

Abstract
Vascular smooth muscle cell (VSMC) hyperplasia plays an important role in both chronic and acute vascular pathologies. Considerable work has focused on the mechanisms regulating VSMC growth and the search for agents that could suppress VSMC hyperproliferation. One of the several inhibitors studied is the glycosaminoglycan heparin, which inhibits VSMC proliferation and migration both in cell culture and in animal models (Mishra-Gorur K, Delmolino LM, Castellot Jr JJ: Biological functions of heparan sulfate and heparan sulfate proteoglycans. Trends Glycosci Glycotechnol 1998, 10:193–210). To aid our understanding of the anti-proliferative mechanism of action of heparin, we used a subtractive hybridization approach to isolate and characterize a novel growth arrest-specific (gas) gene induced in VSMCs exposed to heparin (Delmolino LM, Stearns NA, Castellot Jr JJ: Heparin induces a member of the CCN family which has characteristics of a growth arrest specific gene. Mol Biol Cell 1997, 8:287a and Delmolino LM, Stearns NA, Castellot Jr JJ: COP-1, a member of the CCN family, is a heparin-induced growth arrest specific gene in vascular smooth muscle cells. J Cell Physiol 2001, 188:45–55). This gene is a member of the cysteine-rich 61/connective tissue growth factor/nephroblastoma-overexpressed (CCN) family and has been given the name CCN5. In this report, we provide functional evidence that CCN5 can inhibit VSMC proliferation, motility, and invasiveness. In contrast, adhesion and apoptosis are unaffected by CCN5 in this cell type. We also significantly extend previous data from our laboratory that suggests CCN5 is a growth arrest-specific (gas) gene. Furthermore, we map for the first time the cellular localization of CCN5 protein in cultured VSMCs. We also examine uninjured and balloon-injured rat carotid arteries for CCN5 expression. The results from the in vitro and in vivo localization studies show that CCN5 is temporally and spatially expressed in a manner consistent with a role in regulating proliferation, motility, and invasiveness of VSMCs.
Vascular smooth muscle cell (VSMC) migration into the intima and subsequent proliferation is the hallmark of many vascular pathologies, including arteriosclerosis and restenosis after angioplasty and coronary artery bypass grafts. Considerable work has focused on the mechanisms regulating VSMC hyperproliferation and on the search for agents that can suppress VSMC mitogenesis. One of the inhibitors studied is the glycosaminoglycan heparin, which inhibits VSMC proliferation and migration both in cell culture and in animal models. 1 To aid our understanding of the anti-proliferative mechanism of heparin action, we used a subtractive hybridization approach to isolate and characterize a novel growth arrest-specific (gas) gene induced in VSMCs exposed to heparin. 2,3 This gene is a member of the cysteine-rich 61/connective tissue growth factor/nephroblastoma-overexpressed (CCN) family and has been named CCN5. 4
Several laboratories have described essentially the same gene and protein in mice, rats, and humans. Zhang and colleagues 26 used differential display polymerase chain reaction and found CCN5 (rCop-1) expressed by untransformed Rat-1 embryonic fibroblasts; production ceased on oncogenic transformation. Pennica and colleagues 5 used a suppression-subtractive hybridization strategy and found that CCN5 (WISP-2) was up-regulated by Wnt-1 transformation of a mouse mammary epithelial cell line. Interestingly, in vivo CCN5 mRNA levels are relatively low in human colon carcinoma tumors and significantly higher in normal tissue surrounding the tumors. 5 In contrast, CCN5 mRNA and protein levels are up-regulated in human breast carcinoma MCF-7 cells as compared to normal breast epithelium. 6 Kumar and colleagues 29 found CCN5 (CTGF-L) expressed in murine osteoblasts in a manner that promoted adhesion and inhibited osteocalcin expression. Although these studies provide a good foundation for further exploration of the biological activities of CCN5, little is known about the functional roles that this gene might play in cell proliferation and motility, as well as invasiveness.
The CCN family members are secreted, cell- and matrix-associated proteins that appear to play diverse and important roles in cell function. 7-10 They have been implicated in cell differentiation and survival, wound repair, vascular disease, fibrosis, and progression of certain cancers. Two CCN family members, CCN1 (CYR61) and CCN2 (CTGF), have been shown to induce angiogenesis. All CCN proteins contain 38 conserved cysteine residues and share a homologous modular structure containing four distinct domains, with the exception of CCN5, which lacks the carboxy-terminal domain. Combined with its gas gene expression pattern, this observation led us to hypothesize that CCN5 plays a role in suppressing VSMC proliferation and motility.
In this study, we test our hypothesis using an adenovirus construct that permits the delivery and expression of CCN5 in cultured VSMCs (AdCCN5). Using this approach, we show that AdCCN5 infection of VSMCs inhibits proliferation, motility, and invasiveness. In contrast, AdCCN5 infection has no significant effect on VSMC adhesion or apoptosis. We demonstrate that CCN5 protein expression follows the same temporal expression pattern as the mRNA, thus placing the protein at the right time and place to control cell proliferation and motility. Finally, using a rat carotid artery balloon injury model, we observe a protein expression pattern in uninjured and injured vessels that is consistent with the functions observed for CCN5 in cultured VSMCs. Taken together, the data presented here suggest a complex tissue-specific picture for CCN5 expression and function.
Experimental Procedures
Materials
All tissue culture plastic ware, including growth factor reduced-Matrigel chambers, were obtained from BD Biosciences (Lincoln Park, NJ). Eight-μm polycarbonate filter membranes for use in modified Boyden assays were purchased from NeuroProbe Inc. (Cabin John, MD). Premium fetal calf serum (FCS) was purchased from HyClone (Logan, UT). RPMI base media, trypsin-ethylenediaminetetraacetic acid, glutamine, and penicillin-streptomycin were purchased from Life Technologies, Inc. (Grand Island, NY). Heparin, obtained from Glycomed (Alameda, CA) was derived from porcine mucosa (sodium salt; average molecular mass, 15 kd). Precast sodium dodecyl sulfate (SDS)-gels and running system were obtained from Bio-Rad (Hercules, CA). The ECL Renaissance kit was purchased from New England Nuclear (Boston, MA). Anti-HA rat monoclonal antibody was purchased from Boehringer-Mannheim (Indianapolis, IN). Anti-CCN5 affinity-purified rabbit polyclonal antibody was obtained from FibroGen (San Francisco, CA). This antibody was raised against a synthetic peptide with the following sequence taken from the human CCN5 protein: NGRLYREGETFQPHC. This peptide sequence was selected for its low homology to other CCN family members. We have observed no cross-reactivity with other CCN family members in rat cell lysates or conditioned medium. Horseradish peroxidase- and rhodamine-conjugated donkey anti-rat and anti-rabbit secondary antibodies were purchased from Jackson ImmunoResearch Laboratories Inc. (West Grove, PA). All other chemicals and reagents were purchased from Sigma Chemical Co. (St. Louis, MO).
Cell Culture
Primary Cell Cultures
Aortic muscle cells (VSMCs) were obtained from aortae of Sprague-Dawley rats (Charles River Breeding Laboratories, Inc., Wilmington, MA). They were isolated, cultured, and characterized as previously described. 11 Briefly, the abdominal segment of the aorta was removed and the fascia cleaned away under a dissecting microscope. The aorta was cut longitudinally and small pieces of the media were carefully stripped from the vessel wall. Two or three such strips were placed in 60-mm dishes. Within 1 to 2 weeks, VSMCs migrated from the explants; they were capable of being passaged ∼1 week after the first appearance of cells. They were identified as VSMCs by their characteristic hill-and-valley growth pattern and indirect immunofluorescence staining for VSMC-specific α-actin. All cultures were maintained in RPMI 1640 medium containing 10% FCS (10% FCS/RPMI) at 37°C in a humidified, 5% CO2/95% air atmosphere.
