastomas appeared on the cerebellar surface of ptc1-deficient mice, 17,18 and inactivation of the p53/Rb genes in the EGL caused medulloblastomas in mice. 22 Math1 is tightly regulated in a developing cerebellum for correct coordination of proliferation and differentiation of GCPs. 35,38 Based on the expression of neuronal markers and Math1 in tumors, we conclude that GCPs retained at EGL are the cellular origin of medulloblastomas in PARP-1 knockout mice.
The high frequency of medulloblastomas in PARP-1-deficient p53 null mice suggests a cooperation of DNA end-processing and cell cycle checkpoint molecules as suppressors of malignant transformation of neuronal cells. This finding is surprising since p53 null mice rarely develop brain tumors while approximately 14% of patients with Li-Fraumeni syndrome, caused by germline mutations of p53, develop CNS neoplasms. In agreement with a previous study, 22 our results indicate that p53 null mutation alone is not sufficient to induce brain tumors and that additional genetic mutations, eg, genome caretakers, are required for malignant transformation. PARP-1 is involved in double-strand break (DSB) repair pathways since PARP-1 deficiency renders mice hypersensitive to whole body γ-irradiation 39-42 and facilitates a partial rescue of the V(D)J recombination process in SCID mice. 29 In addition, patients suffering from chromosome instability syndromes, such as Nijmegen breakage syndrome (NBS), 43 Bloom syndrome (BLM), 44 and Fanconi anemia 45 are predisposed to malignancy, including occasional medulloblastomas. Secondly, the brain is constantly exposed to oxidative stress and damage. Full activity of the DNA DSB repair machinery is required to repair these DNA lesions, failure of which, together with the compromised checkpoints, allows accumulation and propagation of genetic mutations. Finally, it recently has been shown that neuronal cells in developing mouse cerebellum are susceptible to DNA damage-induced cell death. 46 PARP-1-deficient neuronal cells are resistant to ischemia or neurotoxic cell death, 47,48 and inhibition of PARP-1 activity abrogates programmed cell death of human medulloblastoma cells. 49 It is possible that this feature may facilitate propagation of damaged cells that can be synergistically promoted by p53-mediated checkpoint failure.
Cytogenetic analysis revealed a high degree of chromosomal abnormalities and aneuploidy in medulloblastoma cells derived from PARP-1/p53 double null mice. This finding is consistent with our previous observation that PARP-1 and p53 synergistically minimize chromosomal aberrations. 31 In addition, triradial and quadriradial chromosomes are often observed (Figure 5) ▶ . While these chromosomal aberrations may be caused by the telomere dysfunction in the absence of PARP-1 and p53, 31 formation of radial chromosomes is most likely attributable to an increased activity of mitotic recombination that promotes chromatid exchange between sister chromatids or interchromosomes. This finding suggests a role of PARP-1 in suppressing mitotic recombination, and is consistent with the hypothesis that PARP-1 may be an antirecombinogenic factor. 26 The radial chromosomes are deleterious since they often cause chromosome breaks and translocations when cells enter the next round of cell division. In somatic cells, mitotic recombination is the major cause of the loss of heterozygosity (LOH). 50 In agreement with this mechanism, fusions (and perhaps also translocations) and breakage/fragmentations in tumor cells are the most prominent chromosomal abnormalities in these medulloblastomas. Interestingly, radial chromosomes are a typical feature of Bloom syndrome, caused by mutations in BLM, leading to chromosomal hyper-recombination. 44 Bloom patients have been reported to develop medulloblastomas, albeit rarely. These data further support the notion that DNA (single- or double-strand) break-processing molecules stabilize chromosomes and thereby prevent tumorigenesis.
Although chromosomal fusions or translocations represent a mechanism for overexpression of transcription factors and activation of oncogenes in human malignancies, 51 the cause for the reactivation of the transcription factor Math1 in medulloblastomas in PARP-1−/−p53−/− mice remains elusive. Given its active role in proliferation, it is possible that Math1 transcription factor acts as an oncogene. Up-regulation of math1 in early and advanced medulloblastomas of PARP-1/p53 double null mice strongly suggests that reactivation of math1 is likely to promote proliferation of neuronal cells, which may represent a molecular event for neuronal oncogenic transformation and tumor progression.
