

Abstract
Forbidden CD4+ββ T cells, which produce interleukin (IL)-4 predominantly, are a pathological subset in the development of colitis in T-cell receptor α chain (TCRα)-deficient mice. Stimulation of naive CD4+ T cells with IL-4 induces Th2 development via the activation of signal transducers and activators of transcription (STAT) 6. In the present study, we had found that IL-4 enhanced the expression of STAT6 in CD4+ββ T cells isolated from TCRα−/− mice with colitis, suggesting that the IL-4 signal in the CD4+ββ T cells is mediated by STAT6. To further investigate the role of STAT6 in the development of colitis induced by TCRα deficiency, we generated double-deficient mice by crossing TCRα−/− mice and STAT6−/− mice. Surprisingly, STAT6 deficiency did not result in decreased severity of colitis in TCRα−/− mice. STAT6-deficient CD4+ββ T cells produced IL-4 and intraperitoneal injection of anti-IL-4 monoclonal antibody in the nondiseased TCRα−/− and STAT6 double-deficient mice prevented the colitis formation, thus indicating that the cells differentiated into the Th2 phenotype have the ability to mediate the development of the colitis in the absence of STAT6.
Human inflammatory bowel disease (IBD) consists of two distinctive types, ulcerative colitis and Crohn’s disease, on the basis of clinical and pathological features. 1-3 Distinct cytokine imbalance is considered important for the induction of these two types of disease. 4-6 Th1 cytokine [interferon-γ, interleukin (IL)-2, and tumor necrosis factor-α] seems to be predominant in Crohn’s disease, whereas Th2 cytokine (IL-4, IL-5, and IL-6) tends to be associated with ulcerative colitis. 7-9
Colitis spontaneously develops in various gene-manipulated murine models: T-cell receptor α chain-deficient (TCRα−/−) mice, IL-2-deficient mice, IL-7-transgenic mice, IL-10-deficient mice, macrophage-specific signal transducers and activators of transcription 3 (STAT3)-deficient mice, Gαi2-deficient mice, and severe combined immunodeficient mice restored with CD45RBhiCD4+ T cells. 3,10-16 Most of these rodent IBD models exhibit disruption of T-cell regulatory networks, with cytokine imbalance of pathogenic CD4+ T cells predominantly shifted to the Th1 type. In contrast, the Th2 pathway is a major causative agent for the development of colitis in TCRα−/− mice, 17-20 hapten-induced murine colitis, 21 and oxazolone-induced colitis. 22
TCRα−/− mice spontaneously develop chronic colitis under specific pathogen-free conditions at ∼10 weeks of age. 18 We have previously shown that CD4+ββ T cells, which produce predominantly IL-4, are increased in the colons of TCRα−/− mice with the disease. 17,18,23 Furthermore, administration of a monoclonal antibody (mAb) against IL-4 suppressed the onset of IBD in TCRα−/− mice. 19 It has been reported also that TCRα−/− × IL-4−/− double-mutant mice have a decreased propensity to develop IBD. 20 Thus, Th2-driven, IL-4-producing CD4+ββ T cells seem to be a fundamental pathological element for the induction of colitis in TCRα−/− mice.
STAT6 are members of the STAT family of proteins, which have been cloned and characterized as IL-4-activated transcription factors. 24,25 The binding of IL-4 to its receptor leads to the tyrosine phosphorylation of STAT6 by JAK1 and JAK3. 26 The phosphorylated STAT6 then forms homodimers, translocates to the nucleus, and binds to the promoter regions of IL-4-responsive genes to initiate the gene expression. 26 From analysis of mice lacking the STAT6 gene it has been concluded that the signal transducer is essential for the biological functions of IL-4, including the development of Th2 cells from naive CD4+ T cells, the class switching of B cells to the production of IgE and IgG1, induction of antigen-dependent airway hyperresponsiveness, and IL-4-mediated up-regulation of cell-surface molecules such as MHC class II and CD23. 24,25 These observations indicate that STAT6 is an essential signal transduction molecule for IL-4 signaling in T lymphocytes. 24,25
To investigate the role of STAT6 in the development of IBD mediated by Th2-type pathological CD4+ββ T cells, we crossed TCRα−/− mice with STAT6−/− mice. Surprisingly, we found that the TCRα−/− × STAT6−/− mice spontaneously developed IBD, with characteristic clinical and histopathological abnormalities. STAT6-deficient CD4+ββ T cells isolated from the diseased double-knockout mice retained the ability to produce IL-4, suggesting that the cells possessed the typical Th2-type phenotype. In addition, anti-IL-4 antibody (Ab) treatment in double-deficient mice prevented the development of colitis indicating that the STAT6-independent IL-4-signaling pathway is critical for the pathogenesis.
Materials and Methods
Mice
TCRα−/− mice, with a background of C57Bl/6, were obtained from the Jackson Laboratory (Bar Harbor, ME). TCRα and STAT6 double-knockout mice were generated by backcrossing TCRα−/− mice into STAT6−/− mice with a background of C57Bl/6. 25 The animals were maintained in the Experimental Animal Facility at the Research Institute for Microbial Diseases, Osaka University, under specific pathogen-free conditions and given sterilized food and autoclaved distilled water ad libitum. To determine the genotype of the mice, polymerase chain reaction (PCR) analysis was performed by using tail DNA. 25,27
Anti-IL-4 mAb Treatment
In this study, a standard protocol was used for mAb in vivo treatment. 18,19 TCRα−/− × STAT6−/− mice from the beginning of 4 weeks to 10 weeks of age were intraperitoneally injected with rat anti-mouse IL-4 mAb (1 mg/mouse) prepared from hybridoma (11B11; American Type Culture Collection, Manassas, VA) or rat IgG1 (R3-34, 1 mg/mouse; PharMingen, San Diego, CA) as mock Ab in 100 μl of phosphate-buffered saline (PBS) twice a week. These treatments did not induce any signs of serum sickness.
Histopathological Analysis
For the assessment of the severity of IBD, the colons of TCRα−/− × STAT6−/− mice and TCRα−/− mice were examined histologically. Tissue samples obtained from the proximal, middle, and distal colon were fixed in 4% paraformaldehyde in PBS for 4 hours, embedded in paraffin, and sectioned at a thickness of 6 μm. 19 The tissue sections were stained with hematoxylin and eosin. The histopathological score shown in Table 1 ▶ was used for the assessment of the severity of the colitis. It is a modification of the scoring system described previously. 21,28 The scoring was performed blindly by two independent investigators (YO and YK). The thickness of the colonic mucosa was determined to compare with the mucosal layer of the equivalent colonic segment in normal C57Bl/6 mice using a micrometer. The total histological score represents the sum of each item score.
Table 1.
