

Abstract
The effect of anti-vascular agents on the growth of experimental tumors is well studied. Their impact on tumor vasculature, the primary therapeutic target of these agents, is not as well characterized, even though this primarily determines treatment outcome. Hypothesizing that the response of vessels to therapy is influenced by their stage of maturation, we studied vascular development and the vascular effects of therapy in several transplanted murine tumor models. Based on size, perfusion, endothelial cell (EC) proliferation, and the presence of pericytes, tumor vessels segregated into three categories. Least mature were highly proliferative, nonperfused EC sprouts emanating from functional vessels. Intermediate were small, perfused vessels which, like the angiogenic sprouts, were not covered by pericytes. Most mature were larger vessels, which were predominantly pericyte-covered with quiescent ECs and few associated sprouts. Thus, a developmental order, similar to that described during physiological neovascularization, was evident among vessels in growing tumors. This order markedly influenced tumor vessel response to anti-vascular therapy with recombinant interleukin-12. Therapy reduced tumor vessel density, which was attributable to a decrease in angiogenic sprouts and induction of EC apoptosis in pericyte-negative vessels. Although the great majority of vessels in growing tumors lacked pericyte coverage, selective loss of less mature vessels with therapy significantly increased the fraction of pericyte-positive vessels after therapy. These data indicate that the therapeutic susceptibility of tumor vasculature to recombinant murine IL-12 and, potentially, other anti-vascular agents is limited by its level of maturation. An implication is that tumor susceptibility is similarly limited, making pericyte coverage of tumor vasculature a potential indicator of tumor responsiveness.
Inhibition of tumor angiogenesis is a recent approach to cancer treatment that has been shown to be effective in many tumor models. 1,2 Unlike other forms of cancer therapy, anti-angiogenic strategies do not target the tumor cell directly. Rather, they target the endothelial cells (ECs) comprising the tumor vasculature, and control of tumor growth is a derivative effect resulting from reduced delivery of oxygen and nutrients 3 and, possibly, decreased production of tumor cell growth and survival factors by the vasculature 4,5 as well as other mechanisms. This strategy has several conceptual advantages, one of which is that the tumor vasculature, derived from normal host cells rather than mutable tumor cells, is less likely to develop therapeutic resistance.
As originally conceived, anti-angiogenesis therapy would inhibit the formation of new blood vessels without affecting existing vessels. 4 However, the dramatic tumor regression observed with some angiogenesis inhibitors 6 implies that they may also affect existing tumor vessels and, indeed, studies have demonstrated that anti-angiogenic therapy induces tumor EC apoptosis, even when tumor growth is slowed but not stopped. 7 Our own studies of murine tumors treated with the angiogenesis inhibitor recombinant murine interleukin-12 (rmIL-12) showed that control of tumor growth is associated with induction of tumor ischemia and ischemic cell death. 3 The reduction in tumor vascularity observed during treatment resulted predominantly from loss of small-caliber vessels, 8 which suggested that the pattern of vessel loss was not essentially stochastic. Rather, it seemed likely to be related to the process by which tumor vessels develop and mature.
The developmental process underlying new blood vessel formation and maturation has been well-studied, although it remains incompletely understood. 9 Neovascularization in adult life is characterized by angiogenesis, a process in which new vessels are formed by cells derived from nearby pre-existing vessels. Early studies of angiogenesis describe an initial phase of EC mitosis associated with expansion of the existing vascular network and formation of new capillary sprouts. 10,11 Subsequently, newly-formed vessels become invested with pericytes (also known as periendothelial cells or Rouget cells), which coincides with cessation of the angiogenic response. 12 The developmental progression of angiogenesis has been visualized best in the postnatal rodent retina. 13,14 Here, sprouting angiogenesis produces an expanding endothelial plexus during the first 2 weeks of postnatal development. A programmed lag in pericyte recruitment allows the nascent vasculature to undergo extensive remodeling, with pericyte investment serving later to preserve the vasculature in its final configuration and provide resistance to regression. The importance of this endothelial-pericyte interaction and vessel maturation process is shown by transgenic mice lacking platelet-derived growth factor B, in which embryonic lethality is associated with pericyte deficiency and capillary rupture. 15
We postulated that vascular development in tumors would essentially parallel the process in normal tissues and give rise to a diversity of vessels with respect to age and maturation. Understanding this process in the murine tumors commonly used in preclinical studies would lead to a better appreciation of the primary therapeutic target of anti-vascular agents that, in turn, might reveal their therapeutic potential and limitations. Accordingly, in this study we characterized the development and maturation of blood vessels in transplanted murine tumors and in these tumors after they were subjected to anti-vascular therapy. Our results indicate that vessels develop in transplanted murine tumors in an ordered process with features that can be identified by immunostaining of conventional histological sections. The level of vessel maturation is strongly correlated with their response to anti-vascular therapy, which leads us to predict that the maturation profile of vessels within tumors will influence or determine vascular and tumor response to anti-vascular agents.
Materials and Methods
Mice and Cell Lines
C57Bl/6 and BALB/c mice were purchased from Jackson Laboratories (Bar Harbor, ME), and C3H/HeN mice were purchased from Harlan Sprague-Dawley (Indianapolis, IN). All mice were 6- to 8-week-old females maintained in microisolator cages under sterile conditions. The K1735 16 and B16F10 17 murine melanoma cell lines (syngeneic with C3H/HeN and C57Bl/6 strains, respectively), along with the RENCA renal cell carcinoma line 18 were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum and penicillin/streptomycin. All three cell lines were tested and demonstrated to be mycoplasma-free.
Tumor Studies
For tumor growth studies, 10 6 tumor cells were injected subcutaneously into the lower left flank of syngeneic mice. Injected cells were derived from low-passage frozen stocks that had been established in culture less than 1 week before injection. When established tumors reached a diameter of 1 to 2 mm, rmIL-12 or phosphate-buffered saline (PBS) vehicle was administered intraperitoneally at or under the maximum tolerated dose for each strain on a five dose per week schedule (five daily injections followed by 2 days of rest) for up to 3 weeks. C3H/HeN mice received their maximum tolerated dose of 125 ng per injection, and C57Bl/6 and BALB/c mice received 500 ng per injection. Tumors were measured bidirectionally by calipers at regular intervals, and tumor volume was calculated using the formula for approximating the volume of a spheroid [0.52 × (width) 2 × (length)]. Mice were euthanized according to guidelines established by the Institutional Animal Care and Use Committee.