The Shh/Ptc1 signaling pathway is involved in cerebellar development and the alteration of this signaling contributes to the etiology of medulloblastomas in humans. 16 Ptc1 represses Shh signaling resulting in down-regulation of its targeted genes, including transcriptional activator gli1 and inactivation of ptc1 in human tumors, which results in the overexpression of gli1. 52 Consistent with this notion, heterozygous ptc1 knockout mice are predisposed to medulloblastomas associated with gli1 overexpression. 17,18 Moreover, activation of gli1 induces hyperproliferation of neuronal precursors. 53 Along these same lines, we found that a significant down-regulation of ptc1 is associated with the overexpression of gli1 in medulloblastomas, indicating an involvement of the Shh/Ptc1 signaling pathway in the formation of this tumor type in PARP-1−/− mice lacking p53.
Chromosomal alterations are prevalent in human medulloblastomas. 1 PARP-1 is localized in the human chromosome 1q41–42. Although alterations in chromosome 1q are found in a certain number of human medulloblastomas, 1,54,55 and loss of genetic materials has been mapped to 1q31–32, 56 the status of the PARP-1 locus has not been documented in this type of tumor. Therefore, further genetic and molecular analyses are required to elucidate the role of PARP-1 and poly(ADP-ribosy)lation activity in the development of human medulloblastomas.
The present study has provided genetic evidence that defects in the DNA double-strand break-sensing molecule PARP-1 and failure in p53-mediated checkpoints render neuronal cells to malignant transformation, leading to medulloblastomas. Given the anatomical location of tumors, age of onset, sex prevalence, predominant neuronal differentiation, and genetic and molecular alterations, all of which are reminiscent of human medulloblastomas, PARP-1/p53 double null mice represent a novel mouse model for this disease. Further characterization of medulloblastomas derived from these animals may lead to the identification of new genes that are instrumental in the evolution of human cerebellar medulloblastoma as well as related primitive neuroectodermal tumors and ultimately, to preclinical valuation of new therapies.
Acknowledgments
We thank D. Galendo for the maintenance of the animal colonies, N. Lyandrat for technical assistance, and G. Mollon for the preparation of photographs. We also thank Dr. E.J. Johnson for the Math1 antibody, and Drs. H. Zogbi (math1), M.P. Scott (ptc1) and T. Curran (gli1) for providing cDNA probes. Further thanks are due to Dr. Zdenko Herceg for his critical reading and discussion of the manuscript.
Footnotes
Address reprint requests to Dr. Zhao-Qi Wang, Ph.D., International Agency for Research on Cancer (IARC), 150, cours Albert Thomas, 69008 Lyon, France. E-mail: zqwang@iarc.fr.
Supported in part by grants from the Association for International Cancer Research (AICR 99–050), United Kingdom and from the Association pour la Recherche contre le Cancer (ARC, A00–2-42), France.
References
- 1.Kleihues P, Cavenee WK: World Health Organization Classification of Tumours. Pathology and Genetics: Tumours of the Nervous System. 2000:pp 129-137 IARC Press, Lyon
- 2.Lantos PL, Louis DN, Rosenblum MK, Kleihues P: Greenfield’s Neuropathology ed 7 2002:pp 117-132 Arnold, London
- 3.Kleihues P, Schauble B, zur Hausen A, Esteve J, Ohgaki H: Tumors associated with p53 germline mutations: a synopsis of 91 families. Am J Pathol 1997, 150:1-13 [PMC free article] [PubMed] [Google Scholar]
- 4.Malkin D: The Li-Fraumeni syndrome. Vogelstein B Kinzler KW eds. The Genetic Basis of Human Cancer. 1998:pp 393-407 McGraw-Hill, New York