Histological Scoring System (Colon)
| Histological changes | Score | |||
|---|---|---|---|---|
| 0 | 1 | 2 | 3 | |
| Loss of goblet cells | None | A few | Numerous and focal | Numerous and diffuse |
| Crypt abscesses | 0 | 1–4 | >5 | - |
| Epithelial erosion | None | Focal | Diffuse | - |
| Hyperemia | None | A few | Numerous and focal | Numerous and diffuse |
| Cellular infiltration in the lamina propria | None | A few | Numerous and focal | Numerous and diffuse |
| Thickness of colonic mucosa | <100% | 101–140% | 141–180% | >181% |
| Increase of the colonic gland | None | A few | Numerous and focal | Numerous and diffuse |
Preparation of Cell Suspensions
Mice anesthetized with ketamine (Sigma Chemical Co., St. Louis, MO) were sacrificed at 10 weeks age. The spleens and mesenteric lymph nodes (MLNs) were aseptically removed, and single-cell suspensions were prepared by a standard mechanical procedure. 18,29,30 Mononuclear cells from the lamina propria (LP) of the colon were dissociated using type IV collagenase (Sigma) to obtain single-cell preparations as described. 18,29,30
Flow Cytometric Analysis and Cell Sorting
For analysis of the distribution of CD4+ββ T cells by flow cytometry, single-cell suspensions of the mononuclear cells (106/sample) prepared from various tissues were stained with optimal concentrations of phycoerythrin-conjugated anti-CD4 mAb (L3T4) and fluorescein isothiocyanate (FITC)-conjugated anti-TCR-β mAb (H57-597). The samples were then subjected to flow cytometric analysis by using a FACScan (Becton Dickinson, Mountain View, CA). Data were analyzed by using CellQuest software (Becton Dickinson). For the analysis of the cytokine mRNA expression by the CD4+ββ T cells, the cells were purified by FACS Vantage (Becton Dickinson) as described previously. 19
Quantitative Reverse-Transcriptase-PCR
To analyze the cytokine-specific mRNA expression by CD4+ββ T cells isolated from colonic LP of the diseased TCRα−/− × STAT6−/− mice, a highly sensitive, quantitative RT-PCR was performed. 31-33 Total RNA was isolated from fluorescence-activated cell sorting-purified CD4+ββ T cells by using TRIzol reagent (Invitrogen, Carlsbad, CA). The RNA was reverse-transcribed into cDNA using Superscript II reverse transcriptase (Invitrogen), RNase inhibitor (Toyobo, Tokyo, Japan), oligo(dT)12-18 primer (Invitrogen), and dNTPs (Amersham Pharmacia Biotech, Arlington Heights, IL). The mixture was incubated at 42°C for 120 minutes and heated to 90°C for 5 minutes. 34 After treatment with RNase H (Toyobo), the synthesized cDNA was extracted by phenol/chloroform. Then, the cytokine-specific cDNA was quantified with LightCycler (Roche Diagnostics, Mannheim, Germany) technology by using LightCycler-DNA Master Hybridization Probes (Roche Diagnostics). For the amplification of cDNA, 20 μl of PCR mix was added to each tube to give a final concentration of 0.05 μmol/L 5′ primer, 0.05 μmol/L 3′ primer, 0.2 μmol/L FITC-labeled probe, 0.2 μmol/L LightCycler Red 640-labeled probe, 2 mmol/L MgCl2, and 1× LightCycler-DNA master hybridization probes mix (Roche Diagnostics). The oligoprimers specific for the IL-4 (sense, 5′-ATGGGTCTCAACCCCCAGCTAGT-3′; anti-sense, 5′-GCTCTTTAGGCTTTCCAGGAAGTC-3′), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (sense, 5′-TTCACCACCATGGAGAAGGC-3′; anti-sense, 5′-GGCATGGACTGTGGTCATGA −3′) were used. 35,36 For detection of the target molecule, FITC-labeled hybrid probe and LightCycler Red 640 (LCR)-labeled hybrid probe to IL-4 (FITC, 5′-CGTTTGGCACATCCATCTCCGT-3′; LCR, 5′-CATGGCGTCCCTTCTCCTGTG −3′), and GAPDH (FITC, 5′-TGGGTGTGAACCACCAGAAATATGAC-3′; LCR, 5′-ACTCACTCAAGATTGTCAGCAATGCA-3′) were prepared according to instructions provided by the manufacturer. After heating at 94°C for 2 minutes, cDNA were amplified for 40 cycles, each cycle consisting of 95°C for 10 seconds, 55°C for 30 seconds, and 72°C for 30 seconds. 33 Once during the cycle in which the log-linear signal could be distinguished from background, it was possible to compare the target concentrations (external standard) in samples with an internal standard in the same samples. 33 After the PCR had been completed, the LightCycler software (Roche Diagnostics) automatically converted the raw data into copies of target molecules. In this study, the relative quantitative expression of cytokine-specific mRNA in each sample was expressed as the amount of cytokine mRNA divided by the amount of mRNA GAPDH. 37
Cytokine Enzyme-Linked Immunosorbent Assay
Purified CD4+ T cells were co-cultured with mAb anti-CD28 (2 μg/ml, 37.51; PharMingen) in anti-CD3 mAb (10 μg/ml, 145-2C11; PharMingen) precoated 96-well tissue culture plates (5 × 10 5 cells/well) for 60 hours. 38 Culture supernatants were then harvested for analysis of cytokine production by cytokine enzyme-linked immunosorbent assay kit (Amersham Pharmacia Biotech) for IL-4.
Western Blotting Analysis for the Expression of Phosphorylated STAT6
Purified CD4+ T cells isolated from MLNs of wild-type C57Bl/6 mice and TCRα−/− mice were stimulated in vitro with IL-4 (20 ng/ml) for 15 minutes, then lysed in cell lysis buffer (5% sodium dodecyl sulfate, 0.5 mol/L Tris-HCl, pH 6.8, 0.5 mol/L ethylenediaminetetraacetic acid, 1 mmol/L dithiothreitol, 1 mmol/L phenylmethyl sulfonyl fluoride, and 10 μg leupeptin) for 30 minutes at 4°C, and centrifuged at 10,000 rpm for 30 minutes. The supernatants were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to a nitrocellulose membrane, and probed with anti-phospho STAT6Ab (Phospho-STAT Antibody Sampler; Cell Signaling, Beverly, MA) and anti-STAT6Ab (Santa Cruz Biotechnology, Santa Cruz, CA). The membrane was finally developed by use of an enhanced chemiluminescence kit (ECL plus; Amersham Pharmacia Biotech).
Statistical Analysis
Data were statistically analyzed by Student’s two-tailed t-test or Pearson’s correlation coefficient test with P < 0.05 considered statistically significant.
Results
STAT6 Mediated the IL-4 Signaling in CD4+ββT Cells
It has been shown that IL-4-mediated differentiation of naive CD4+ T cells into Th2 cells is dependent on STAT6. 24,25 Thus, it was important to examine whether the STAT6-mediated signaling pathway was involved in the activation of IL-4-producing pathological CD4+ββ T cells clonally expanded in the diseased TCRα−/− mice. According to the method described previously, 18,19,23 the CD4+ββ T cell-enriched fraction was prepared by MACS separation. Western blotting analysis revealed that phosphorylated STAT6 was induced in MLN-derived ββ T cell-enriched CD4+ T cells after stimulation with IL-4, thus indicating that STAT6 was involved in the IL-4 signaling event in the cells (Figure 1) ▶ .
Figure 1.

Expression of phosphorylated STAT6 in CD4+ββ T cells from TCRα−/− mice and CD4+αβ T cells from wild-type C57Bl/6 mice. MACS-purified CD4+ T cells from MLNs were stimulated in vitro with recombinant IL-4 (20 ng/ml) for 15 minutes, and then phosphorylated STAT6 protein was examined by Western blotting analysis. Top, Anti-phospho STAT6 Ab; middle, anti-STAT6 Ab; bottom, Coomassie brilliant blue staining for the confirmation of that equivalent amounts of cellular protein were present. The data are representative from one of three independent experiments.