Immunohistochemistry
Four-μm sections from formalin-fixed, paraffin-embedded tumors were stained for ECs and pericytes using anti-von Willebrand factor (vWF; DAKO, Carpinteria, CA) and anti-α-smooth muscle actin (SMA; DAKO) antibodies, respectively. Deparaffinized sections underwent antigen retrieval by incubation in 0.08% pronase (Roche, Indianapolis, IN) followed by incubation in PBS and 10% goat serum. Anti-vWF antibody diluted 1:800 was added for 2 hours followed by ALEXA488-conjugated goat anti-rabbit Ig antibody (Molecular Probes, Eugene, OR) diluted 1:200 for 1 hour. Blocking of endogenous mouse tissue immunoglobulin was achieved using the Mouse-On-Mouse kit (Vector Laboratories, Burlingame, CA). Anti-SMA antibody was applied at 1:60 dilution for 60 minutes, followed by Texas Red-conjugated goat anti-mouse Ig antibody (Molecular Probes) for 1 hour. Proliferating cells were detected by staining with a peroxidase-conjugated antibody against proliferating cell nuclear antigen (PCNA, DAKO) along with a biotinyl tyramide amplification kit (New England Nuclear, Boston, MA) and 3-amino-9-ethylcarbonazole (AEC) substrate (Vector Laboratories). Apoptotic cells were detected using the Apoptag indirect fluorescein detection kit (Intergen, Purchase, NY). Three-color staining for ECs, pericyte, and proliferating/apoptotic nuclei was performed on the same sections. For Hoechst dye perfusion studies, 200 μl of 10 mmol/L Hoechst 33342 (Sigma-Aldrich, St. Louis, MO) in PBS was administered intravenously 5 minutes before tumor excision. Excised tumors were then embedded in OCT compound (Sakura Finetek, Torrance, CA), frozen in liquid nitrogen, and cut with a cryostat. Ten-μm sections were fixed in 4% paraformaldehyde in PBS (pH 7.4) and stained for ECs as described below. All histological specimens were viewed under a Microphot-FX light microscope (Nikon, Melville, NY) equipped with a Photometrics Coolsnap digital camera and image acquisition software (Roper Scientific, Trenton, NJ).
Confocal Microscopy of Tumor Vasculature
Tumor-bearing mice were injected with 150 μl of 2 mg/ml fluorescein isothiocyanate-conjugated tomato (Lycopersicon esculentum) lectin (Vector Laboratories) in PBS intravenously into the tail vein. Ten minutes later, the mice were anesthetized and then perfused with 4% paraformaldehyde in PBS (pH 7.4) intracardially. For confocal imaging of the vasculature alone, tumors were excised and sectioned essentially as described previously. 8 For histological staining of thick tumor sections, tumors were embedded in OCT compound, frozen, and cut with a cryostat. Thick (60 μm) tumor sections were stained for the endothelial marker CD31 (PECAM) and SMA according to a method adopted from Hashizume and colleagues. 19 Sections were blocked with Mouse-on-Mouse reagent and incubated overnight at room temperature in anti-SMA antibody diluted 1:200 in PBS and 0.3% Triton X-100, washed, and then incubated in Cy5-conjugated goat anti-mouse Ig (Amersham Pharmacia, Piscataway, NJ). Subsequently, sections were incubated overnight in rat anti-mouse PECAM antibody (Pharmingen, San Diego, CA) diluted 1:500, washed, and incubated in Texas Red anti-rat Ig (Molecular Probes) diluted 1:200 for 3 hours.
All thick sections were imaged using an upright Nikon (Augusta, GA) E-600 Eclipse microscope equipped with a Bio-Rad (Hercules, CA) 1024-ES confocal system. Fluorescein isothiocyanate, Texas Red, and Cy5 fluorescence were detected by a three-line, 15 mW argon-krypton laser system (American Laser, Fraser, MI). For each image set acquired, individual fluorescence channel data were saved along with composite data sets indicating regions of fluorescence overlap. Image stacks were integrated to create a composite maximum intensity projection of tumor vasculature imaged in three dimensions.
Image Analysis
For microvessel density measurements, slides were scanned at low power (×40) to identify areas of highest vascularity. Ten to 20 high-powered (×200) fields were then selected randomly within these areas, and microvessel densities were calculated based on the number of vWF-positive structures. In addition, vessel lumen cross-sectional areas were determined for all counted vessels automatically (ImageTool; University of Texas, San Antonio, TX) based on spatial calibration parameters established with a slide micrometer. Microvessel counting was performed by multiple blinded observers in conjunction with a pathologist. A pericyte-positive vessel was defined as a vWF-positive vessel surrounded by at least one cell staining positive for α-SMA. Proliferating and apoptotic vessels were defined as vessels in which at least one vWF-positive cell contained a nucleus that stained positive for PCNA and terminal dUTP nick-end labeling (TUNEL), respectively. Two to three sections were examined per tumor, and at least five tumors were examined per treatment group.
Statistical Analysis
Assessment of statistical significance was performed either by Student’s t-test (for normally distributed data sets) or Mann-Whitney U-test (for nonnormally distributed data sets). Correlation coefficients were derived from Pearson’s correlation method. All statistical analysis was performed using Instat software for the Macintosh version 2.0 (Graphpad Software, Philadelphia, PA).