- 5.Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, Vorechovsky I, Holmberg E, Unden AB, Gillies S, Negus K, Smyth I, Pressman C, Leffell DJ, Gerrard B, Goldstein AM, Dean M, Toftgard R, Chenevix-Trench G, Wainwright B, Bale AE: Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996, 85:841-851 [DOI] [PubMed] [Google Scholar]
- 6.Johnson RL, Rothman AL, Xie J, Goodrich LV, Bare JW, Bonifas JM, Quinn AG, Myers RM, Cox DR, Epstein EH, Jr, Scott MP: Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 1996, 272:1668-1671 [DOI] [PubMed] [Google Scholar]
- 7.Hamilton SR, Liu B, Parsons RE, Papadopoulos N, Jen J, Powell SM, Krush AJ, Berk T, Cohen Z, Tetu B, Burger PC, Wood PA, Taqi F, Booker SV, Petersen GM, Offerhaus GJA, Tersmette AC, Giardiello FM, Vogelstein B, Kinzler KW: The molecular basis of Turcot’s syndrome. N Engl J Med 1995, 332:839-847 [DOI] [PubMed] [Google Scholar]
- 8.Ohgaki H, Eibl RH, Schwab M, Reichel MB, Mariani L, Gehring M, Petersen I, Holl T, Wiestler OD, Kleihues P: Mutations of the p53 tumor suppressor gene in neoplasms of the human nervous system. Mol Carcinog 1993, 8:74-80 [DOI] [PubMed] [Google Scholar]
- 9.Pietsch T, Waha A, Koch A, Kraus J, Albrecht S, Tonn J, Sorensen N, Berthold F, Henk B, Schmandt N, Wolf HK, von Deimling A, Wainwright B, Chenevix-Trench G, Wiestler OD, Wicking C: Medulloblastomas of the desmoplastic variant carry mutations of the human homologue of Drosophila patched. Cancer Res 1997, 57:2085-2088 [PubMed] [Google Scholar]
- 10.Raffel C, Jenkins RB, Frederick L, Hebrink D, Alderete B, Fults DW, James D: Sporadic medulloblastomas contain ptc1h mutations. Cancer Res 1997, 57:842-845 [PubMed] [Google Scholar]
- 11.Xie J, Johnson RL, Zhang X, Bare JW, Waldman FM, Cogen PH, Menon AG, Warren RS, Chen LC, Scott MP, Epstein EH, Jr: Mutations of the PATCHED gene in several types of sporadic extracutaneous tumors. Cancer Res 1997, 57:2369-2372 [PubMed] [Google Scholar]
- 12.Wolter M, Reifenberger J, Sommer C, Ruzicka T, Reifenberger G: Mutations in the human homologue of the Drosophila segment polarity gene patched (PTCH) in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res 1997, 57:2581-2585 [PubMed] [Google Scholar]
- 13.Zurawel RH, Chiappa S, Allen C, Raffel C: Sporadic medulloblastomas contain oncogenic β-catenin mutations. Cancer Res 1998, 58:896-899 [PubMed] [Google Scholar]
- 14.Huang H, Mahler-Araujio BM, Sankila A, Chimeli L, Yonekawa Y, Kleihues P, Ohgaki H: APC mutations in sporadic medulloblastomas. Am J Pathol 2000, 156:433-437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dahmen RP, Koch A, Denkhaus D, Tonn JC, Sorensen N, Berthold F, Behrens J, Birchmeier W, Wiestler OD, Pietsch T: Deletions of axin1, a component of the wnt/wingless pathway, in sporadic medulloblastomas. Cancer Res 2001, 61:7039-7043 [PubMed] [Google Scholar]
- 16.Wechsler-Reya RJ, Scott MP: The developmental biology of brain tumors. Annu Rev Neurosci 2001, 24:385-428 [DOI] [PubMed] [Google Scholar]
- 17.Goodrich LV, Milenkovic L, Higgins KM, Scott MP: Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 1997, 277:1109-1113 [DOI] [PubMed] [Google Scholar]
- 18.Wetmore C, Eberhart DE, Curran T: The normal patched allele is expressed in medulloblastomas from mice with heterozygous germline mutation of patched. Cancer Res 2000, 60:2239-2246 [PubMed] [Google Scholar]
- 19.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, Bradley A: Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumors. Nature 1992, 356:215-221 [DOI] [PubMed] [Google Scholar]
- 20.Harvey M, McArthur MJ, Montagomery CA, Jr, Butel JS, Bradley A, Donehower LA: Spontaneous and carcinogen-induced tumorigenesis in p53-deficient mice. Nat Genet 1993, 5:225-229 [DOI] [PubMed] [Google Scholar]