Growth Arrest of Cells
Cells were routinely plated at various densities, washed with serum-free RPMI, and placed in 0.4% FCS/RPMI for 72 hours. Flow cytometry and determination of [3H] thymidine-labeled nuclei indicated that >95% of the cells were arrested in G0(G1). 12 Cells were released from quiescence by replacing the low serum medium with 10% FCS/RPMI. Cultures were used at passage 8 or lower.
Adenovirus Production and Infection 13
An adenovirus, expressing both green fluorescent protein (GFP) and CCN5 tagged by a nine amino acid HA epitope on the C-terminus, was produced using the Ad-Easy system provided and described by He and colleagues. 13 The two genes in the construct are under the control of two separate CMV promoters. A control virus expressing only GFP was also produced. Polymerase chain reaction primers were constructed to amplify the CCN5 construct from cDNA, introduce Xba-1 sites for insertion into the adenovirus shuttle vector, and attach the HA epitope tag sequence. They are: forward, TCTAGATTAGAAAGCACTGTTCCATGAGCT; reverse, TAGACCATGTACCCCTACGACGTGCCCGACTACGCCAGGGGCAGCCCACTGATCCATCTT.
VSMCs are routinely infected in our laboratory by the following method. Cells are first washed and exposed to various amounts of virus for 2 hours in serum-free RPMI 1640 with occasional agitation. Ten percent FCS/RPMI-containing medium is then added back to the cells. After 1 to 2 days, infected cells can be trypsinized and used for experiments as usual. Infection can be monitored quickly by observing the fraction of cells expressing GFP in the population.
Protein Analysis
Western Blotting
Proteins from the various conditions described above were harvested from 100-mm culture dishes, with all procedures performed at 4°C, as follows. Cells were rinsed twice with cold TBS (20 mmol/L Tris, pH 7.6, 137 mmol/L NaCl) and lysed with 100 μl of RIPA lysis buffer. Lysates were transferred to Eppendorf tubes, rocked for 20 minutes and spun at 12,000 rpm for 10 minutes in an Eppendorf microfuge (Hamburg, Germany). Five ml of serum-free conditioned medium from VSMCs was also collected from 100-mm culture dishes of growth-arrested cells. Supernatant and conditioned medium were stored at −20°C until use. Protein estimations were performed using the Pierce BCA method adapted for microtiter plates. Extracts containing 40 μg of protein or 40 μl of conditioned medium were boiled in 2× SDS sample loading buffer, resolved by SDS-polyacrylamide gel electrophoresis (PAGE) (either 10% or 4→20% gradient gels were used), and blotted onto 0.2-μm pore size Immun-Blot polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA) in Towbin buffer without methanol (25 mmol/L Trizma base, 192 mmol/L glycine) at 200 mA for 5 hours. The blots were dried and rewetted. Membranes were blocked for 1 hour in TBS containing 5% milk and Western blots were performed using the primary antibodies described (1:500) and horseradish peroxidase-conjugated anti-rat or anti-rabbit IgG (1:10,000) in TBST (TBS + 0.2% Tween 20). Bands were visualized using the NEN (Boston, MA) Renaissance enhanced chemiluminescence (ECL) detection reagents and autoradiography as described by the vendor. Prestained protein standard markers (Bio-Rad) were used as molecular weight markers. Densitometry analysis of films was performed using the Stratagene (La Jolla, CA) Eagle Eye II system. Blots were stained with Amido Black stain to confirm equal loading and proper transfer of proteins to the membrane.
Rat Model of Balloon Injury
Acute endothelial denudation of the left common carotid artery was performed essentially as described by Clowes and colleagues. 14 Briefly, The bifurcation of the left common carotid artery of anesthetized 400 to 500 g male Sprague-Dawley rats was exposed through a midline incision, and the left common, internal, and external carotid arteries were temporarily ligated. A 2F embolectomy catheter (Baxter Edwards Health Care Corp.) was introduced into the external carotid, and advanced to the distal ligation of the common carotid artery, inflated with saline, and drawn toward the arteriotomy site three times to produce a distending, de-endothelializing injury. After the catheter was withdrawn, the proximal external carotid artery was ligated, and blood flow was restored to the common carotid artery by release of the ligatures. The right uninjured carotid artery was used as control tissue. At the indicated times after injury, rats were euthanized and both injured and uninjured common carotid arteries were perfused with saline and dissected free of the surrounding tissue. For immunohistochemistry, tissue specimens were fixed by immersion in methanol overnight and then embedded in paraffin. Paraffin sections were prepared and processed as described previously. 15
Immunocytochemistry
VSMCs were plated onto 18-mm coverslips in 24-well dishes at a density of 8 × 103 cells/well. Cells were growth-arrested and subsequently reintroduced to normal growth medium for various times. Cells previously infected with AdCCN5 were also plated onto coverslips. They were then fixed in 1% paraformaldehyde, blocked, and exposed to the affinity-purified rabbit anti-peptide antibody specific to CCN5 or anti-HA epitope tag antibody. Permeabilized, nonpermeabilized, and membrane-extracted cells were all prepared for analysis. Secondary antibody, donkey anti-rabbit (in the case of anti-peptide antibody), or donkey anti-rat (in the case of the anti-HA epitope tag antibody), conjugated to Texas Red, was used to visualize CCN5 on the cells. Hoechst 33258 stain was also added to the mounting medium to visualize cell nuclei.
Paraffin sections of uninjured and balloon-injured rat carotid arteries were prepared as described 15 and probed for CCN5 using the anti-peptide rabbit polyclonal antibody. Briefly, sections were deparaffinized in xylene and graded ethyl alcohol, blocked, and then exposed to primary antibody (rabbit anti-peptide) followed by washing and exposure to donkey anti-rabbit Texas Red-conjugated secondary antibody.
For both cells and sections digital images were obtained with an RT Color Spot camera (Diagnostic Instruments Inc.) on a Nikon inverted fluorescence microscope. Figures were prepared with Adobe Photoshop v.6.0.
Cell Proliferation Assay
Cell proliferation assays were performed as described previously. 16 Briefly,8 × 103 cells were plated into 16-mm multiwell dishes. In some cases, cells were previously infected with adenovirus as described above. Cells were then growth-arrested as above. Cultures were released from G0 by exposure to 10% FCS/RPMI. In some cases cells were also treated with 10% FCS/RPMI containing 300 μg/ml of heparin. Cells were allowed to grow for the indicated time and then counted in duplicate using a Coulter counter (Fullerton, CA) after trypsinization. The net growth of VSMCs was obtained by subtracting the starting cell number from the above count results (at the time of release from G0). Degree of inhibition is determined as follows:
 |
Cell Motility and Invasiveness
Modified Boyden Chamber Motility Assay 11,17,18
The effect of CCN5 expression on VSMC migration was assessed using a 96-well modified Boyden chamber (NeuroProbe Inc.). VSMCs infected with AdGFP or AdCCN5 as well as uninfected VSMCs were trypsinized and then resuspended at 7 × 105 cells/ml. The lower chambers of the plate were then loaded with RPMI 1640 containing various amounts of FCS and overlaid with an 8-μm polycarbonate filter coated with collagen I (Vitrogen). Three hundred μl of the above cell suspensions were then added to the upper chambers and allowed to migrate through the membrane for 2.5 hours at 37°C. On completion filters were removed, fixed at 4°C in 10% trichloroacetic acid (TCA) for 30 minutes, and then stained with sulfarhodamine B in 4% acetic acid. Cells on the top of the filter were removed by gentle swabbing and the remaining cells on the bottom side of the filter were counted under a light microscope with an eyepiece grid to visualize set fields. At least four fields in triplicate wells were counted for each condition.