Results
Heterogeneity among Tumor Vessels
We began by examining blood vessels in untreated K1735 tumors. Examination of thin (4 μm) tumor sections stained with anti-vWF antibody (Figure 1A) ▶ revealed that tumor vessels varied greatly in size, with small-caliber (<15 μm diameter) vessels predominating (Figure 1B) ▶ . At the smallest end of the spectrum were individual vWF-positive cells lacking a visible lumen. These stained for other EC markers (CD31 and Tie2) but not for the melanocyte-specific antigen S100 expressed by K1735 cells (data not shown) and accounted for 18% of total K1735 tumor vascular structures. To assess whether these single endothelial cells (SECs) were functional vessels, tumor-bearing mice were injected with Hoechst 33342 intravenously to label the nuclei of cells exposed to blood flow. 20,21 It failed to stain SECs (Figure 1C ▶ , arrowheads) or the cells around them (Figure 1C ▶ , top row) but did stain the vast majority (>90%) of larger vessels (Figure 1C ▶ , bottom row), indicating that SECs were not perfused. This result also showed that SECs were discrete endothelial structures rather than tangentially cut sections of larger vessels. Dual staining for vWF and PCNA revealed that a much greater percentage of SECs (54 ± 15%) were proliferating compared to ECs in other vessels (18 ± 6%; P < 0.01, Mann-Whitney U-test). To test the idea that SECs were related to tumor angiogenesis, K1735 cells were engineered to overexpress the proangiogenic factor, vascular endothelial growth factor (VEGF164). These K1735.VEGF cells, which produce 20-fold more VEGF in 21% O2 cultures and induce 62% more angiogenesis in in vivo Matrigel neovascularization assays than control cells (data not shown), gave rise to tumors with a microvessel density (MVD) of 42.6 ± 15.5 vessels/high-power field compared to 14.5 ± 4.3 vessels/high-power field for control tumors. SECs were four to five times more abundant in these tumors, constituting 26.6 ± 5.3% of all vessels in K1735.VEGF tumors compared to 17.9 ± 2.4% of vessels in K1735 tumors (Figure 1D) ▶ . To place SECs in the context of overall tumor vasculature, thick (60 μm) sections from tumors whose perfused vessels had been previously labeled in situ with a fluorochrome-conjugated tomato lectin, 8 were stained for CD31. Laser-scanning confocal imaging of these sections revealed numerous thin tendrils of unperfused (lectin unstained) ECs sprouting from larger perfused vessels (Figure 1E ▶ , arrows). These structures were undoubtedly angiogenic sprouts, which suggested that the corresponding SECs seen in thin tumor sections also reflected tumor angiogenic activity.
Figure 1.
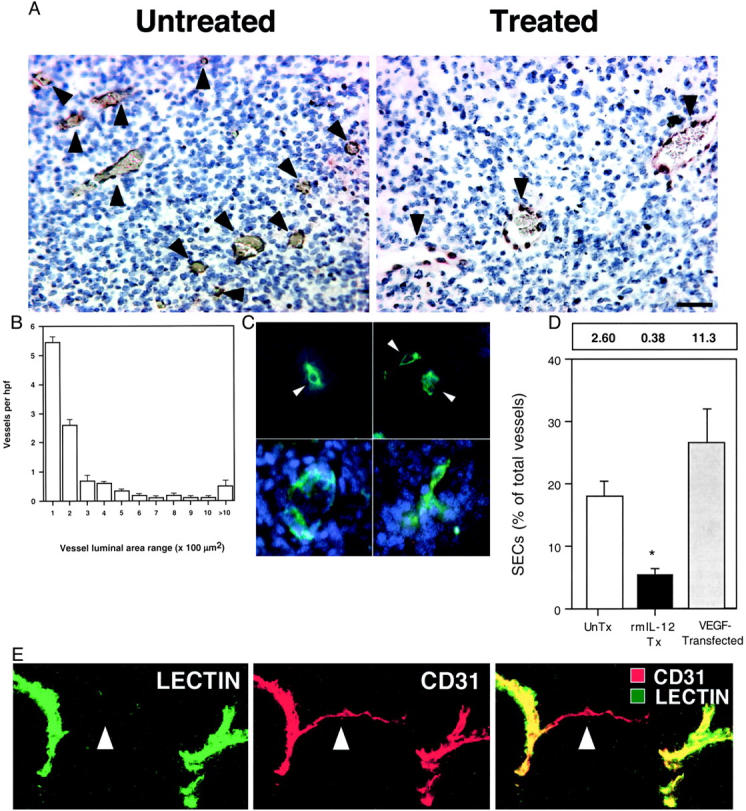
Characterization of endothelial sprouts in growing tumors. A: Histological sections from K1735 tumors were stained for vWF to detect ECs. Representative high-powered fields from untreated (left) or rmIL-12 (right) K1735 tumor sections are shown. B: Vessel size profile of untreated K1735 tumors. The histogram depicts the overall density of vessels per high-powered (×200) field categorized by cross-sectional area range. The data shown represent 10 tumors, with 30 to 60 high-powered fields analyzed per tumor. SE bars are displayed. C: Thin section histological staining revealed single ECs (top row, arrowheads) that did not bear any detectable blood flow (which would appear blue). The bottom row shows larger vessels with detectable perfusion for comparison. D: Comparison of SEC proportion in K1735 tumors in the absence (white bar) or presence (black bar) of rmIL-12 therapy, as well as K1735 tumors engineered to overexpress VEGF164 (gray bar). The data were obtained from five tumors in each group. Numbers above graph indicate mean absolute SEC density/high-power field. Error bars indicate SD, asterisk indicates statistical significance (P < 0.01, Student’s t-test). E: Confocal microscopy of thick (60 μm) sections from fluorescent lectin (green)-perfused tumors stained for CD31 (red) reveals numerous fine EC processes (arrowheads) devoid of lectin perfusion extending from larger perfused vessels. Scale bars: 50 μm (A); 20 μm (C); 75 μm (E).
Pericyte Investment Denotes Tumor Vessel Maturation
K1735 tumor vessels were also characterized with respect to pericyte coverage by staining tumor sections with anti-vWF and anti-α-SMA antibodies; pericyte-positive vessels were defined as vWF-positive structures associated with at least one SMA-positive cell. Analysis of 10 untreated K1735 tumors ranging in size from 4 to 13 mm in diameter revealed that overall, 38 ± 5% of vessels were pericyte covered. Because of the possibility that pericytes surrounding some vessels might be missed because of the thinness of the histological sections, we traced 100 apparently pericyte-negative vessels through five sequential 4-μm sections (20 μm of tumor depth) to determine how many were pericyte covered on subsequent sections. Pericytes were only detected in 6 of these 100 vessels, indicating that the absence of pericyte coverage could be determined on the basis of one tumor section with 94% accuracy. Pericyte-negative vessels tended to be smaller (mean vessel cross-sectional area, 110 ± 75 μm2) than their pericyte-invested counterparts (mean area, 430 ± 215 μm2) (Figure 2A) ▶ .
Figure 2.