TCRα−/− × STAT6−/− Mice Developed Colitis
To assess the role of STAT6 in the development of colitis in the TCRα-deficient mice, we generated TCRα- and STAT6-double-mutant (TCRα−/− × STAT6−/−) mice. The expression of STAT6 protein was not detected in CD4+ββ T cells isolated from MLNs of TCRα−/− × STAT6−/− mice (Figure 1) ▶ . Unexpectedly, the TCRα−/− × STAT6−/− mice, like TCRα−/− mice, lost significant body weight from 11 weeks of age when compared with C57Bl/6 wild-type mice and STAT6−/− mice (Figure 2) ▶ . The average body weight at the end of observation was: wild-type mice, 26.42 ± 0.62 g; STAT6−/− mice, 26.19 ± 0.62 g; TCRα−/− mice, 21.17 ± 0.93 g; and TCRα−/− × STAT6−/− mice, 20.63 ± 0.73 g. TCRα−/− × STAT6−/− mice also displayed the physical changes typical of those in TCRα-deficient mice with colitis, ie, hunched posture, anorectal prolapse, and diarrhea. Some TCRα−/− × STAT6−/− mice had died after 8 to 10 weeks of age, and all were dead by 15 weeks. Histologically, colons of the TCRα−/− × STAT6−/− mice at age 10 weeks were thickened, eroded, and infiltrated with inflammatory mononuclear cells, just as in TCRα−/− mice with IBD (Figure 3A) ▶ . In contrast, the colons of STAT6−/− mice and wild-type mice had no evident inflammatory changes. We further compared the degree of histopathological changes present in the colonic tissues of TCRα−/− × STAT6−/− mice with those in other mice by using the histological scoring system as described (Table 1) ▶ . 21,28 The scores in TCRα−/− × STAT6−/− mice (8.7 ± 1.1) were comparable to those in TCRα−/− mice with colitis (9.4 ± 0.5) (Figure 3B) ▶ , whereas the scores in C57Bl/6 wild-type and STAT6−/− mice were both 0.
Figure 2.

Decreased body weight in TCRα−/− × STAT6−/− mice and TCRα−/− mice but not in STAT6−/− mice or wild-type C57Bl/6 mice. Body weight of the mice was measured weekly for 14 weeks. All of the mice used in this study have the same inbred C57Bl/6 background. Statistical analysis was performed using Student’s two-tailed t-tests to compare TCRα−/− × STAT6−/− mice, TCRα−/− mice, and STAT6−/− mice with C57Bl/6 mice. The results are expressed as mean ± SEM of three independent experiments (10 mice/group). *, P < 0.05.
Figure 3.
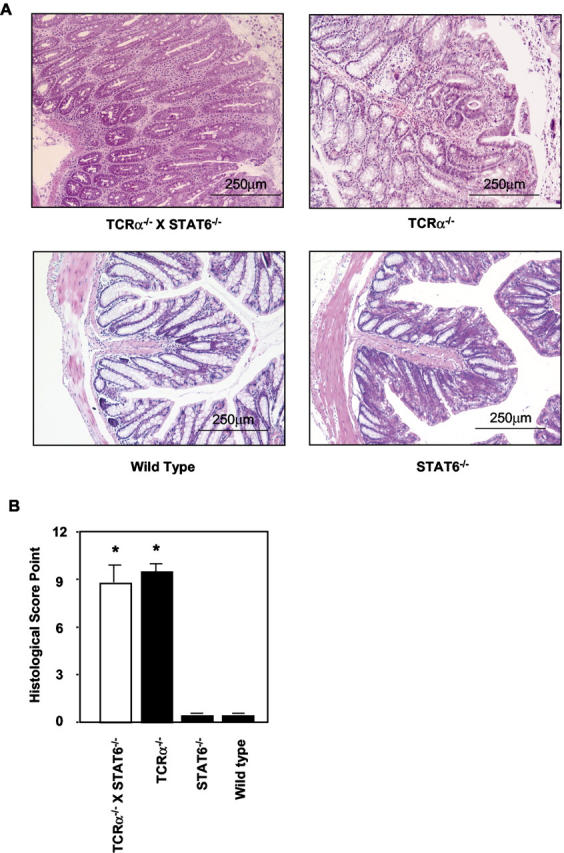
Histological analysis of colons isolated from TCRα−/− × STAT6−/− mice, TCRα−/− mice, STAT6−/− mice, and wild-type C57Bl/6 mice. All of the mice were sacrificed at 10 weeks of age. A: The tissue sections prepared from paraffin-embedded fixed colon were stained with H&E. B: The severity of the colitis was also examined by using the histological disease scoring system (Table 1) ▶ as described previously. 21,28 The histological score shown in each strain of mice (n = 7) was determined according to the diagnosis and grading of the colitis and was expressed as mean ± SD. Statistical analysis was performed for the comparison of TCRα−/− × STAT6−/− mice, TCRα−/− mice, and STAT6−/− mice. *, P < 0.05.
STAT6-Deficient CD4+ββT Cells Were Present in TCRα−/− × STAT6−/− Mice with Colitis
Because CD4+ββ T cells reportedly are crucial for the induction of IBD in TCRα−/− mice, 18,23,39 we next assessed the presence of this unique subset of T cells in TCRα−/− × STAT6−/− mice with IBD. When mononuclear cells isolated from the colonic LP and MLNs of the TCRα−/− × STAT6−/− mice with IBD were examined for the subset of CD4+ββ T cells, increased numbers of the cells were present (MLN, 1.89 ± 0.41%; colonic LP, 19.24 ± 3.90%). The degree of increase was similar to those in TCRα−/− mice with IBD [MLN, 1.67 ± 0.44%; colonic lamina propria lymphocytes (LPLs), 19.86 ± 2.22%] (Figure 4, A and B) ▶ .
Figure 4.