Scratch Wound Motility Assay 19,20
Infected VSMCs were plated at confluence onto glass chamber slides previously coated with collagen. The following day a uniform straight scratch was made in the monolayer using a 200-μl yellow plastic pipette tip (Fisher Scientific, Pittsburgh, PA). Monolayers were washed gently, marked (for reference), and photographed using Nomarski optics on an inverted microscope. After incubation for 30 hours at 37°C, the cells were fixed in 1% paraformaldehyde and mounted. Nuclei were visualized using Hoechst 33258 stain and then photographed with a fluorescence microscope. Calculations are based on the number of nuclei that have moved into the middle half of the wound. Four counts were made at various points along each wound that were photographed initially and marked.
Invasion Assay 21-23
VSMC invasiveness was measured using chambers (BD Biosciences, New Bedford, MA) containing an 8-μm pore-size polycarbonate filter insert that divides the chamber into upper and lower portions. The filters were uniformly coated with growth factor reduced-Matrigel. We used the protocol provided by the supplier. Briefly, 3 × 104 cells were added to the upper chamber of the insert while FCS, platelet-derived growth factor (PDGF)-AB (the generous gift of Dr. Brent Cochran, Tufts University), or PDGF-BB (R&D Systems, Minneapolis, MN) were added in RPMI 1640 to the lower chambers. After allowing invasion to proceed for 24 hours, cells that passed through the filter were counted in a similar manner to the Boyden chamber assay outlined above.
Measurement of VSMC Adhesion
AdCCN5- or AdGFP-infected VSMCs were trypsinized and subsequently plated into 24-well dishes at a density of 3 × 105 cells per well in 300 μl. Dishes were either tissue culture plastic, or collagen coated. Cells were allowed to attach for various amounts of time, then fixed and assayed as described. 24 Briefly, 1% methylene blue was used for staining, and OD650 measurements reflecting the extent of staining were taken on a 96-well plate reader.
Detection of Apoptosis
The ApoAlert Caspase-3 Colorimetric Assay kit by Clontech (Palo Alto, CA) was used according to the manufacturer’s instructions to determine the amount of apoptosis in VSMC cultures. Briefly, 2 × 106 AdGFP-infected, AdCCN5-infected, or uninfected control cells were growth-arrested then collected after overnight incubation in 10% FCS, washed in phosphate-buffered saline, and then the lysates were assayed for caspase-3 activity. Exposure to staurosporine at 2.5 μmol/L in 10% FCS/RPMI overnight was used as a positive control for apoptosis. 25
Results
CCN5 Is Expressed by VSMCs during Growth Arrest
Previous work in our laboratory has demonstrated that VSMCs express CCN5 mRNA in a growth arrest-specific pattern. 3 CCN5 mRNA expression is absent in exponentially growing VSMCs and is up-regulated on entry into G0. Expression of the mRNA is rapidly lost on re-entry into the cell cycle. Others have shown that CCN5 is a secreted protein that is tightly associated with the cell surface. 26 To determine whether CCN5 protein expression follows the previously observed pattern of mRNA expression in growth-arrested and exponentially growing VSMCs and to ascertain if the protein is membrane-associated, we used two independent approaches: immunocytochemical staining methods to visualize CCN5 on VSMCs and Western blotting of cell lysates and conditioned medium for CCN5 to observe the expression pattern.
Using an affinity-purified anti-peptide CCN5 antibody, we demonstrate that VSMCs express CCN5 on the cell surface during G0 (Figure 1, A and F) ▶ . When VSMCs were fixed but not permeabilized the staining appeared as diffuse staining over the entire cell surface (Figure 1A) ▶ . To confirm that the diffuse pattern of CCN5 staining was cell surface-associated, we examined the pattern of immunofluorescence in membrane-extracted cells. When cells were stripped of their plasma membrane before fixing and staining, the diffuse staining disappeared and a distinct perinuclear localization of CCN5 was seen, suggesting a possible Golgi localization of the protein (Figure 1B) ▶ . This pattern is also observed in cells along with surface staining in VSMCs permeabilized after fixing (Figure 1B, G ▶ 0, arrows). Fluorescein isothiocyanate-conjugated phalloidin staining showed a pattern of microfilament stress fibers typical of growth-arrested VSMCs (Figure 1B) ▶ . After reintroduction of 10% FCS/RPMI the cells rapidly lost CCN5 cell-surface staining. The loss was first observed from the surface within 2 hours, followed by loss of all staining throughout the cell; by 18 hours CCN5 was completely undetectable (Figure 1F) ▶ . As negative controls, fixed cells were incubated with antibody preincubated with the specific peptide antigen used to immunize the rabbits (Figure 1C) ▶ , or secondary antibody alone (Figure 1D) ▶ ; no staining was observed in either case.
Figure 1.
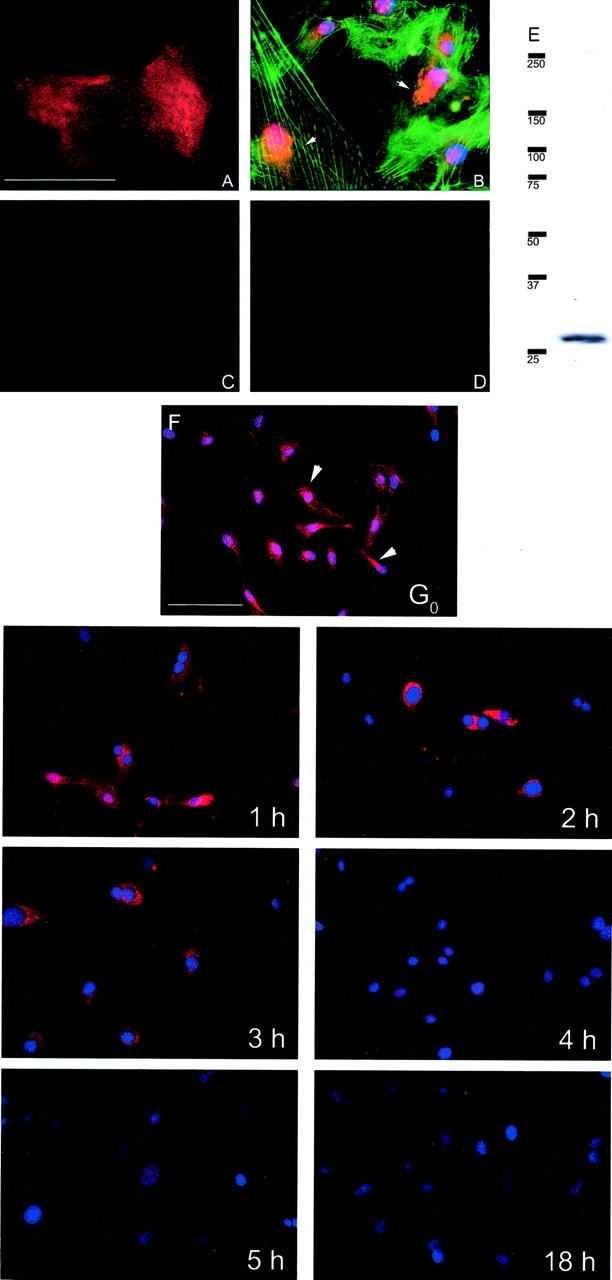
CCN5 is expressed on the surface of VSMCs during growth arrest and lost on reintroduction to serum. VSMCs were plated onto 18-mm coverslips, growth-arrested by serum starvation for 72 hours, fixed, and subjected to immunocytochemistry as described in Experimental Procedures. A: Nonpermeabilized VSMCs stained for CCN5 (anti-CCN5; Texas Red secondary; red). B: VSMCs were Triton X-100 treated during fixing to extract the cellular membrane yet leave the cell intact. They were subsequently stained for CCN5 (anti-CCN5; Texas Red secondary; red), actin (fluorescein isothiocyanate-conjugated phalloidin; green), and nuclei (Hoechst 33258 stain; blue). C and D: Negative controls: peptide preincubated and secondary alone, respectively. E: Western blot showing the presence of CCN5 in conditioned medium from growth-arrested VSMCs. F: G0 to 18 hours: VSMCs on coverslips were growth-arrested by serum starvation for 72 hours and subsequently reintroduced to RPMI medium containing 10% FCS. Hoechst 33258 stain was added to mounting medium to visualize nuclei. Panels are labeled according to time course (G0 to 18 hours). Arrows denote perinuclear staining. Scale bar, 50 μm.