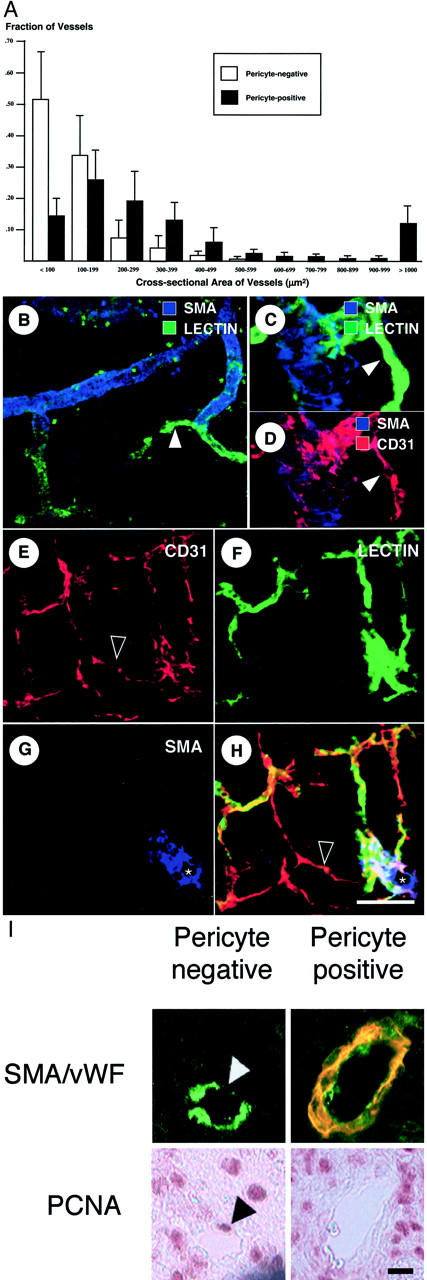
Pericyte investment of tumor vessels denotes maturation. A: K1735 tumor histological sections were stained for the ECs (vWF) and pericytes (SMA). Tumor vessel number, size, and pericyte coverage were determined as described in Materials and Methods. The histogram depicts the proportion of pericyte-negative (white) or pericyte-positive (black) vessels in each size range. n = 10 tumors analyzed. Error bars indicate SD. B–H: Thick tumor sections were stained for SMA, CD31, and lectin perfusion. B: A representative large pericyte-covered vessel with a smaller branch (arrowhead) devoid of pericytes. C and D: A higher magnification view of a small pericyte-negative vessel (arrowhead) seen branching off of a larger, pericyte-covered vessel. E–H: A representative confocal image set with the individual CD31 (E), lectin (F), and SMA (G) fluorescent channels, along with the composite image (H). Note the large pericyte-covered vessel (asterisk) with pericyte-negative arborization, and endothelial sprouts (hollow arrowheads) only seen originating from vessels devoid of pericytes. I: Simultaneous staining for vWF (green), SMA (red), and PCNA (purple) to assess vessel proliferation. The arrowheads indicate an EC (white) containing a PCNA-positive nucleus (black). Scale bars: 100 μm (B); 10 μm (C, D); 50 μm (E–H); 20 μm (I).
To examine the relationship between these different vascular structures, confocal imaging of thick tumor sections was performed. Larger, pericyte-covered vessels were seen to branch off frequently into smaller, pericyte-negative vessels (Figure 2B) ▶ , which appeared at higher magnification as thin, lectin-perfused EC tubes (Figure 2, C and D) ▶ . Angiogenic sprouts frequently (Figure 2E) ▶ emanated from perfused vessels (Figure 2F) ▶ , with the vast majority (>95%) sprouting from distal pericyte-negative vessels rather than proximal pericyte-covered vessels (Figure 2, G and H) ▶ . This is consistent with our observation on thin tumor sections that SECs were rarely associated with pericytes. When EC proliferative status was examined as a function of pericyte coverage by sequential staining for SMA/vWF and PCNA (Figure 2I) ▶ , 34.8 ± 6.5% of pericyte-negative tumor vessels contained one or more PCNA-positive ECs, compared with 14.7 ± 3.9% of pericyte-positive vessels (P < 0.01, Mann-Whitney U-test). Thus, from the standpoints of vessel size, endothelial sprouting, and proliferation status, pericyte coverage marks more established tumor vessels, just as pericyte coverage of normal vessels denotes maturation. 13
To investigate whether the spectrum of vessels in tumors changed as a function of tumor growth, we analyzed the vasculature of 27 K1735 tumors ranging from 3 to 14 mm in diameter (Figure 3) ▶ . Compared to large tumors (5 to 14 mm), small tumors (<5 mm) had a higher mean MVD (13.4 ± 1.9 versus 11.3 ± 1.4, P < 0.01), a larger fraction of vessels that were SECs (26.0 ± 3.6 versus 18.7 ± 4.4, P < 0.01), and a lower fraction of pericyte-covered vessels (29.5 ± 5.7 versus 34.5 ± 4.5, P < 0.05). Thus, the vasculature of small K1735 tumors exhibited a more angiogenic and less mature phenotype. However, differences were not large, overlap was considerable, and vascular characteristics could not be predicted based on individual tumor size (Figure 3) ▶ . Furthermore, significant differences in vascular parameters were not found among the larger tumors.
Figure 3.

Tumor vessel characteristics as a function of size. K1735 tumors of varying sizes were excised, sectioned, and analyzed for vessel number and pericyte coverage according to the Materials and Methods. Tumors were categorized as small (<5 mm diameter, n = 7), medium (5 to 10 mm, n = 13), or large (>10 mm, n = 7). Individual tumor values are displayed as a scattergram. Horizontal bars indicate mean values for each parameter measured.
rmIL-12 Therapy Decreases Endothelial Sprouting and Selectively Induces Apoptosis in Vessels Devoid of Pericytes
We next characterized the pattern of K1735 vascular change with anti-angiogenesis therapy. Mice bearing either untreated or rmIL-12-treated tumors were injected with fluorescein isothiocyanate-tomato lectin, and tumor vasculature was imaged in thick sections (Figure 4, A and B) ▶ . In untreated tumors, the vasculature consisted of a fine, reticulated network of small diameter vessels (mean diameter, 11 μm). Therapy led to a decrease in vessel density, with the remaining vessels being predominantly of larger size (mean diameter, 24 μm). When thin sections from size-matched untreated and rmIL-12-treated tumors were stained for vWF and SMA (Figure 4, C and D) ▶ , treatment was seen to reduce overall MVD (Figure 4E ▶ ; 11.5 ± 2.8 versus 6.8 ± 1.8 vessels/high-power field; P < 0.05, t-test) but not the density of pericyte-positive vessels (Figure 4E ▶ , shaded bars; 4.41 ± 0.92 versus 4.35 ± 0.57 vessels/high-power field, respectively). Rather, a decrease in the density of pericyte-negative tumor vessels during treatment (from 7.1 to 2.5 vessels/high-power field; P < 0.05 t-test) accounted for the decrease in overall MVD. The numbers of endothelial sprouts (Figure 4, F and G) ▶ and SECs (5.41 ± 0.55% versus 17.9 ± 2.41% of total vessels, P < 0.01 Student’s t-test; Figure 1D ▶ ) were also markedly reduced, indicating that rmIL-12 therapy inhibited angiogenesis. Together, these changes resulted in a marked increase in the proportion of pericyte-covered vessels in treated tumors (62% in treated versus 38% in untreated tumors, P < 0.01).