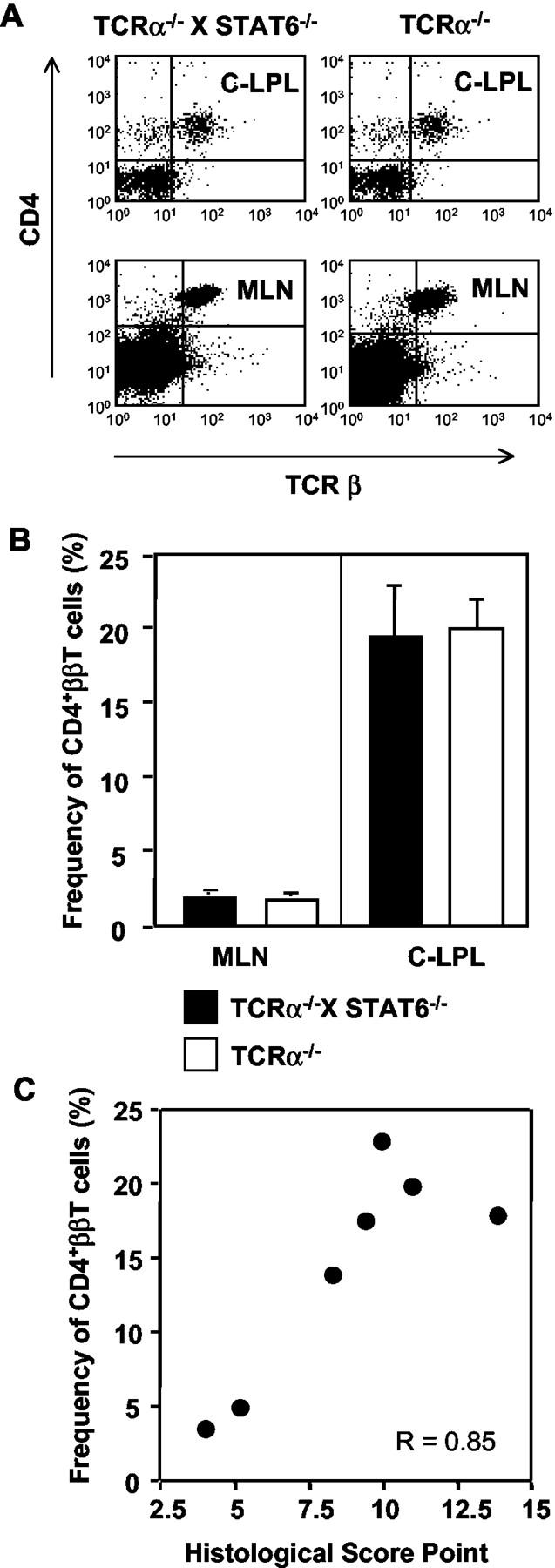
Flow cytometric analysis of CD4+ββ T cells in TCRα−/− × STAT6−/− mice with IBD. A: Lymphocytes were isolated from the colonic LP (C-LPLs) and MLNs of the double-mutant and TCRα−/− mice with IBD and co-stained with appropriate fluorescence-conjugated anti-CD4 (L3T4) and anti-TCR-β (H57-597) mAb for fluorescence-activated cell sorting analysis. B: The frequency of CD4+ββ T cells in the C-LPLs and MLNs of TCRα−/− × STAT6−/− mice (▪) and TCRα−/− mice (□) was calculated. C: The increase of CD4+ββ T cells and the disease score in each TCRα−/− × STAT6−/− mouse was correlated (P = 0. 016, r = 0.85, n = 7). The data are representative of three independent experiments.
It has been reported also that the severity of IBD in TCRα−/− mice correlates with the increase of colonic CD4+ββ T cells. 18 We observed a similar and significant correlation in the TCRα−/− × STAT6−/− mice with IBD when the correlation coefficient between histopathological changes and increase of CD4+ββ T cells was calculated (r = 0.85, P < 0.05) (Figure 4C) ▶ .
STAT6-Deficient Intestinal CD4+ββT Cells Produced IL-4 in the Double-Mutant Mice with Colitis
Th2-type pathological CD4+ββ T cells from TCRα−/− mice produce predominantly IL-4, 17,18 and the administration of mAb against IL-4 reportedly suppressed the onset of IBD in TCRα−/− mice. 19 Because CD4+ββ T cells were increased in the colons of our TCRα−/− × STAT6−/− colitic mice, we next investigated cytokine production by CD4+ββ T cells isolated from the mice. Total RNA isolated from purified pathogenic CD4+ββ T cells of colonic LP and MLNs was subjected to cytokine-specific quantitative RT-PCR. An equivalent or slightly increased amount of IL-4-specific mRNA was found in the cells of these diseased animals as compared with amounts expressed in conventional CD4+ββ T cells isolated from TCRα−/− mice with IBD (Figure 5A) ▶ . We also examined, by enzyme-linked immunosorbent assay, the levels of the cytokine in the culture supernatant of purified CD4+ T cells from TCRα−/− × STAT6−/− mice (Figure 5B) ▶ , and found levels similar to those produced by cells from TCRα−/− mice. Thus, even in the absence of STAT6, CD4+ββ T cells in the TCRα−/− × STAT6−/− mice with colitis produced much IL-4.
Figure 5.

IL-4 production by CD4+ββ T cells isolated from TCRα−/− × STAT6−/− mice with colitis. A: Relative quantity of cytokine-specific mRNA expression by CD4+ββ T cells in the mucosal compartment of TCRα−/− × STAT6−/− mice and TCRα−/− mice de novo. CD4+ββ T cells in the C-LPLs (▪) and MLNs (□) were purified by flow cytometry, and cytokine-specific mRNA expression was analyzed by quantitative RT-PCR. Cytokine-specific mRNA production was expressed as the amounts of relative quantity against GAPDH. (C-LPL: TCRα−/− × STAT6−/−, 5.39 ± 2.42; TCRα−/−, 4.16 ± 0.81; wild-type, 3.01 ± 1.08; MLN: TCRα−/− × STAT6−/−, 14.94 ± 5.72; TCRα−/−, 6.58 ± 1.53; wild-type, 0.75 ± 0.13). B: Cytokine production by MACS-purified CD4+ T cells isolated from TCRα−/− × STAT6−/− mice and TCRα−/− mice with IBD. The T lymphocytes from the C-LPLs (▪) and MLNs (□) were cultured in vitro with precoated anti-CD3 mAb in the presence of anti-CD28 mAb for 60 hours. Then culture supernatants were collected and cytokine production was analyzed by cytokine enzyme-linked immunosorbent assay. (C-LPL: TCRα−/− × STAT6−/−, 634.2 ± 47.6 pg/ml; TCRα−/−, 542.2 ± 51.4 pg/ml; wild-type, 219.5 ± 28.2 pg/ml; MLN: TCRα−/− × STAT6−/−, 682.6 ± 35.8 pg/ml; TCRα−/−, 784.0 ± 36.3 pg/ml; wild-type, 249.9 ± 16.5 pg/ml). The results are expressed as mean ± SEM of three independent experiments. *, P < 0.05.
Anti-IL-4 mAb Administration Suppressed the Onset of Colitis in TCRα−/− × STAT6−/− Mice
To clarify that IL-4 plays an essential role for the development of IBD in TCRα−/− × STAT6−/− mice, the double-mutant mice were treated intraperitoneally with anti-IL-4 mAb from the beginning of 4 weeks to 10 weeks of age. The mice treated with mock IgG developed IBD, including anorectal prolapse, diarrhea, hemorrhagic stool, and the weight loss. In contrast, the mice treated with anti-IL-4 mAb showed no sign for the development of IBD. Histological examination of the colon demonstrated anti-IL-4 mAb treatment prevented the formation of colonic inflammation (Figure 6A) ▶ . We further compared the degree of histopathological changes present in the colonic tissues of TCRα−/− × STAT6−/− mice with and without anti-IL-4 mAb by using the histological scoring system (Table 1) ▶ . The score in anti-IL-4 mAb treated mice (0.6 ± 0.4) was significantly less than that in mock IgG-treated mice (8.3 ± 1.2) (Figure 6B) ▶ . We next assessed the alteration of CD4+ββ T cells in TCRα−/− × STAT6−/− mice with and without the anti-IL-4 mAb treatment. A significant difference was not observed between anti-IL-4-treated (0.91 ± 0.20%) and untreated (or mock IgG treated, 1.78 ± 0.38%) groups in the frequency of CD4+ββ T cells in MLNs (P = 0.26). However, the frequency of the pathogenic CD4+ββ T cells was significantly decreased in the colon of double-knockout mice treated with anti-IL-4 mAb (anti-IL-4 mAb, 4.60 ± 0.87%; mock IgG, 18.59 ± 3.76%) (Figure 6C) ▶ . These findings indicated that IL-4 plays a critical role for the onset of IBD in TCRα−/− × STAT6−/− mice despite in the absence of STAT6.