Because CCN5 is a secreted protein in other cell types, we also looked for its presence in conditioned medium taken from growth-arrested VSMCs. In the 20- to 250-kd range we observe a single band corresponding to full-size CCN5 at the expected molecular weight (Figure 1E) ▶ . The presence of full-length CCN5 in VSMC conditioned medium as well as on the cell surface suggests that it could exert its effects in both an autocrine and paracrine manner.
The disappearance of CCN5 expression shown by immunocytochemistry was corroborated by a Western blot of cell lysates (Figure 2) ▶ . There was a fivefold decrease in CCN5 levels 2 hours after reintroduction of 10% FCS/RPMI as shown by densitometry. These results, along with the immunofluorescence data, demonstrate that the temporal expression pattern of CCN5 protein closely follows its mRNA counterpart, and that CCN5 is a secreted, plasma membrane-associated protein.
Figure 2.

Western blot depicting disappearance of CCN5 from VSMC lysates after release from G0. Cell lysates were collected from VSMCs after growth arrest by serum starvation for 72 hours and reintroduction to 10% FCS/RPMI. Forty μg of each lysate were then used for SDS-PAGE and transferred to polyvinylidene difluoride membranes for Western blotting with the anti-CCN5 antibody as described in Experimental Procedures. Amido Black staining (not shown) insured equal loading of protein on the gel. An ECL film from this experiment is shown in the inset. The graph depicts relative densitometry of the bands from ECL. The band representing CCN5 appears at ∼28 kd.
CCN5 Expression in Rat Carotid Artery Balloon Injury Model
Earlier studies in our laboratory revealed that CCN5 mRNA is present in tissue taken from uninjured artery. 3 To observe the expression pattern of CCN5 protein in uninjured, as well as injured artery we used the rat carotid artery balloon injury model. Sections from uninjured carotids and vessels 2 and 14 days after injury were probed for CCN5 protein. Uninjured carotid sections show the strongest expression of CCN5 protein in the medial portion of the vessel, in which the VSMCs are quiescent. The specificity of the CCN5 staining is shown by the observation that it can be competed away by addition of the antigenic peptide (Figure 3) ▶ . On day 2 after injury, vessels that have the highest fraction of cycling VSMCs, 15 show greatly reduced levels of CCN5 in the media (Figure 3) ▶ . In the day 14 after injury vessels, myointimal lesion formation is essentially complete and the VSMCs have stopped proliferating. CCN5 staining is observed throughout the fully formed lesion as well as in the media of the vessel (Figure 3) ▶ . This data demonstrates an expression pattern for CCN5 in the carotid artery that is consistent with a role for CCN5 in regulating the migration and proliferation of VSMCs in uninjured and injured artery wall.
Figure 3.

CCN5 expression pattern in rat carotid artery balloon injury model. The common carotid artery of a male rat was subjected to a de-endothelializing injury with an inflated balloon catheter inserted. Rats were sacrificed just before injury and at 2 and14 days after injury. The injured segment of the artery was dissected free of surrounding tissue, fixed, and imbedded in paraffin. Sections were cut, dewaxed, and stained for CCN5 using the anti-CCN5 rabbit polyclonal antibody described earlier. Texas Red-conjugated donkey anti-rabbit antibody was used as secondary. A: Uninjured carotid artery. B: Day 2 after injury. C: Day 14 after injury. D: Uninjured carotid artery, preincubated with blocking peptide. E: Day 14 after injury, preincubated with blocking peptide. F: Day 14 after injury, secondary antibody alone.
Heparin Maintains CCN5 Expression in VSMCs
Treatment with heparin has been shown to maintain high levels of CCN5 mRNA expression in VSMCs after release from growth arrest. 3 To determine whether CCN5 protein expression follows the same pattern, we used Western blotting of cell lysates from growth-arrested cells treated with 10% FCS/RPMI with and without heparin (Figure 4) ▶ . CCN5 protein levels were high in growth-arrested cells and dropped significantly within 4 hours in the presence of 10% FCS. Growth-arrested VSMCs exposed for 4 hours to 10% FCS/RPMI containing 300 μg/ml of heparin had CCN5 levels essentially identical to growth-arrested cells. Our results demonstrate that heparin is able to maintain the expression of CCN5 protein in the presence of 10% FCS, in agreement with observations made for CCN5 mRNA. 3
Figure 4.

Heparin maintains expression of CCN5 protein after release from G0. VSMCs were growth-arrested by serum starvation for 72 hours and subsequently reintroduced to medium containing 10% FCS/RPMI with and without heparin for 4 hours. Forty μg of each cell lysate were used for SDS-PAGE, Western blotting, and probed with anti-CCN5 peptide antibody (top). Lane 1: Lysate from G0 VSMCs. Lane 2: Lysate from cells treated with 10% FCS. Lane 3: Lysate from cells treated with 10% FCS/RPMI and 300 μg/ml of heparin. Amido Black staining (not shown) insured equal loading of protein on gel. The band representing CCN5 appears at ∼28 kd. Bottom: Northern blot analysis of CCN5 mRNA levels in VSMCs treated under exactly the same conditions used for the Western blot in the top panel. The Northern blot data are taken from Delmolino and colleagues 3 and is included here to permit direct comparison of CCN5 mRNA and protein levels.
Cells Infected with AdCCN5 Express CCN5 Protein
In our hands, VSMCs are relatively resistant to retroviral or DNA transfection methods. To circumvent this problem, we constructed a nonreplicating adenovirus containing an epitope-tagged version of the rat CCN5 cDNA sequence driven by a CMV promoter. In addition, the adenovirus construct also contains the green fluorescent protein (gfp) gene driven by a separate CMV promoter. By using GFP as a marker it is possible to quickly ascertain the extent of infection produced by adenovirus treatment. The relative GFP fluorescence level provides a quick measure to compare levels of infection between controls and AdCCN5-infected cells.
We observed rates of infection approaching 90 to 95% in VSMCs as a result of adenoviral gene transfer. Using our AdCCN5 virus to infect VSMCs resulted in strong expression of the tagged CCN5 protein (Figure 5 ▶ ; A to C) and high levels of fluorescence (Figure 5D) ▶ . The band corresponding to CCN5 cross-reacted with the specific anti-CCN5 anti-peptide antibody (Figure 5A) ▶ . When equivalent blots were probed with either the anti-CCN5 antibody or the HA tag-specific antibody a single band was observed (Figure 5, A and B) ▶ .
Figure 5.