Figure 4.
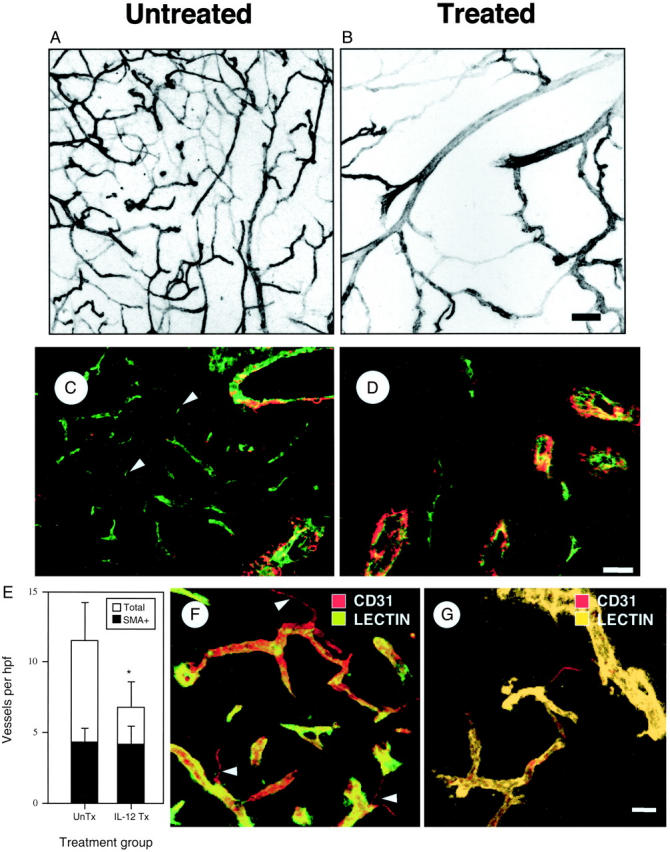
Effect of anti-vascular therapy on tumor vasculature. Representative confocal microscopic images of the vasculature from size-matched lectin-perfused K1735 tumors in the absence (A) or presence (B) of rmIL-12 therapy. C and D: Size matched untreated (C) or rmIL-12-treated (D) K1735 tumors were stained for vWF and SMA to detect ECs and pericytes. Arrowheads indicate SECs. E: Total microvessel density (open bars) and density of pericyte-covered vessels (filled bars) were determined for size-matched K1735 tumors that were either untreated or rmIL-12 treated. Error bars indicate SD, asterisks indicate statistical significance (*, P < 0.05) by Student’s t-test. Six tumors were analyzed from each group, with multiple sections assessed for each tumor. F and G: Confocal images from thick tumor sections from untreated (F) and rmIL-12-treated (G) tumors that were lectin-perfused (green) and stained for ECs (CD31, red). Note the decrease in vessel density and endothelial sprouts (arrowheads) after therapy. Scale bars: 50 μm (B, G); 30 μm (D).
To assess EC apoptosis directly and the pericyte status of regressing vessels, tumor sections were stained for apoptotic cells by TUNEL (Figure 5; A to F) ▶ . Apoptotic vessels were defined as those in which at least one vWF-positive cell contained a TUNEL-positive nucleus (Figure 5, B and C) ▶ . Few, if any, vessels were apoptotic in untreated K1735 tumors (Figure 5G) ▶ . rmIL-12 treatment increased apoptosis among pericyte-negative vessels (1.05 ± 0.25 vessels/high-power field or 26% of vessels treated versus 0.27 ± 0.15 or 3.2% of vessels untreated; P < 0.05 Mann-Whitney U-test) but not among pericyte-positive vessels (0.15 ± 0.16 vessels/high-power field treated versus 0.14 ± 0.10 vessels/high-power field untreated). Additionally, we did not observe pericyte apoptosis in treated tumors. The apoptosis in pericyte-negative vessels accompanying their selective decrease with treatment indicated that rmIL-12 reduced K1735 tumor vascularity by inducing vessel regression as well as by inhibiting new vessel formation.
Figure 5.
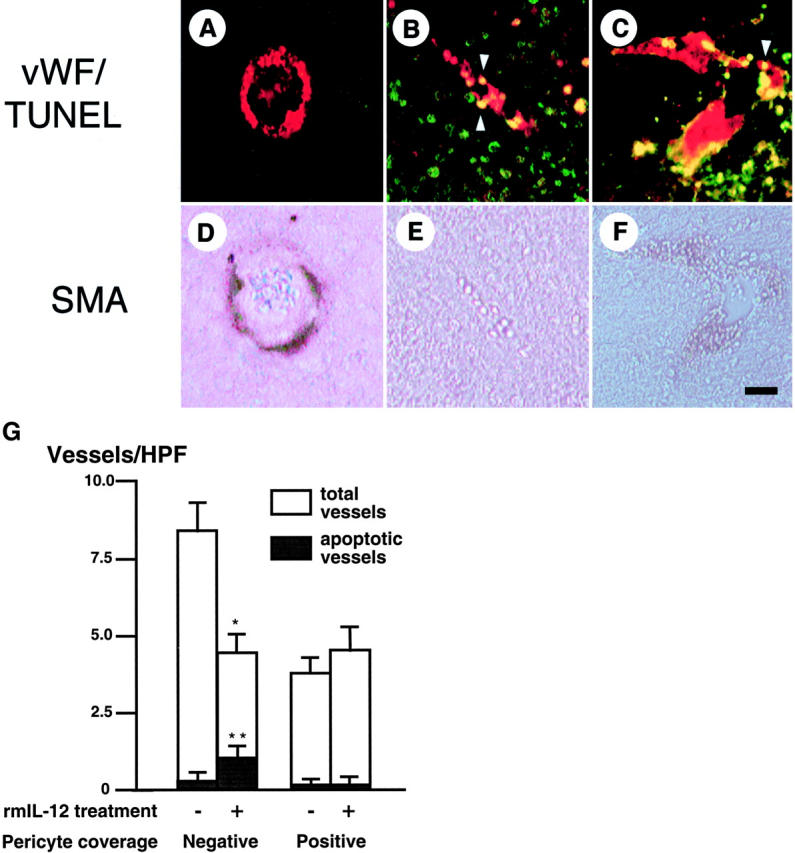
Selective apoptosis of tumor vessels devoid of pericytes. A–F: Histological assessment of vessel pericyte coverage and apoptosis in K1735 tumors was performed by fluorescent staining (A–C) for vWF (red) and TUNEL (green), followed by histochemical staining (D–F) for SMA. A nonapoptotic, pericyte-positive (A and D) and two apoptotic, pericyte-negative (B and E, C and F) vessels are depicted. Arrowheads denote apoptotic EC nuclei. G: Summary of effect of rmIL-12 treatment on K1735 tumor vessel apoptosis and vessel density, categorized according to pericyte status. Five tumors (two sections per tumor) were analyzed per group. Asterisks indicate statistical significance (*, P < 0.05, Student’s t-test; **, P < 0.05, Mann-Whitney U-test). Scale bar, 25 μm (F).