Figure 6.
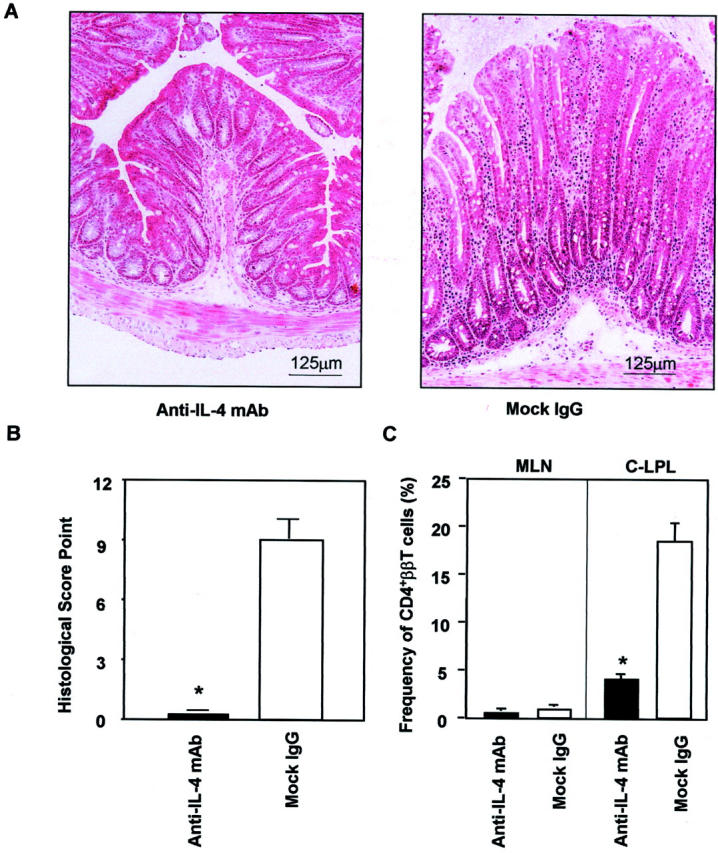
The suppressive effect of anti-IL-4 mAb treatment for the development of colitis in TCRα−/− × STAT6−/− mice. A: Histological analysis of the colon in TCRα−/− × STAT6−/− mice treated intraperitoneally with anti-IL-4 mAb or mock Ab (three mice/group). The treatment was initiated at 4 weeks of age and all of the mice were then sacrificed at 10 weeks of age. The tissue sections were prepared from colon and stained with H&E. B: The severity of the colitis was also determined in each group of mice (n = 3) by using the histological disease scoring system (Table 1) ▶ and was expressed as mean ± SD. The administration of anti-IL-4 Ab ameliorated the severity of the colitis. C: Flow cytometric analysis of CD4+ββ T cells in TCRα−/− × STAT6−/− mice with and without anti-IL-4 mAb. Lymphocytes were isolated from the C-LPLs and MLNs, then co-stained with anti-CD4 (L3T4) and anti-TCRβ (H57-597) mAbs. The results are expressed as mean ± SEM, *, P < 0.05.
Discussion
IL-4 produced by CD4+ββ T cells is critical for the development of IBD in TCRα−/− mice. 18-20 Previously we reported that the administration of anti-IL-4 antibody changed the pattern of cytokine production in CD4+ββ T cells from dominant Th2 to Th1 and resulted in reduced ability to induce IBD in TCRα−/− mice. 19 Others have reported that TCRα−/− and IL-4−/− double-knockout mice, but not TCRα−/− and interferon-γ−/− double-knockout mice, have a decreased frequency of IBD when compared to TCRα−/− mice. 20 STAT6 is a transcriptional molecule that is a constitutive ingredient for IL-4 functions; 24,25,40 in the absence of STAT6, IL-4 failed to activate naive T cells to differentiate into Th2 cells or to enhance the proliferation of differentiated Th2 cells. 24,25,40,41 Further, STAT6 was shown to be an essential transcriptional molecule for IL-4-driven Th2 differentiation and cell expansion in CD4+ T cells. 42 Also, in a murine model of allergy, the loss of STAT6 abrogated antigen-induced respiratory hyperresponsiveness. 43,44 Thus, it was logical to postulate that the removal of the STAT6-specific gene from the pathological CD4+ββ T cells would result in diminished development of colitis in TCRα−/− mice.
Contrary to this postulate, we found that TCRα−/− × STAT6−/− mice developed IBD similar in severity to that seen in TCRα−/− mice (Figures 2 and 3) ▶ ▶ . Because anti-IL-4 mAb treatment suppressed the induction of IBD in TCRα−/− × STAT6−/− mice, a STAT6-independent signaling pathway responsible for the induction of IL-4-producing CD4+ββ T cells in murine colitis was implied. Such a possibility seems reasonable especially because several additional molecules reportedly are critical for IL-4 signaling in Th2-type cells. 26,45 Ouyang and colleagues 46 reported that Th2 development is dependent on GATA-3 expression independent of IL-4 and STAT6, and activation of GATA-3 is a central player in Th2 differentiation. 46,47 GATA-3 has been shown to express naive T cells, 48 followed by a substantial increase during Th2 development with down-regulation of Th1 development. 49 Thus, GATA-3 can inhibit Th1 development by repressing IL-12Rβ expression. 49,50 Further, GATA-3 provides an instructive signal for the development of Th2 type cells, 47 and GATA-3 generates stability of Th2 commitment via the chromatin remodeling of Th2-specific cytokine loci. 51,52 Our preliminary results indicated that the levels of expression of GATA-3 were enhanced in CD4+ββ T cells purified from the diseased TCRα−/− × STAT6−/− mice, implying that GATA-3 is at least one alternative component responsible for Th2 development of CD4+ββ T cells in the double-knockout mice with colitis (data not shown). This point is now carefully addressed in a separate study.
In addition to GATA-3, STAT6-independent IL-4 signaling reportedly is mediated via several other molecules, such as phosphotyrosine-binding domain proteins; 26 insulin receptor substrate; 53 src homology 2 domain-containing α2 collagen-related protein (Shc); 54 IL-4 receptor-interacting protein FRIP; 55 and Th2-related transcription factors such as GATA-3 48,56 c-Maf, 57 NF-AT, 58 and BCL-6. A proto-oncogene, BCL-6, binds to the same DNA-binding motifs of STAT transcription factors as a transcriptional repressor. 59,60 The removal of the BCL-6 gene resulted in the overproduction of Th2 cells, leading to severe inflammation of the heart and lung 59,61 and creation of double-mutant mice in both BCL-6 and either STAT6 or IL-4 unexpectedly resulted in the development of lethal Th2-type inflammation. 62 These findings further support our present result in which pathological Th2 type CD4+ββ T cells developed in the absence of STAT6. Thus, there might be multiple redundant pathways for IL-4 production by CD4+ββ T cell, exhibiting equivalent effects on the differentiation of the unique CD4+ββ T cells into the Th2-biased phenotype.