AdCCN5 produces CCN5 protein. Forty μg of cell lysates from VSMCs infected with AdGFP or AdCCN5 (300 moi) were used for SDS-PAGE and Western blotting to probe for CCN5 using either anti-CCN5 rabbit polyclonal or anti-HA epitope-tag antibodies as described in Experimental Procedures. A: Blot probed with rabbit polyclonal anti-CCN5 peptide antibody. Lane 1: Lysate from AdGFP-infected VSMCs. Lane 2: Cell lysate from AdCCN5-infected VSMCs. B: Blot probed with anti-HA epitope-tag antibody. Lanes 1 and 2 contain same lysates as in A. C: Western blot (probed with anti-HA epitope-tag antibody) and densitometric analysis depicting change in CCN5 protein levels (right axis) and percent GFP-expressing cells (left axis) relative to moi. Lane 1: Uninfected VSMC lysate. Lane 2: AdGFP 300 moi lysate. Lane 3: AdCCN5 10 moi lysate. Lane 4: AdCCN5 30 moi lysate. Lane 5: AdCCN5 100 moi lysate. Lane 6: AdCCN5 300 moi lysate. D: GFP expression in cells infected with 100 moi AdCCN5 visualized by fluorescence microscope (green). E: AdCCN5-infected, nonpermeabilized VSMCs stained for HA epitope-tag (anti-HA epitope-tag; Texas Red secondary; red). F: AdGFP-infected, nonpermeabilized VSMCs stained for HA epitope-tag (anti-HA epitope-tag; Texas Red secondary; red).
In addition to detection of the expressed CCN5 in Western blots we also observe cell-surface expression of HA-tagged CCN5 in cells infected with the AdCCN5 virus (Figure 5E) ▶ . The staining pattern observed is the same as that observed for the native protein (Figure 1A) ▶ . AdGFP-infected cells show no staining (Figure 5F) ▶ . This result indicates that virally expressed protein is secreted from the cell and remains membrane associated as observed in growth-arrested VSMCs.
It is also possible to modulate the level of CCN5 expression by changing the multiplicity of infection (moi) (Figure 5C) ▶ . Based on Western blot analysis, there was a dose-dependent increase in CCN5 protein levels in cell lysates as the moi increased, with 300 moi producing approximately sevenfold more CCN5 than 10 moi. There was also a dose-dependent increase in the percentage of cells expressing GFP corresponding directly to the relative amount of CCN5 present in the lysate (Figure 5C) ▶ . AdGFP controls and AdCCN5-treated cells displayed the same amount of GFP at each respective moi, confirming approximately equal rates of infection. Our observations indicate that infection in the range of 30 to 60 moi produces CCN5 levels similar to that of a growth-arrested cell, whereas moi in the range of 100 to 300 represent modest overexpression of CCN5 (threefold to fivefold increase). These results indicate that the adenovirus vector described above is a useful tool for studying CCN5 protein expression in VSMCs.
AdCCN5 Inhibits VSMC Proliferation
Earlier studies in our laboratory showed that CCN5-conditioned medium is capable of inhibiting VSMC proliferation. 3 To provide more definitive evidence for an anti-proliferative function of CCN5, we infected VSMCs with AdCCN5 and measured changes in cell number after release from G0; AdGFP was used as a negative control (Figure 6) ▶ , as were uninfected cells. VSMCs showed a dose-dependent decrease in proliferation on infection with AdCCN5 as compared to AdGFP-infected controls. At maximal infection (300 moi), there was >50% inhibition of cell number after 48 hours in 10% FCS/RPMI (Figure 6) ▶ . At 100 moi, the reduction in cell number was less but still significant (∼28%; Figure 6 ▶ ). Lower moi (30 or below) did not have a significant inhibitory effect consistent with the greatly reduced level of CCN5 protein expressed at these moi (Figure 5C) ▶ . These data strongly support the idea that CCN5 is capable of inhibiting VSMC proliferation and is consistent with other observations that suggest it might play a role in regulating VSMC mitogenesis.
Figure 6.

Infection with AdCCN5 inhibits VSMC proliferation. Cells (8 × 103 VSMCs/well), previously infected with either AdCCN5 or AdGFP control virus, were plated overnight into 24-well dishes, washed with RPMI, and placed in RPMI containing 0.4% FCS/RPMI for 72 hours. Cells were released from quiescence by replacing the low serum medium with normal growth medium, RPMI 1640 containing 10% FCS. Cell proliferation was determined by direct cell counting using a Coulter counter. The net growth of VSMCs was obtained by subtracting the starting cell number from the final count results (at the time of release from G0).
CCN5 Suppresses VSMC Motility
VSMC migration from the media into the intimal surface of the blood vessel is an important step during restenosis after vascular surgical procedures. To determine whether CCN5 affects VSMC motility, we used two independent approaches: a modified Boyden assay and a monolayer scratch wound assay.
Boyden chamber assays are well established for measurement of cell motility. 11,17,18 As positive controls, we placed different concentrations of FCS (a strong VSMC attractant) in the lower chambers, while AdCCN5-infected VSMCs in 0.1% FCS/RPMI were placed on top of the filter. FCS/RPMI (0.1%) was used in the lower chamber as a negative control. Inhibition of motility was observed within 2 to 3 hours in VSMCs infected with 100 moi AdCCN5 over a wide range of FCS concentrations (0.1 to 10%; Figure 7 ▶ ). Significant inhibition of motility was seen at FCS concentrations up to 5%, with the greatest degree of inhibition observed at 0.5% FCS. Under these conditions, motility was reduced by nearly half in VSMCs (Figure 7) ▶ . AdGFP-infected controls showed no change in motility compared to uninfected cells at either level of infection. When higher concentrations of FCS were used, there was a steady decrease in the ability of AdCCN5 to inhibit motility, suggesting that high levels of serum provide a bolus of motility-stimulating factors that can override the CCN5 effect, especially during the short time period (2 to hours) allowed for the assay (Figure 7) ▶ . Thirty moi produced a moderately lower level of inhibited cell movement (unpublished data).
Figure 7.

VSMC motility is inhibited by CCN5 in Boyden chamber assays. VSMCs previously infected with 100 moi AdGFP (control) or AdCCN5 as well as uninfected VSMCs were trypsinized and then resuspended at 7 × 105 cells/ml. The lower chambers of the 96-well Boyden chamber were then loaded with 0.1, 1, 2, 5, and 10% FCS/RPMI and overlaid with an 8-μm polycarbonate filter, which was previously coated with collagen I (Vitrogen). Three hundred μl of the above cell suspensions were then added to the upper chambers and allowed to migrate through the membrane at 37°C. After 2.5 hours, filters were removed, fixed, and then stained with sulfarhodamine B. Cells on the bottom side of the filter were counted under a light microscope with an eyepiece grid to visualize set fields. Each condition included triplicate wells counted in quadruplicate fields. Similar results were obtained using 30 moi, although the magnitude of the decrease was somewhat less (unpublished data).
Monolayer scratch wound assays have also been used to study VSMC motility. 19,20 Using Hoechst staining to visualize nuclei 30 hours after wounding revealed that significantly fewer cells had migrated into the wound in monolayers composed of AdCCN5 (300 moi)-treated VSMCs compared to AdGFP-infected or uninfected (Figure 8; A to F) ▶ . The anti-motility effect of AdCCN5 in this assay system was observed over a range of moi (Figure 8G) ▶ . A 32% reduction in wound filling was seen at 3 moi; this rose to a maximum of 66% at 300 moi. There was no significant change in cell motility for AdGFP-infected VSMCs versus uninfected VSMCs over the entire range of moi tested (Figure 8 ▶ ; A to F; unpublished data). It is possible that CCN5 inhibition of cell division could account for the differences in the scratch wound assay. Contribution of cell division to the counts was minimized by using a 30-hour time point. We have previously shown that contact inhibited (G0) VSMCs cultured in these precise conditions require 32 to 36 hours before a significant increase in cell number are seen. 12,27 Using Hoechst stain we observed mitotic figures in <1% of the cells in both control and AdCCN5-infected cultures at the 30-hour time point. Taken together, the results of the Boyden chamber assay and the monolayer scratch wound assay support the idea of an anti-motility function for CCN5 in VSMCs.