To see whether the vascular changes induced in K1735 tumors by rmIL-12 treatment were generalizable to other therapy-responsive tumors. RENCA renal cell carcinomas 18 in BALB/c mice and B16F10 melanomas 17 in C57Bl/6 mice were studied. As in K1735 tumors, the growth (Table 1) ▶ and vascularity 8 of these other two tumor types were consistently suppressed by rmIL-12 treatment. Untreated RENCA tumors had an overall MVD of 9.56 ± 1.23 vessels/high-power field, of which 27% were pericyte-covered (Table 2) ▶ . After rmIL-12 treatment, MVD was reduced by 33% while the density of pericyte-covered vessels was not significantly altered. Untreated B16F10 tumors had a similar MVD and level of pericyte coverage as untreated RENCA tumors, and rmIL-12 therapy led to a 58% reduction in overall MVD. There was a 33% reduction in the density of pericyte-covered vessels but a more marked 68% reduction in the density of pericyte-negative vessels (P < 0.005). A feature of pericyte-covered vessels in treated B16F10 tumors was the occasional finding of TUNEL staining in both pericytes and ECs, notably in vessels located in regions of massive tumor cell death (data not shown). This indicates that pericyte-positive tumor vessels are not invulnerable and will regress under certain conditions, as may exist when the vessel microenvironment is disrupted. Angiogenesis was decreased in both RENCA and B16F10 tumors after rmIL-12 treatment, with SECs decreasing from 11.2% of total vessels to 4.6% in RENCA (P < 0.05) and from 19.5% to 5.5% in B16F10 (P < 0.005). As in K1735 tumors, the preferential loss of pericyte-negative vessels in RENCA and B16F10 tumors with rmIL-12 treatment resulted in a higher percentage of pericyte-covered vessels after therapy (48% and 47%, respectively).
Table 1.
Effect of Angiogenesis Inhibitor Therapy on Tumor Growth
| Tumor | Treatment | Mean tumor volume (mm3) ± SD during treatment | |||
|---|---|---|---|---|---|
| Day 0 | Day 7 | Day 14 | Day 21 | ||
| K1735 | Vehicle | 1.41 ± 0.62 | 120.64 ± 49.39 | 700.94 ± 179.73 | E |
| rmIL-12 | 5.86 ± 2.83 | 27.06 ± 12.60 | 78.24 ± 29.08 | 170.72 ± 71.04 | |
| RENCA | Vehicle | 1.60 ± 1.02 | 23.89 ± 15.09 | 95.65 ± 44.68 | 498.86 ± 146.04 |
| rmIL-12 | 4.04 ± 3.02 | 13.16 ± 6.50 | 40.42 ± 20.64 | 55.04 ± 26.77 | |
| B16F10 | Vehicle | 1.14 ± 0.71 | 523.85 ± 201.65 | E | E |
| rmIL-12 | 10.50 ± 8.56 | 98.81 ± 54.54 | 298.56 ± 194.48 | 204.63 ± 154.03 | |
Established K1735, B16F10, and RENCA tumors were treated either with rmIL-12 or vehicle. Tumor volumes were obtained by bidirectional caliper measurement according to the Materials and Methods. E, tumors had grown to sufficient size to warrant mouse euthanasia according to Institutional Animal Care and Use Committee guidelines.
Table 2.
Effects of rmIL-12 Therapy on RENCA and B16F10 Tumor Vasculature
| Tumor | Treatment | MVD (vessels/hpf) | Pericyte-covered vessels (per hpf) | SEC density (per hpf) |
|---|---|---|---|---|
| RENCA | Vehicle | 9.56 ± 1.23 | 2.58 ± 0.58 | 1.07 ± 0.39 |
| rmIL-12 | 6.41 ± 0.99** | 3.07 ± 0.55 | 0.29 ± 0.27* | |
| B16F10 | Vehicle | 9.33 ± 2.50 | 2.72 ± 0.59 | 1.82 ± 0.56 |
| rmIL-12 | 3.93 ± 1.31** | 1.81 ± 0.45* | 0.22 ± 0.23** |
Size-matched tumors from mice treated with either rmIL-12 or PBS vehicle were sectioned and stained for endothelial cells and pericytes according to the Materials and Methods. The data are based on analysis of multiple tumor sections (10 to 20 high-powered fields per section) from at least five tumors per treatment group. Asterisks indicate statistical significance (
*, P < 0.05;
**, P < 0.005 Student’s t-test).
MVD, micro-vessel density; hpf, high-powered field.