STAT6 deficiency did not affect the ability of CD4+ββ T cells to secrete IL-4 and proliferate. These results imply that STAT6 is not an absolute requirement for Th2 differentiation of CD4+ββ T cells, although it has been reported that STAT6 is necessary and sufficient to mediate both IL-4-driven Th2 differentiation and cell expansion in naive CD4+ T cells in normal mice. 42 It should be noted that the concept of a primary role of IL-4 and STAT6 signaling in the induction of Th2-type cells has recently been put in doubt: in mice genetically deficient in IL-4, IL-4R, and STAT6, Th2-type cells and those associated immune responses emerged in the absence of the IL-4/IL-4R and STAT6 signaling cascade, 63-66 and classical IL-4- and IL-5-producing Th2-type cells developed in helminth-infected STAT6-deficient mice. 67 Further, repetitive anti-CD3 stimulation of memory type CD4+CD62Lhigh T cells from STAT6-deficient mice resulted in the generation of IL-4-producing Th2-type cells. 68 Moreover, the number of CD4+ββ T cells was not different in TCRα−/− × IL-4−/− and TCRα−/− mice with IBD. 20 Taken together, the previous and present findings strongly suggest that the STAT6 signaling pathway is not essential for the development of pathological Th2-type CD4+ββ T cells.
Our previous data also showed that anti-IL-4 treatment did not influence the number of CD4+ββ T cells in TCRα−/− mice. 19 Attempts to culture CD4+ββ T cells isolated from TCRα−/− mice failed to replicate these aberrant T cells under in vitro Th2-skewing-conditions (data not shown). Because IL-4 did not act as a trophic factor for CD4+ββ T cells it probably does not directly affect the development of CD4+ββ T cells. This implies that a unique environment exists in vivo that is capable of driving Th2 responses independent of IL-4-STAT6 signaling. Interestingly, CD4+ββ T cells were not detected in TCRα−/− × MHC class II−/− mice. 30 Therefore, CD4+ββ T cells may develop and expand in response to unidentified MHC class II-restricted antigens. TCRα−/− mice with IBD showed humoral immune responses to food, self antigens, 18 and intestinal bacteria, such as Bacteroides vulgatus. 38 Perhaps then CD4+ββ T cells exhibit Ag specificities against innocuous luminal antigens for the production of Th2-type cytokines, eventually leading to the development of IBD.
The results of the present study illustrate the complexity of mechanisms involved in the pathogenesis of colitis in TCRα-deficient condition. Further investigation of signaling molecules other than STAT6 involved in IL-4 signaling may clarify the molecular mechanism behind Th2-type colitis. Moreover, the TCRα−/− × STAT6−/− mouse strain is a useful animal model for investigating the mechanism of STAT6-independent Th2 differentiation of the unique pathogenic CD4+ββ T cells.
Acknowledgments
We thank Dr. William R. Brown (Denver VA Medical Center) for his helpful comments and editorial assistance.
Footnotes
Address reprint requests to Ichiro Takahashi, Ph.D., Department of Preventive Dentistry and Host Defense, Hiroshima University Graduate School of Biomedical Sciences, 1-2-3 Kasumi, Minami, Hiroshima 734-8553, Japan. E-mail: snatum@hiroshima-u.ac.jp.
Supported in part by grants-in-aid for scientific research from the Ministry of Education, Science, Sports, and Culture of Japan, and the Ministry of Health and Welfare of Japan.
References
- 1.Powrie F: T cells in inflammatory bowel disease: protective and pathogenic roles. Immunity 1995, 3:171-174 [DOI] [PubMed] [Google Scholar]
- 2.Blumberg RS, Saubermann L, Strober W: Animal models of mucosal inflammation and their relation to human inflammatory bowel disease. Curr Opin Immunol 1999, 11:648-656 [DOI] [PubMed] [Google Scholar]
- 3.Strober W, Ehrhardt RO: Chronic intestinal inflammation: an unexpected outcome in cytokine or T cell receptor mutant mice. Cell 1993, 75:203-205 [DOI] [PubMed] [Google Scholar]
- 4.Sartor RB: Cytokines in intestinal inflammation: pathophysiological and clinical considerations. Gastroenterology 1994, 106:533-539 [DOI] [PubMed] [Google Scholar]
- 5.Podolsky DK: Inflammatory bowel disease (1). N Engl J Med 1991, 325:928-937 [DOI] [PubMed] [Google Scholar]
- 6.Niessner M, Volk BA: Altered Th1/Th2 cytokine profiles in the intestinal mucosa of patients with inflammatory bowel disease as assessed by quantitative reversed transcribed polymerase chain reaction (RT-PCR). Clin Exp Immunol 1995, 101:428-435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.West GA, Matsuura T, Levine AD, Klein JS, Fiocchi C: Interleukine 4 in inflammatory bowel disease and mucosal immune reactivity. Gastroenterology 1996, 110:1683-1695 [DOI] [PubMed] [Google Scholar]
- 8.Fuss IJ, Neurath M, Boirivant M, Klein JS, de la Motte C, Strong SA, Fiocchi C, Strober W: Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-γ, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunol 1996, 157:1261-1270 [PubMed] [Google Scholar]
- 9.Parronchi P, Romagnani P, Annunziato F, Sampognaro S, Becchio A, Giannarini L, Maggi E, Pupilli C, Tonelli F, Romagnani S: Type 1 T-helper cell predominance and interleukin-12 expression in the gut of patients with Crohn’s disease. Am J Pathol 1997, 150:823-832 [PMC free article] [PubMed] [Google Scholar]
- 10.Mombaerts P, Mizoguchi E, Grusby MJ, Glimcher LH, Bhan AK, Tonegawa S: Spontaneous development of inflammatory bowel disease in T cell receptor mutant mice. Cell 1993, 75:274-282 [DOI] [PubMed] [Google Scholar]
- 11.Sadlack B, Merz H, Schorle H, Schimpl A, Feller AC, Horak I: Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell 1993, 75:253-261 [DOI] [PubMed] [Google Scholar]
- 12.Watanabe M, Ueno Y, Yajima T, Okamoto S, Hayashi T, Yamazaki M, Iwao Y, Ishii H, Habu S, Uehira M, Nishimoto H, Ishikawa H, Hata J, Hibi T: Interleukin 7 transgenic mice develop chronic colitis with decreased interleukin 7 protein accumulation in the colonic mucosa. J Exp Med 1998, 187:389-402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W: Interleukin-10-deficient mice develop chronic enterocolitis. Cell 1993, 75:263-274 [DOI] [PubMed] [Google Scholar]
- 14.Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, Forster I, Akira S: Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity 1999, 10:39-49 [DOI] [PubMed] [Google Scholar]