Figure 8.
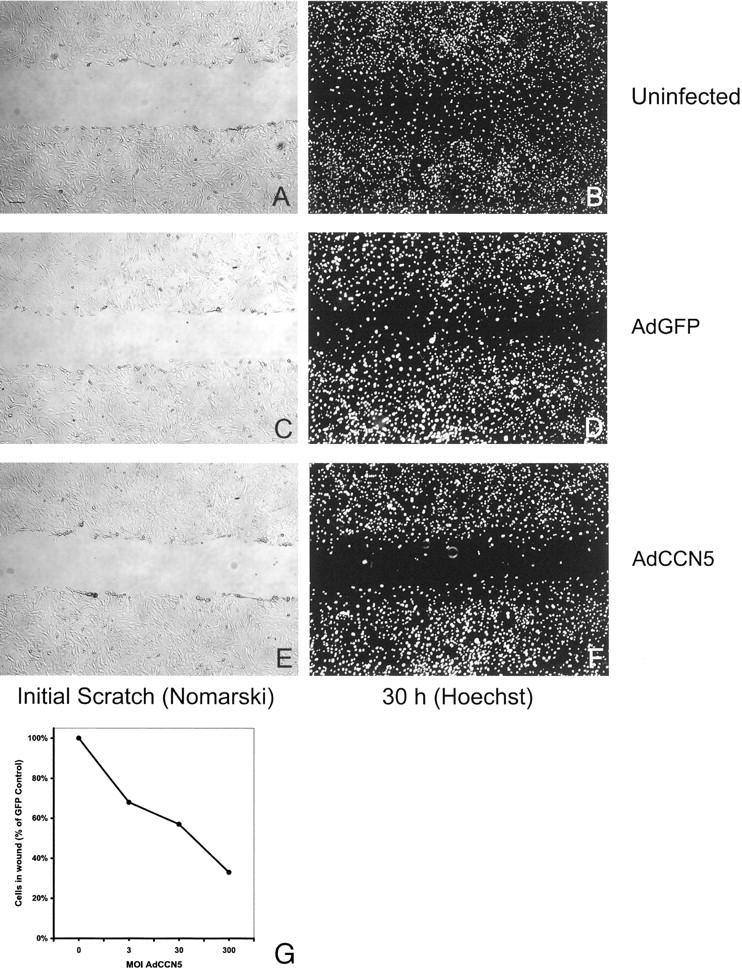
CCN5 suppresses VSMC migration in a monolayer scratch wound assay. Representative photographs from monolayer scratch wound assay. Infected (AdGFP or AdCCN5 300 moi) or uninfected VSMCs were plated at confluence onto glass chamber slides previously coated with collagen I. The following day a uniform straight scratch was made in the monolayer using a 200-μl yellow plastic pipette tip (Fisher). Monolayers were washed gently, marked (for reference), and photographed using Nomarski optics on an inverted microscope. After incubation for 30 hours at 37°C, the cells were fixed in 1% paraformaldehyde, and mounted. Hoechst 33258 was used stain to visualize nuclei. Stain was photographed using a fluorescence microscope. A: Uninfected, immediately after scratch. B: Uninfected, 30 hours. C: AdGFP 300 moi, immediately after scratch. D: AdGFP, 30 hours. E: AdCCN5 300 moi, immediately after scratch. F: AdCCN5, 30 hours. G: VSMC monolayers were infected with AdGFP or AdCCN5 (0, 3, 30, or 300 moi) and scratched. Nuclei of VSMCs that migrated into the wound were counted using images of Hoechst 33258-stained monolayers. AdCCN5 VSMCs were compared to AdGFP counterparts infected at similar moi. Scale bar, 50 μm.
CCN5 Expression Inhibits VSMC Invasiveness
Although VSMCs are generally not thought of as invasive, they do need to pass though the basement membrane of the endothelial cell layer to reach the intimal surface of the artery wall. In addition, nonvascular SMCs occasionally form leiomyosarcomas, which are invasive cancers. To model the in vivo process, we used invasion chambers coated with growth factor-reduced Matrigel (BD Biosciences), which may be thought of as a partially reconstituted basement membrane. This assay has been used extensively to assay invasiveness. 21-23 Similar to the Boyden assay, VSMCs must pass though a polycarbonate filter with 8-μm pores. In contrast to the Boyden assay, which represents short-term (2 to 3 hours) movement of cells through open 8-μm pores with an essentially monomolecular coating of collagen I, this assay represents a long-term (48 hours) invasion of VSMCs through occluded pores in a relatively thick layer of Matrigel. Interestingly, 5% FCS, although a good inducer of invasiveness, did not provide the maximal invasive response in this assay. Both PDGF-AB and PDGF-BB induced a much higher invasive response than FCS. For example, 5% FCS-induced invasion was threefold less than 5 ng/ml of PDGF-AB (5.2 versus 16.7 cells/field; Figure 9 ▶ ) and fivefold less than 25 ng/ml PDGF-AB (5.2 versus 25.8 cells/field; Figure 9 ▶ ) in uninfected control cells. In AdGFP-infected controls, both PDGF-AB- and PDGF-BB-induced invasion was similar to that of uninfected VSMCs (29 and 25.8 cells/field, respectively, at 25 ng/ml; 18.2 versus 16.7 cells/field, respectively, at 5 ng/ml; Figure 9 ▶ ). In all cases, AdCCN5-infected cells were much less invasive when compared to their control AdGFP-infected or uninfected control counterparts. For example, invasiveness stimulated by FCS was reduced by 85% in AdCCN5-infected cells. In AdCCN5-infected cells exposed to either PDGF-AB or PDGF-BB, invasiveness was reduced by >80% at both 5 ng/ml and 25 ng/ml (Figure 9) ▶ . These results support the notion that CCN5 is capable of inhibiting invasion of cells through a basement membrane rather than simply altering motility.
Figure 9.

VSMC invasiveness is inhibited by CCN5. VSMC (3 × 104 cells per well in 500 μl) were plated into the upper portion of growth factor reduced-Matrigel chambers (BD Biosciences) and allowed to invade for 24 hours in response to either PDGF (AB or BB, 5 or 25 ng/ml) or 10% FCS. Filters were cleaned, fixed, stained, and counted as described in Experimental Procedures.
CCN5 Does Not Alter VSMC Adhesiveness
Other CCN family members have been implicated in altering the adhesiveness of various cell types. 24,28-30 We therefore considered the possibility that the presence of CCN5 on the surface of VSMCs could alter adhesion of the cell to its substrate, because this could account (at least in part) for the decrease in cell number observed in the proliferation and motility assays already described. We examined the ability of uninfected control, AdGFP-infected, and AdCCN5-infected VSMCs to adhere to both collagen I and tissue culture plastic, using a standard cell adhesion assay system (Figure 10) ▶ . 24 Although these assays do not address the contribution of CCN5 to adhesion in a complex matrix environment, they may offer some insight into the adhesion properties of VSMCs expressing CCN5. On plastic, cell attachment was essentially the same in all four cell conditions at both 30 and 100 moi over a range of times (30 minutes to 4 hours; Figure 9A ▶ ), as demonstrated by virtually identical slopes in all of the conditions tested. The results observed for attachment to collagen I were virtually identical to those obtained for plastic (Figure 10B) ▶ . These data show that overexpression of CCN5 on the VSMC surface does not alter adhesiveness to tissue culture surfaces in vitro.
Figure 10.