- 21.Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA: Tumor spectrum analysis in p53-mutant mice. Curr Biol 1994, 4:1-7 [DOI] [PubMed] [Google Scholar]
- 22.Marino S, Vooijs M, van Der Gulden H, Jonkers J, Berns A: Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev 2000, 14:994-1004 [PMC free article] [PubMed] [Google Scholar]
- 23.Wetmore C, Eberhart DE, Curran T: Loss of p53 but not ARF accelerates medulloblastoma in mice heterozygous for patched. Cancer Res 2001, 61:513-516 [PubMed] [Google Scholar]
- 24.Khanna KK, Jackson SP: DNA double-strand breaks: signaling, repair, and the cancer connection. Nat Genet 2001, 27:247-254 [DOI] [PubMed] [Google Scholar]
- 25.Pierce AJ, Stark JM, Araujo FD, Moynahan ME, Berwick M, Jasin M: Double-strand breaks and tumorigenesis. Trends Cell Biol 2001, 11:S52-S59 [DOI] [PubMed] [Google Scholar]
- 26.Lindahl T, Satoh MS, Poirier GG, Klungland A: Post-translational modification of poly(ADP-ribose) polymerase induced by DNA strand breaks. Trends Biochem Sci 1995, 20:405-411 [DOI] [PubMed] [Google Scholar]
- 27.Herceg Z, Wang Z-Q: Functions of poly(ADP-ribose) polymerase (PARP) in DNA repair, genomic integrity, and cell death. Mutat Res 2001, 477:97-110 [DOI] [PubMed] [Google Scholar]
- 28.Tong WM, Cortes U, Wang Z-Q: Poly(ADP-ribose) polymerase: a guardian angel protecting the genome and suppressing tumourigenesis. Biochim Biophys Acta Rev Cancer 2001, 1552:27-37 [DOI] [PubMed] [Google Scholar]
- 29.Morrison C, Smith GCM, Stingl L, Jackson SP, Wagner EF, Wang Z-Q: Genetic interaction between PARP and DNA-PK in V(D)J recombination and tumorigenesis. Nat Genet 1997, 17:479-482 [DOI] [PubMed] [Google Scholar]
- 30.Tsutsumi M, Masutani M, Nozaki T, Kusuoka O, Tsujiuchi T, Nakagama H, Suzuki H, Konishi Y, Sugimura T: Increased susceptibility of poly(ADP-ribose) polymerase-1 knockout mice to nitrosamine carcinogenicity. Carcinogenesis 2001, 22:1-3 [DOI] [PubMed] [Google Scholar]
- 31.Tong WM, Hande MP, Lansdorp PM, Wang Z-Q: DNA strand break-sensing molecule PARP cooperates with p53 in telomere function, chromosome stability, and tumor suppression. Mol Cell Biol 2001, 21:4046-4054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Z-Q, Auer B, Stingl L, Berghammer H, Haidacher D, Schweiger M, Wagner EF: Mice lacking ADPRT and poly(ADP-ribosyl)ation develop normally but are susceptible to skin disease. Genes Dev 1995, 9:509-520 [DOI] [PubMed] [Google Scholar]
- 33.Helms AW, Johnson JE: Progenitors of dorsal commissural interneurons are defined by MATH1 expression. Development 1998, 125:919-928 [DOI] [PubMed] [Google Scholar]
- 34.Tong WM, Bises G, Sheinin Y, Ellinger A, Genser D, Potzi R, Wrba F, Wenzl E, Roka R, Neuhold N, Peterlik M, Cross HS: Establishment of primary cultures from human colonic tissue during tumor progression: vitamin-D responses and vitamin-D-receptor expression. Int J Cancer 1998, 75:467-472 [DOI] [PubMed] [Google Scholar]
- 35.Ben-Arie N, Bellen HJ, Armstrong DL, McCall A, Gordadze PR, Guo Q, Matzuk MM, Zoghbi HY: Math1 is essential for genesis of cerebellar granule neurons. Nature 1997, 390:169-172 [DOI] [PubMed] [Google Scholar]
- 36.Yokota N, Aruga J, Takai S, Yamada K, Hamazaki M, Iwase T, Sugimura H, Mikoshiba K: Predominant expression of human zic in cerebellar granule cell lineage and medulloblastoma. Cancer Res 1996, 56:377-383 [PubMed] [Google Scholar]
- 37.Michiels EM, Oussoren E, Van Groenigen M, Pauws E, Bossuyt PM, Voute PA, Baas F: Genes differentially expressed in medulloblastoma and fetal brain. Physiol Genomics 1999, 1:83-91 [DOI] [PubMed] [Google Scholar]
- 38.Helms AW, Abney AL, Ben-Arie N, Zoghbi HY, Johnson JE: Autoregulation and multiple enhancers control Math1 expression in the developing nervous system. Development 2001, 127:1185-1196 [DOI] [PubMed] [Google Scholar]