Discussion
Our studies delineate vessel development and response to anti-vascular therapy in transplanted mouse tumors. We show that tumor vessels develop through identifiable stages, beginning with lines of ECs emanating from functional blood vessels. Angiogenic sprouts, which have been previously described, 19 correspond to SECs in immunostained thin tumor sections, suggesting that tumor angiogenic activity can be gauged in archival human specimens. These proliferative endothelial sprouts evolve into small caliber tumor vessels, the majority of which are perfused endothelial tubes lacking pericytes. Pericyte coverage comes later, appears to occur by contiguous spread and is associated with larger vessel caliber, fewer proliferating ECs and reduced sprouting. Endothelial sprouts, pericyte-negative and pericyte-positive vessels and their hierarchical relationship were often seen in confocal images of thick K1735 tumor sections (Figure 4; E to H) ▶ . Larger tumors tend to have a lower fraction of nascent vessels and a higher fraction of mature vessels, suggesting that the spectrum of tumor vessels shifts as tumors enlarge. A recent study using anti-desmin and anti-SMA antibodies to stain pericytes and confocal imaging of thick tumor sections suggested that pericytes are associated with the vast majority of blood vessels in tumors and even cover endothelial sprouts. 22 We cannot explain why we saw a more restricted pattern of pericyte coverage both in thin and thick sections, but the tumors studied were different. That pericytes invest only a subset of tumor vessels has been described by others in mouse 7,23 and human 24,25 tumors, and the association between coverage and vessel maturation recalls the process of retinal neovascularization in the neonatal rodent eye. 13 The limited distribution that we observe correlates with vessel development and patterns of vessel therapeutic response and should be useful for discriminating between different categories of tumor vessels.
Given the heterogeneity of tumor vessels based on stage of development or maturation, we examined the latter for determinants of vessel response. In multiple tumor models, the decrease in vessel density with therapy is attributable primarily to loss of pericyte-negative vessels, resulting in a higher proportion of pericyte-positive vessels after therapy. Loss of pericyte-negative vessels is because of both angiogenesis inhibition and regression of existing vessels. Although pericyte-negative vessels regress, some may also be lost through acquisition of pericytes during therapy. Maturation would reduce the number of these vessels and help maintain the density of pericyte-positive vessels during therapy, when there is continued, albeit slow, tumor growth. Preferential survival of pericyte-covered vessels may be because of pericyte production of EC survival factors 26 or cell contacts formed with ECs providing survival signals. It is also possible that pericyte coverage is an indicator of protected vessels and that factors other than pericytes but associated with pericyte coverage actually provide protection. Pericyte coverage marks vessels protected from anti-vascular therapies in addition to rmIL-12, including VEGF withdrawal 24 and anti-vascular thalidomide analogs (MS Gee, unpublished observations).
A tumor’s vascular response to anti-vascular therapy will be the primary determinant of its clinical response, although factors such as tumor cell dependence on vascular supply 21 and response to ischemic stress 27 may influence outcome. Because of this, pericyte coverage and protection of tumor vessels may strongly influence tumor therapeutic response. Transplanted mouse tumors we studied have a relatively low fraction of pericyte-covered vessels, indicating that most of their vessels are newly formed, and angiogenesis is vigorous. Many of their vessels are susceptible to regression, and angiogenesis inhibition would slow or stop the rapid addition of new vessels, so effective anti-vascular agents produce readily detectable effects. Tumors with a high fraction of pericyte-covered vessels have a greater proportion of mature vessels, fewer newly formed vessels, and may be less angiogenic. Active agents may have less effect on these tumors because fewer vessels are susceptible. This may help explain why autochthonous MMTV-induced mouse mammary tumors with a substantial fraction of pericyte-covered vessels do not respond to rmIL-12 therapy, whereas transplanted Mm5MT mammary tumors, derived from similar cells and with a much lower fraction of pericyte-covered vessels, respond well. 28 A correlation between tumor vessel pericyte coverage and response is strengthened by the fact that modulation of pericyte coverage in K1735 tumors alters their response to rmIL-12 therapy in the expected direction (NM Yeilding, unpublished observations). This has implications for human cancers in which some of the most common types, eg, breast, colon, lung, and prostate, have vessels with moderate to extensive pericyte coverage. 25 Although the response of these cancers to anti-vascular agents has not been described in detail, there is an impression of disparity between the striking results seen in mouse studies and the effects obtained in human trials. 29
Clinical application of pericyte information will require reliable identification of pericytes in human tumor specimens. Although pericyte phenotype may be variable, 22 reproducible discrimination between pericyte-positive and pericyte-negative vessels in human tumors has been reported. 25 Quantitation of SECs may be useful because it provides an indicator of tumor angiogenic activity distinct from, but related to, pericyte coverage. The adverse influence of pericyte coverage on anti-vascular efficacy may apply only to certain agents. Efficacy of agents targeting ECs in more mature vessels or targeting other vascular components may not be limited by pericyte coverage. 20,30 For agents that do selectively target angiogenic ECs, pericyte and SEC information may provide additional evidence of therapeutic anti-vascular effect. This requires tumor samples from before and after treatment, but an increase in pericyte coverage and a decrease in SECs would confirm that an effect was achieved. A better understanding of vessel development and maturation in human cancers and of the factors affecting pericyte coverage of their vessels may advance anti-vascular therapy of human cancers.
Acknowledgments
We thank the Genetics Institute (Andover, MA) for supplying recombinant murine IL-12; Dr. Su-jean Seo for providing histological reagents and helpful suggestions; and the GI Division Morphology Core Laboratory at the Hospital of the University of Pennsylvania (Cancer Center grant P30 DK50306) for all histology images.
Footnotes
Address reprint requests to Dr. William Lee, BRB II/III, Room 312, 421 Curie Blvd., Philadelphia, PA 19104. E-mail: leemingf@mail.med.upenn.edu.
Supported by the National Institutes of Health (grants R01 CA 77851 and CA 83042 to W. M. F. L. and an Medical Scientist Training Program (MSTP) grant to M. S. G.), the American Cancer Society (CSM-98759 to N. M. Y.), and the United States Department of Defense (BC 990509 to N. M. Y.).