AdCCN5 infection of VSMCs does not affect adhesion to collagen I or plastic. VSMCs (3 × 105 cells in 300 μl per well) were added to 24-well dishes that were either tissue culture treated or collagen I coated. Cells were washed, fixed, and stained at different time points (0.5, 1, and 4 hours) using methylene blue. OD650 measurements were taken of extracted stain to assess the amount of cell material present. Note that the slopes are virtually identical in all of the conditions tested. A: VSMC plated on plastic. B: VSMC plated on collagen I. For both A and B, maximum SE ± 0.069 OD; no statistically significant differences were observed (P < 0.05).
Neither CCN5 nor Adenoviral Infection Affect Apoptosis in VSMC
It is possible that apoptosis induced by either adenoviral infection or CCN5 itself could account, wholly or in part, for the reduction in cell number observed in the various assays performed above. To test this, we used the Clontech ApoAlert Caspase-3 Colorimetric Assay kit (Figure 11) ▶ . Staurosporine (2.5 μmol/L, 20 hours), which is known to induce apoptosis in VSMC, 25 was used as a positive control. Uninfected VSMC OD405 values representative of background caspase-3 activity were more than threefold lower than the positive control values (0.009 compared to 0.03; Figure 11 ▶ ). Using an inhibitor to caspase-3 abolished the activity in the staurosporine-treated cells, indicating that the observed OD405 values can be attributed to caspase-3. The observed levels of caspase-3 activity in both AdGFP- and AdCCN5-infected VSMC were at or below the levels seen in uninfected cells (ie, below 0.009) at moi up to 300 (Figure 11) ▶ . To confirm that neither CCN5 nor adenoviral infection is able to induce apoptosis in VSMCs, we used a marker for mitochondrial breakdown in AdCCN5-infected cells compared to uninfected VSMCs. No change in the very low fraction of apoptotic cells was observed using this assay (unpublished data). The results of these two independent apoptosis assays indicate that elevated rates of programmed cell death do not play a role in the proliferation or motility assays we use, and further suggest that apoptosis is unlikely to play a significant role in the mechanism of action of CCN5 on VSMCs.
Figure 11.

AdCCN5 infection of VSMCs does not affect apoptosis. VSMCs (2 × 106) infected with 100 or 300 moi AdGFP or AdCCN5 were collected after overnight incubation in 10% FCS, washed in phosphate-buffered saline, and then the lysates were assayed for caspase-3 activity. Exposure to staurosporine at 2.5 μmol/L in 10% FCS/RPMI overnight was used as a positive control for apoptosis. Caspase-3 inhibitor was also added as a control to show that measured OD is because of caspase-3 activity. Values have blank (OD405 for reaction mix without substrate) subtracted. S, staurosporine; S+I, staurosporine plus inhibitor.
Discussion
In this study, we provide functional evidence that CCN5, a member of the CCN family of proteins, can inhibit VSMC proliferation, motility, and invasiveness. In contrast, adhesion and apoptosis are unaffected by CCN5 in this cell type. We also significantly extend previous data from our laboratory that suggests CCN5 is a growth arrest-specific (gas) gene. Furthermore, we map for the first time the cellular localization of CCN5 protein in cultured VSMCs as well as in the arterial wall and show that it is temporally and spatially expressed in a manner consistent with a role in regulating mitogenesis, migration, and invasiveness (Figures 1 to 4) ▶ ▶ ▶ . Using the rat carotid balloon injury model we demonstrate that CCN5 expression is present whenever VSMCs are in a quiescent growth state (uninjured and 14 days after injury) 15 and is greatly reduced when VSMCs are actively cycling (2 days after injury). 15 Of particular interest is the observation that the media strongly expresses CCN5 in uninjured blood vessels (Figure 3) ▶ , an observation corroborated by horseradish peroxidase/diaminobenzidine staining (data not shown). These in vivo results place CCN5 at precisely the right time and place to play an important role in maintaining quiescence of VSMCs in the uninjured artery wall and in myointimal lesion stabilization after injury.
Using two independent approaches, we show that CCN5 can inhibit VSMC motility (Figures 7 to 8) ▶ ▶ . To our knowledge, this is the first demonstration in any cell type of a motility-regulating activity for CCN5. If these activities are present in arterial wall SMC, it is tempting to speculate that CCN5 might serve to maintain the normally nonmotile and quiescent state of VSMCs in the uninjured artery. This possibility is especially interesting because a strong up-regulation of CCN2 has been implicated in the progression of atherosclerosis, 31 and this CCN protein is also induced by PDGF and TGF-β. 32,33 We also show that CCN5 suppresses VSMC invasiveness (Figure 9) ▶ . Interestingly, PDGF was a much stronger inducer of invasiveness than serum, whereas serum stimulated motility to a greater extent than PDGF. Previous work in our laboratory and by others 18,34 has shown that PDGF plays an important role in VSMC migration and invasiveness. We showed that normal expression of CCN5 in VSMCs during growth arrest was ablated by treatment with PDGF-BB. 3 Although VSMCs are not usually considered an invasive cell, they do migrate through an endothelial basement membrane to form myointimal lesions after vascular surgery and in early atherosclerotic plaques. Furthermore, nonvascular SMCs can occasionally become invasive tumors (leiomyosarcomas). Finally, it is possible that the anti-invasive action of CCN5 might be applicable in other metastatic cells, regardless of origin. These possibilities require further experimentation.
Our data also provide the most direct demonstration to date that cell proliferation can be suppressed by a CCN family member (Figure 6) ▶ . This finding is in contrast to the suggestions made for CCN5 function in other cell types and for the functions of other CCN family proteins. For example, Zhang and colleagues 26 showed that CCN5 had no effect on the proliferation of Rat-1 fibroblasts and was expressed during S phase of the cell cycle. This pattern of expression suggests that in Rat-1 cells, CCN5 does not suppress mitogenesis. Kumar and colleagues 29 observed effects of CCN5 on adhesion and osteocalcin production in osteoblasts but did not examine the effect of CCN5 on the proliferation of osteoblasts. Interestingly, two separate groups suggest that CCN5 may stimulate proliferation of MCF-7 breast tumor cells. Inadera and colleagues 6 reported that CCN5 is an estrogen-responsive gene in human breast carcinoma cells (MCF-7), and Zoubine and colleagues 35 showed that CCN5 is serum-inducible and expressed to a much higher extent in MCF-7 cells than cells from normal breast. The differences between our results for VSMCs and observations made in other cell types suggest a complex tissue- and cell-specific set of functions for CCN5 and may reflect different functions of CCN5 in epithelial tissue derivatives compared to mesenchymally derived cells.
Other CCN family members have been implicated in control of cell proliferation as well. Although it is hypothesized that CCN3 may be a negative regulator of proliferation, there is no direct evidence to date supporting this function. In fact, recombinant CCN3 has been unable thus far to inhibit proliferation of cells in culture, including VSMCs. 24 Perhaps the best understood CCN proteins, CCN1 and CCN2, are immediate-early serum-responsive genes expressed by fibroblasts as well as by other cell types. 9,10 They have been shown to stimulate cell proliferation, 36,37 and there is evidence to suggest that the carboxy-terminal domain is responsible for this activity in uterine SMC. 38 Taken together, these observations suggest the intriguing possibility that CCN5 may represent a naturally occurring antagonist to the 4-domain members of the CCN family.
CCN5 does not modulate adhesiveness of VSMCs under the conditions tested in this study (Figure 10) ▶ . In contrast, Kumar and colleagues 29 demonstrate that recombinant CCN5-coated plastic is conducive to osteoblast attachment in a dose-dependent manner. Indeed, other CCN members have also been shown to affect cell adhesion. Ellis and colleagues 24 found that CCN3 increases VSMC cell adhesiveness. CCN1 and CCN2 have also been shown to increase adhesion via an integrin-dependent pathway. 30,37,39 It should be noted, however, that we cannot rule out the possibility that within the complex matrix milieu of a tissue environment CCN5 could have an effect on VSMC adhesion.