- 39.Menessier-de Murcia J, Niedergang C, Trucco C, Ricoul M, Dutrillaux B, Mark M, Oliver FJ, Masson M, Dierich A, LeMeur M, Walztinger C, Chambon P, de Murcia G: Requirement of poly (ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc Natl Acad Sci USA 1997, 94:7303-7307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Z-Q, Stingl L, Morrison C, Jantsch M, Los M, Schulze-Osthoff K, Wagner EF: PARP is important for genomic stability but dispensable in apoptosis. Genes Dev 1997, 11:2347-2358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Masutani M, Nozaki T, Nishiyama E, Shimokawa T, Tachi Y, Suzuki H, Nakagama H, Wakabayashi K, Sugimura T: Function of poly(ADP-ribose) polymerase in response to DNA damage: gene-disruption study in mice. Mol Cell Biochem 1999, 193:149-152 [PubMed] [Google Scholar]
- 42.Tong WM, Galendo D, Wang Z-Q: Role of DNA break-sensing molecule poly(ADP-ribose) polymerase (PARP) in cellular function and radiation toxicity. Cold Spring Harbor Symp Quant Biol 2000, 65:583-591 [DOI] [PubMed] [Google Scholar]
- 43.Van der Burgt I, Chrzanowska KH, Smeets D, Weemaes C: Nijmegen breakage syndrome. J Med Genet 1996, 33:153-156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.German J, Ellis NA: Bloom syndrome. Vogelstein B Kinzler KW eds. The Genetic Basis of Human Cancer. 1998:pp 301-316 McGraw-Hill, New York
- 45.Auerbach AD, Buchwald M, Joenje H: Fanconi anemia. Vogelstein B Kinzler KW eds. The Genetic Basis of Human Cancer. 1998:pp 317-332 McGraw-Hill, New York
- 46.Herzog KH, Chong MJ, Kapsetaki M, Morgan JI, McKinnon PJ: Requirement for Atm in ionizing radiation-induced cell death in the developing central nervous system. Science 1998, 280:1089-1091 [DOI] [PubMed] [Google Scholar]
- 47.Juluri K, Ha HC, Snyder SH: Nervous system functions of poly(ADP-ribose) polymerase-1. Zhang J eds. PARP as a Therapeutic Target. 2002:pp 1-18 CRC Press, London
- 48.Ha HC, Snyder SH: Poly(ADP-ribose) polymerase-1 in the nervous system. Neurobiol Dis 2000, 7:225-239 [DOI] [PubMed] [Google Scholar]
- 49.Fulda S, Friesen C, Los M, Scaffidi C, Mier W, Benedict M, Nunez G, Krammer PH, Peter ME, Debatin KM: Betulinic acid triggers CD95 (APO-1/Fas)- and p53-independent apoptosis via activation of caspases in neuroectodermal tumors. Cancer Res 1997, 57:4956-4964 [PubMed] [Google Scholar]
- 50.Vrieling H: Mitotic maneuvers in the light. Nat Genet 2001, 28:101-102 [DOI] [PubMed] [Google Scholar]
- 51.Sanchez-Garcia I: Consequences of chromosomal abnormalities in tumor development. Annu Rev Genet 1997, 31:429-453 [DOI] [PubMed] [Google Scholar]
- 52.Matise MP, Joyner AL: Gli genes in development and cancer. Oncogene 1999, 18:7852-7859 [DOI] [PubMed] [Google Scholar]
- 53.Dahmane N, Sanchez P, Gitton Y, Palma V, Sun T, Beyna M, Weiner H, Ruiz I, Altaba A: The Sonic hedgehog-Gli pathway regulates dorsal brain growth and tumorigenesis. Development 2001, 128:5201-5212 [DOI] [PubMed] [Google Scholar]
- 54.Kraus JA, Koch A, Albrecht S, von Deimin A, Wiestler OD, Pietsch T: Loss of heterozygosity at locus F13B on chromosome 1q in human medulloblastoma. Int J Cancer 1996, 67:11-15 [DOI] [PubMed] [Google Scholar]
- 55.Reardon DA, Michalkiewicz E, Boyett JM, Sublett JE, Entrekin RE, Ragsdale ST, Valentine MB, Behm FG, Li H, Heideman RL, Kun LE, Shapiro DN, Look AT: Extensive genomic abnormalities in childhood medulloblastoma by comparative genomic hybridization. Cancer Res 1997, 57:4042-4047 [PubMed] [Google Scholar]
- 56.Pietsch T, Koch A, Wiestler OD: Molecular genetic studies in medulloblastomas: evidence for tumor suppressor genes at the chromosomal regions 1q31–32 and 17p13. Klin Padiatr 1997, 209:150-155 [DOI] [PubMed] [Google Scholar]