References
- 1.Giavazzi R, Taraboletti G: Angiogenesis and angiogenesis inhibitors in cancer. Forum (Genova) 1999, 9:261-272 [PubMed] [Google Scholar]
- 2.Folkman J: Angiogenesis research: from laboratory to clinic. Forum (Genova) 1999, 9:59-62 [PubMed] [Google Scholar]
- 3.Gee MS, Koch CJ, Evans SM, Jenkins WT, Pletcher CH, Moore JS, Koblish HK, Lee J, Lord EM, Trinchieri G, Lee WMF: Hypoxia-mediated apoptosis from angiogenesis inhibition underlies tumor control by recombinant interleukin 12. Cancer Res 1999, 59:4882-4889 [PubMed] [Google Scholar]
- 4.Folkman J: Anti-angiogenesis: new concept for therapy of solid tumors. Ann Surg 1972, 175:409-416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kerbel RS: Inhibition of tumor angiogenesis as a strategy to circumvent acquired resistance to anti-cancer therapeutic agents. Bioessays 1991, 13:31-36 [DOI] [PubMed] [Google Scholar]
- 6.Boehm T, Folkman J, Browder T, O’Reilly MS: Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature 1997, 390:404-407 [DOI] [PubMed] [Google Scholar]
- 7.Shaheen RM, Tseng WW, Davis DW, Liu W, Reinmuth N, Vellagas R, Wieczorek AA, Ogura Y, McConkey DJ, Drazan KE, Bucana CD, McMahon G, Ellis LM: Tyrosine kinase inhibition of multiple angiogenic growth factor receptors improves survival in mice bearing colon cancer liver metastases by inhibition of endothelial cell survival mechanisms. Cancer Res 2001, 61:1464-1468 [PubMed] [Google Scholar]
- 8.Gee MS, Saunders HM, Lee JC, Sanzo JF, Jenkins WT, Evans SM, Trinchieri G, Sehgal CM, Feldman MD, Lee WM: Doppler ultrasound imaging detects changes in tumor perfusion during antivascular therapy associated with vascular anatomic alterations. Cancer Res 2001, 61:2974-2982 [PubMed] [Google Scholar]
- 9.Darland DC, D’Amore PA: Blood vessel maturation: vascular development comes of age. J Clin Invest 1999, 103:157-158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Warren BA, Shubik P: The growth of the blood supply to melanoma transplants in the hamster cheek pouch. Lab Invest 1966, 15:464-478 [PubMed] [Google Scholar]
- 11.Ausprunk DH, Knighton DR, Folkman J: Differentiation of vascular endothelium in the chick chorioallantois: a structural and autoradiographic study. Dev Biol 1974, 38:237-248 [DOI] [PubMed] [Google Scholar]
- 12.Crocker DJ, Murad TM, Geer JC: Role of the pericyte in wound healing. An ultrastructural study. Exp Mol Pathol 1970, 13:51-65 [DOI] [PubMed] [Google Scholar]
- 13.Benjamin LE, Hemo I, Keshet E: A plasticity window for blood vessel remodelling is defined by pericyte coverage of the preformed endothelial network and is regulated by PDGF-B and VEGF. Development 1998, 125:1591-1598 [DOI] [PubMed] [Google Scholar]
- 14.Alon T, Hemo I, Itin A, Pe’er J, Stone J, Keshet E: Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat Med 1995, 1:1024-1028 [DOI] [PubMed] [Google Scholar]
- 15.Lindahl P, Johansson BR, Leveen P, Betsholtz C: Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science 1997, 277:242-245 [DOI] [PubMed] [Google Scholar]
- 16.Kripke ML: Speculations on the role of ultraviolet radiation in the development of malignant melanoma. J Natl Cancer Inst 1979, 63:541-548 [DOI] [PubMed] [Google Scholar]
- 17.Fidler IJ: Biological behavior of malignant melanoma cells correlated to their survival in vivo. Cancer Res 1975, 35:218-224 [PubMed] [Google Scholar]
- 18.Murphy GP, Hrushesky WJ: A murine renal cell carcinoma. J Natl Cancer Inst 1973, 50:1013-1025 [DOI] [PubMed] [Google Scholar]
- 19.Hashizume H, Baluk P, Morikawa S, McLean JW, Thurston G, Roberge S, Jain RK, McDonald DM: Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol 2000, 156:1363-1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dark GG, Hill SA, Prise VE, Tozer GM, Pettit GR, Chaplin DJ: Combretastatin A-4, an agent that displays potent and selective toxicity toward tumor vasculature. Cancer Res 1997, 57:1829-1834 [PubMed] [Google Scholar]
- 21.Yu JL, Rak JW, Carmeliet P, Nagy A, Kerbel RS, Coomber BL: Heterogeneous vascular dependence of tumor cell populations. Am J Pathol 2001, 158:1325-1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morikawa S, Baluk P, Kaidoh T, Haskell A, Jain RK, McDonald DM: Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am J Pathol 2002, 160:985-1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tian S, Hayes AJ, Metheny-Barlow LJ, Li LY: Stabilization of breast cancer xenograft tumour neovasculature by angiopoietin-1. Br J Cancer 2002, 86:645-651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Benjamin LE, Golijanin D, Itin A, Pode D, Keshet E: Selective ablation of immature blood vessels in established human tumors follows vascular endothelial growth factor withdrawal. J Clin Invest 1999, 103:159-165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eberhard A, Kahlert S, Goede V, Hemmerlein B, Plate KH, Augustin HG: Heterogeneity of angiogenesis and blood vessel maturation in human tumors: implications for antiangiogenic tumor therapies. Cancer Res 2000, 60:1388-1393 [PubMed] [Google Scholar]
- 26.Reinmuth N, Liu W, Jung YD, Ahmad SA, Shaheen RM, Fan F, Bucana CD, McMahon G, Gallick GE, Ellis LM: Induction of VEGF in perivascular cells defines a potential paracrine mechanism for endothelial cell survival. EMBO J 2001, 15:1239-1241 [DOI] [PubMed] [Google Scholar]
- 27.Yu JL, Rak JW, Coomber BL, Hicklin DJ, Kerbel RS: Effect of p53 status on tumor response to antiangiogenic therapy. Science 2002, 295:1526-1528 [DOI] [PubMed] [Google Scholar]
- 28.Lee JC, Kim DC, Gee MS, Saunders HM, Sehgal CM, Feldman MD, Ross SR, Lee WM: Interleukin-12 inhibits angiogenesis and growth of transplanted but not in situ mouse mammary tumor virus-induced mammary carcinomas. Cancer Res 2002, 62:747-755 [PubMed] [Google Scholar]
- 29.Kerbel RS, Yu J, Tran J, Man S, Viloria-Petit A, Klement G, Coomber BL, Rak J: Possible mechanisms of acquired resistance to anti-angiogenic drugs: implications for the use of combination therapy approaches. Cancer Metastasis Rev 2001, 20:79-86 [DOI] [PubMed] [Google Scholar]
- 30.Luster AD, Greenberg SM, Leder P: The IP-10 chemokine binds to a specific cell surface heparan sulfate site shared with platelet factor 4 and inhibits endothelial cell proliferation. J Exp Med 1995, 182:219-231 [DOI] [PMC free article] [PubMed] [Google Scholar]