- 15.Rudolph U, Finegold MJ, Rich SS, Harriman GR, Srinivasan Y, Brabet P, Boulay G, Bradley A, Birnbaumer L: Ulcerative colitis and adenocarcinoma of the colon in Gαi2-deficient mice. Nat Genet 1995, 10:143-150 [DOI] [PubMed] [Google Scholar]
- 16.Morrissey PJ, Charrier K, Braddy S, Liggitt D, Watson JD: CD4+ T cells that express high levels of CD45RB induce wasting disease when transferred into congenic severe combined immunodeficient mice. Disease development is prevented by cotransfer of purified CD4+ T cells. J Exp Med 1993, 178:237-244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mizoguchi A, Mizoguchi E, Chiba C, Spiekermann GM, Tonegawa S, Nagler-Anderson C, Bhan AK: Cytokine imbalance and autoantibody production in T cell receptor-α mutant mice with inflammatory bowel disease. J Exp Med 1996, 183:847-856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takahashi I, Kiyono H, Hamada S: CD4+ T-cell population mediates development of inflammatory bowel disease in T-cell receptor α chain-deficient mice. Gastroenterology 1997, 112:1876-1886 [DOI] [PubMed] [Google Scholar]
- 19.Iijima H, Takahashi I, Kishi D, Kim JK, Kawano S, Hori M, Kiyono H: Alteration of interleukin 4 production results in the inhibition of T helper type 2 cell-dominated inflammatory bowel disease in T cell receptor α chain-deficient mice. J Exp Med 1999, 190:607-615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mizoguchi A, Mizoguchi E, Bhan AK: The critical role of interleukin 4 but not interferon gamma in the pathogenesis of colitis in T-cell receptor α mutant mice. Gastroenterology 1999, 116:320-326 [DOI] [PubMed] [Google Scholar]
- 21.Dohi T, Fujihashi K, Rennert PD, Iwatani K, Kiyono H, McGhee JR: Hapten-induced colitis is associated with colonic patch hypertrophy and T helper cell 2-type responses. J Exp Med 1999, 189:1169-1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boirivant M, Fuss IJ, Chu A, Strober W: Oxazolone colitis: a murine model of T helper cell type 2 colitis treatable with antibodies to interleukin 4. J Exp Med 1998, 188:1929-1939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takahashi I, Iijima H, Katashima R, Itakura M, Kiyono H: Clonal expansion of CD4+ TCRββ+ T cells in TCR α-chain-deficient mice by gut-derived antigens. J Immunol 1999, 162:1843-1850 [PubMed] [Google Scholar]
- 24.Takeda K, Tanaka T, Shi W, Matsumoto M, Minami M, Kashiwamura S, Nakanishi K, Yoshida N, Kishimoto T, Akira S: Essential role of Stat6 in IL-4 signalling. Nature 1996, 380:627-630 [DOI] [PubMed] [Google Scholar]
- 25.Takeda K, Kishimoto T, Akira S: STAT6: its role in interleukin 4-mediated biological functions. J Mol Med 1997, 75:317-326 [DOI] [PubMed] [Google Scholar]
- 26.Nelms K, Keegan AD, Zamorano J, Ryan JJ, Paul WE: The IL-4 receptor: signaling mechanisms and biologic functions. Annu Rev Immunol 1999, 17:701-738 [DOI] [PubMed] [Google Scholar]
- 27.Hughes DP, Hayday A, Craft JE, Owen MJ, Crispe IN: T cells with γ/δT cell receptors (TCR) of intestinal type are preferentially expanded in TCRα-deficient lpr mice. J Exp Med 1995, 182:233-241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Corozza N, Eichenberger S, Eugster HP, Mueller C: Nonlymphocyte-derived tumor necrosis factor is required for induction of colitis in recombination activating gene (RAG)2−/− mice upon transfer of CD4+CD45RBhi T cells. J Exp Med 1999, 190:1479-1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fujihashi K, Taguchi T, Aicher WK, McGhee JR, Bluestone JA, Eldridge JH, Kiyono H: Immunoregulatory functions for murine intraepithelial lymphocytes: γ/δT cell receptor-positive (TCR+) T cells abrogate oral tolerance, while α/β TCR+ T cells provide B cell help. J Exp Med 1992, 175:695-707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takahashi I, Kiyono H, Jackson RJ, Fujihashi K, Staats HF, Hamada S, Clements JD, Bost KL, McGhee JR: Epitope maps of the Escherichia coli heat-labile toxin B subunit for development of a synthetic oral vaccine. Infect Immun 1996, 64:1290-1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wittwer CT, Ririe KM, Andrew RV, David DA, Gundry RA, Balis UJ: The LightCycler: a microvolume multisample fluorimeter with rapid temperature control. BioTechniques 1997, 22:176-181 [DOI] [PubMed] [Google Scholar]
- 32.Wittwer CT, Herrmann MG, Moss AA, Rasmussen RP: Continuous fluorescence monitoring of rapid cycle DNA amplification. BioTechniques 1997, 22:130-148 [DOI] [PubMed] [Google Scholar]
- 33.Hiroi T, Yanagita M, Ohta N, Sakaue G, Kiyono H: IL-15 and IL-15 receptor selectively regulate differentiation of common mucosal immune system-independent B-1 cells for IgA responses. J Immunol 2000, 165:4329-4337 [DOI] [PubMed] [Google Scholar]
- 34.Yanagita M, Hiroi T, Kitagaki N, Hamada S, Ito H, Shimauchi H, Murakami S, Okada H, Kiyono H: Nasopharyngeal-associated lymphoreticular tissue (NALT) immunity: fimbriae-specific Th1 and Th2 cell-regulated IgA responses for the inhibition of bacterial attachment to epithelial cells and subsequent inflammatory cytokine production. J Immunol 1999, 162:3559-3565 [PubMed] [Google Scholar]
- 35.Hiroi T, Fujihashi K, McGhee JR, Kiyono H: Polarized Th2 cytokine expression by both mucosal γδ and αβ T cells. Eur J Immunol 1995, 25:2743-2751 [DOI] [PubMed] [Google Scholar]
- 36.Overbergh L, Valckx D, Waer M, Mathieu C: Quantification of murine cytokine mRNAs using real time quantitative reverse transcriptase PCR. Cytokine 1999, 11:305-312 [DOI] [PubMed] [Google Scholar]
- 37.Yoneyama H, Harada A, Imai T, Baba M, Yoshie O, Zhang Y, Higashi H, Murai M, Asakura H, Matsushima K: Pivotal role of TARC, a CC chemokine, in bacteria-induced fulminant hepatic failure in mice. J Clin Invest 1998, 102:1933-1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kishi D, Takahashi I, Kai Y, Tamagawa H, Iijima H, Obunai S, Nezu R, Ito T, Matsuda H, Kiyono H: Alteration of Vβ usage and cytokine production of CD4+ TCR ββ homodimer T cells by elimination of Bacteroides vulgatus prevents colitis in TCR α-chain-deficient mice. J Immunol 2000, 165:5891-5899 [DOI] [PubMed] [Google Scholar]
- 39.Takahashi I, Iijima H, Kishi D, Kiyono H: Oligoclonal Th2-biased ββ T cells induce murine inflammatory bowel disease. Immunol Res 1999, 20:237-242 [DOI] [PubMed] [Google Scholar]
- 40.Kaplan MH, Schindler U, Smiley ST, Grusby MJ: STAT6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity 1996, 4:313-319 [DOI] [PubMed] [Google Scholar]
- 41.Kaplan MH, Wurster AL, Smiley ST, Grusby MJ: Stat6-dependent and -independent pathways for IL-4 production. J Immunol 1999, 163:6536-6540 [PubMed] [Google Scholar]
- 42.Zhu J, Guo L, Watson CJ, Hu-Li J, Paul WE: Stat6 is necessary and sufficient for IL-4’s role in Th2 differentiation and cell expansion. J Immunol 2001, 166:7276-7281 [DOI] [PubMed] [Google Scholar]
- 43.Tomkinson A, Kanehiro A, Rabinovitch N, Joetham A, Cieslewicz G, Gelfand EW: The failure of STAT6-deficient mice to develop airway eosinophilia and airway hyperresponsiveness is overcome by interleukin-5. Am J Respir Crit Care Med 1999, 160:1283-1291 [DOI] [PubMed] [Google Scholar]
- 44.Miyata S, Matsuyama T, Kodama T, Nishioka Y, Kuribayashi K, Takeda K, Akira S, Sugita M: STAT6 deficiency in a mouse model of allergen-induced airways inflammation abolishes eosinophilia but induces infiltration of CD8+ T cells. Clin Exp Allergy 1999, 29:114-123 [DOI] [PubMed] [Google Scholar]
- 45.Murphy KM, Ouyang W, Farrar JD, Yang J, Ranganath S, Asnagli H, Afkarian M, Murphy TL: Signaling and transcription in T helper development. Annu Rev Immunol 2000, 18:451-494 [DOI] [PubMed] [Google Scholar]
- 46.Ouyang W, Lohning M, Gao Z, Assenmacher M, Ranganath S, Radbruch A, Murphy KM: Stat6-independent GATA-3 autoactivation directs IL-4-independent Th2 development and commitment. Immunity 2000, 12:27-37 [DOI] [PubMed] [Google Scholar]
- 47.Farrar JD, Ouyang W, Lohning M, Assenmacher M, Radbruch A, Kanagawa O, Murphy KM: An instructive component in T helper cell type 2 (Th2) development mediated by GATA-3. J Exp Med 2001, 193:643-649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zheng W, Flavell RA: The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell 1997, 89:587-596 [DOI] [PubMed] [Google Scholar]
- 49.Ouyang W, Ranganath SH, Weindel K, Bhattacharya D, Murphy TL, Sha WC, Murphy KM: Inhibition of Th1 development mediated by GATA-3 through an IL-4-independent mechanism. Immunity 1998, 9:745-755 [DOI] [PubMed] [Google Scholar]
- 50.Ferber IA, Lee HJ, Zonin F, Heath V, Mui A, Arai N, O’Garra A: GATA-3 significantly downregulates IFN-γ production from developing Th1 cells in addition to inducing IL-4 and IL-5 levels. Clin Immunol 1999, 91:134-144 [DOI] [PubMed] [Google Scholar]
- 51.Bird JJ, Brown DR, Mullen AC, Moskowitz NH, Mahowald MA, Sider JR, Gajewski TF, Wang CR, Reiner SL: Helper T cell differentiation is controlled by the cell cycle. Immunity 1998, 9:229-237 [DOI] [PubMed] [Google Scholar]
- 52.Agarwal S, Rao A: Modulation of chromatin structure regulates cytokine gene expression during T cell differentiation. Immunity 1998, 9:765-775 [DOI] [PubMed] [Google Scholar]
- 53.Sun XJ, Wang LM, Zhang Y, Yenush L, Myers MGJ, Jr, Glasheen E, Lane WS, Pierce JH, White MF: Role of IRS-2 in insulin and cytokine signaling. Nature 1995, 377:173-177 [DOI] [PubMed] [Google Scholar]
- 54.Wery S, Letourneur M, Bertoglio J, Pierre J: Interleukin-4 induces activation of mitogen-activated protein kinase and phosphorylation of shc in human keratinocytes. J Biol Chem 1996, 271:8529-8532 [DOI] [PubMed] [Google Scholar]
- 55.Nelms K, Snow AL, Hu-Li J, Paul WE: FRIP, a hematopoietic cell-specific rasGAP-interacting protein phosphorylated in response to cytokine stimulation. Immunity 1998, 9:13-24 [DOI] [PubMed] [Google Scholar]
- 56.Zhang DH, Yang L, Ray A: Differential responsiveness of the IL-5 and IL-4 genes to transcription factor GATA-3. J Immunol 1998, 161:3817-3821 [PubMed] [Google Scholar]
- 57.Ho IC, Hodge MR, Rooney JW, Glimcher LH: The proto-oncogene c-maf is responsible for tissue-specific expression of interleukin-4. Cell 1996, 85:973-983 [DOI] [PubMed] [Google Scholar]
- 58.Rincon M, Flavell RA: Transcription mediated by NFAT is highly inducible in effector CD4+ T helper 2 (Th2) cells but not in Th1 cells. Mol Cell Biol 1997, 17:1522-1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dent AL, Shaffer AL, Yu X, Allman D, Staudt LM: Control of inflammation, cytokine expression, and germinal center formation by BCL-6. Science 1997, 276:589-592 [DOI] [PubMed] [Google Scholar]
- 60.Hartatik T, Okada S, Okabe S, Arima M, Hatano M, Tokuhisa T: Binding of BAZF and Bc16 to STAT6-binding DNA sequences. Biochem Biophys Res Commun 2001, 284:26-32 [DOI] [PubMed] [Google Scholar]
- 61.Ye BH, Cattoretti G, Shen Q, Zhang J, Hawe N, de Waard R, Leung C, Nouri-Shirazi M, Orazi A, Chaganti RS, Rothman P, Stall AM, Pandolfi PP, Dalla-Favera R: The BCL-6 proto-oncogene controls germinal-centre formation and Th2-type inflammation. Nat Genet 1997, 16:161-170 [DOI] [PubMed] [Google Scholar]
- 62.Dent AL, Hu-Li J, Paul WE, Staudt LM: T helper type 2 inflammatory disease in the absence of interleukin 4 and transcription factor STAT6. Proc Natl Acad Sci USA 1998, 95:13823-13828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hogan SP, Matthaei KI, Young JM, Koskinen A, Young IG, Foster PS: A novel T cell-regulated mechanism modulating allergen-induced airways hyperreactivity in BALB/c mice independently of IL-4 and IL-5. J Immunol 1998, 161:1501-1509 [PubMed] [Google Scholar]
- 64.Pearce EJ, Cheever A, Leonard S, Covalesky M, Fernandez-Botran R, Kohler G, Kopf M: Schistosoma mansoni in IL-4-deficient mice. Int Immunol 1996, 8:435-444 [DOI] [PubMed] [Google Scholar]
- 65.Brewer JM, Conacher M, Satoskar A, Bluethmann H, Alexander J: In interleukin-4-deficient mice, alum not only generates T helper 1 responses equivalent to Freund’s complete adjuvant, but continues to induce T helper 2 cytokine production. Eur J Immunol 1996, 26:2062-2066 [DOI] [PubMed] [Google Scholar]
- 66.Noben-Trauth N, Shultz LD, Brombacher F, Urban JFJ, Gu H, Paul WE: An interleukin 4 (IL-4)-independent pathway for CD4+ T cell IL-4 production is revealed in IL-4 receptor-deficient mice. Proc Natl Acad Sci USA 1997, 94:10838-10843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kaplan MH, Whitfield JR, Boros DL, Grusby MJ: Th2 cells are required for the Schistosoma mansoni egg-induced granulomatous response. J Immunol 1998, 160:1850-1856 [PubMed] [Google Scholar]
- 68.Jankovic D, Kullberg MC, Noben-Trauth N, Caspar P, Paul WE, Sher A: Single cell analysis reveals that IL-4 receptor/Stat6 signaling is not required for the in vivo or in vitro development of CD4+ lymphocytes with a Th2 cytokine profile. J Immunol 2000, 164:3047-3055 [DOI] [PubMed] [Google Scholar]