Based on two separate assay systems, we have shown that CCN5 does not induce apoptosis in growth-arrested VSMC exposed to serum (Figure 11 ▶ ; unpublished data). Again, this observation stands in counterpoint to the observations of others that CCN proteins, including CCN5, are proapoptotic. For example, CCN5 has been shown to induce apoptosis in transformed fibroblasts, 26 and overexpression of CCN2 is thought to be responsible for apoptosis in VSMCs. 40 Although CCN2 is also proapoptotic in MCF-7 cells, 41 it is protective against this effect in endothelial cells. 30 A possible explanation for these paradoxical results is that CCN5 may actually be able to function as both a gas gene (thus participating in maintaining or returning the cell to the quiescent state) and as a proapoptotic stimulus. In normal cells, which possess the ability to exit the cell cycle and enter G0, the gas function permits the cell to become quiescent and survive, thus circumventing the proapoptotic mechanism. Transformed cells, however, lack the ability to exit the cell cycle, and in this setting CCN5 exposure activates a programmed cell death pathway. Alternatively, CCN5 may possess only a gas gene activity in both normal and transformed cells; the sensitivity of transformed cells to CCN5-induced apoptosis could be an indirect effect. For example, if CCN5 prevents the transformed cell from traversing the cell cycle it would be subject to other apoptotic stimuli within a relatively short time whereas the normal cell escapes into G0.
In this study we provide temporal, spatial, and functional data to support the hypothesis that CCN5 is capable of inhibiting three processes in cultured VSMCs that are thought to be important in the pathogenesis of arteriosclerosis and restenosis: proliferation, motility, and transmigration through the intimal basement membrane. These effects are not because of alterations in cell adhesion or induction of apoptosis. Corroborating the hypothesis is the observation that high levels of CCN5 mRNA are present in uninjured rat aortic tissue. 3 We extend this observation by showing that CCN5 protein levels are high in uninjured arterial wall, markedly reduced in the injured arterial wall, and return after stabilization of the myointimal lesion (Figure 3) ▶ . Although it is clear that the biological functions as well as the physiological roles, if any, for CCN5 are complex and likely to be tissue-specific, these data suggest a role for CCN5 in the control of VSMC motility and proliferation in both the uninjured and injured artery wall. Experiments to examine this possibility are currently underway and should help shed light on the role of CCN5 in VSMCs in vivo.
Footnotes
Address reprint requests to John J. Castellot, Jr., Ph.D., Department of Anatomy and Cell Biology, Tufts University School of Medicine, 136 Harrison Ave., Boston, MA 02111. E-mail: john.castellot@tufts.edu.
Supported by the National Institutes of Health (grant HL49973 to J. J. C.).
References
- 1.Mishra-Gorur K, Delmolino LM, Castellot JJ, Jr: Biological functions of heparan sulfate and heparan sulfate proteoglycans. Trends Glycosci Glycotechnol 1998, 10:193-210 [Google Scholar]
- 2.Delmolino LM, Stearns NA, Castellot JJ, Jr: Heparin induces a member of the CCN family which has characteristics of a growth arrest specific gene. Mol Biol Cell 1997, 8:287a. [DOI] [PubMed] [Google Scholar]
- 3.Delmolino LM, Stearns NA, Castellot JJ, Jr: COP-1, a member of the CCN family, is a heparin-induced growth arrest specific gene in vascular smooth muscle cells. J Cell Physiol 2001, 188:45-55 [DOI] [PubMed] [Google Scholar]
- 4.Ayer-Lelievre C, Brigstock D, Lau L, Pennica D, Perbal B, Yeger H: Report and abstracts on the first international workshop on the CCN family of genes. Mol Pathol 2001, 54:105-120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pennica D, Swanson TA, Welsh JW, Roy MA, Lawrence DA, Lee J, Brush J, Taneyhill LA, Deuel B, Lew M, Watanabe C, Cohen RL, Melhem MF, Finley GG, Quirke P, Goddard AD, Hillan KJ, Gurney AL, Botstein D, Levine AJ: WISP genes are members of the connective tissue growth factor family that are up-regulated in wnt-1-transformed cells and aberrantly expressed in human colon tumors. Proc Natl Acad Sci USA 1998, 95:14717-14722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Inadera H, Hashimoto S, Dong HY, Suzuki T, Nagai S, Yamashita T, Toyoda N, Matsushima K: WISP-2 as a novel estrogen-responsive gene in human breast cancer cells. Biochem Biophys Res Commun 2000, 275:108-114 [DOI] [PubMed] [Google Scholar]
- 7.Bork P: The modular architecture of a new family of growth regulators related to connective tissue growth factor. FEBS Lett 1993, 327:125-130 [DOI] [PubMed] [Google Scholar]
- 8.Oemar BS, Luscher TF: Connective tissue growth factor. Friend or foe? Arterioscler Thromb Vasc Biol 1997, 17:1483-1489 [DOI] [PubMed] [Google Scholar]
- 9.Lau LF, Lam SC: The CCN family of angiogenic regulators: the integrin connection. Exp Cell Res 1999, 248:44-57 [DOI] [PubMed] [Google Scholar]
- 10.Brigstock DR: The connective tissue growth factor/cysteine-rich 61/nephroblastoma overexpressed (CCN) family. Endocr Rev 1999, 20:189-206 [DOI] [PubMed] [Google Scholar]
- 11.Castellot JJ, Jr, Favreau LV, Karnovsky MJ, Rosenberg RD: Inhibition of vascular smooth muscle cell growth by endothelial cell-derived heparin. Possible role of a platelet endoglycosidase. J Biol Chem 1982, 257:11256-11260 [PubMed] [Google Scholar]
- 12.Castellot JJ, Jr, Pukac LA, Caleb BL, Wright TC, Jr, Karnovsky MJ: Heparin selectively inhibits a protein kinase C-dependent mechanism of cell cycle progression in calf aortic smooth muscle cells [published erratum appears in J Cell Biol 1990,110:863].J Cell Biol 1989, 109:3147-3155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B: A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA 1998, 95:2509-2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clowes AW, Reidy MA, Clowes MM: Kinetics of cellular proliferation after arterial injury. I Smooth muscle growth in the absence of endothelium. Lab Invest 1983, 49:327-333 [PubMed] [Google Scholar]
- 15.Wei GL, Krasinski K, Kearney M, Isner JM, Walsh K, Andres V: Temporally and spatially coordinated expression of cell cycle regulatory factors after angioplasty. Circ Res 1997, 80:418-426 [PubMed] [Google Scholar]
- 16.Castellot JJ, Jr, Cochran DL, Karnovsky MJ: Effect of heparin on vascular smooth muscle cells. I Cell metabolism. J Cell Physiol 1985, 124:21-28 [DOI] [PubMed] [Google Scholar]
- 17.Postlethwaite AE, Snyderman R, Kang AH: The chemotactic attraction of human fibroblasts to a lymphocyte-derived factor. J Exp Med 1976, 144:1188-1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grotendorst GR, Seppa HE, Kleinman HK, Martin GR: Attachment of smooth muscle cells to collagen and their migration toward platelet-derived growth factor. Proc Natl Acad Sci USA 1981, 78:3669-3672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Majack RA, Clowes AW: Inhibition of vascular smooth muscle cell migration by heparin-like glycosaminoglycans. J Cell Physiol 1984, 118:253-256 [DOI] [PubMed] [Google Scholar]
- 20.Lemire JM, Merrilees MJ, Braun KR, Wight TN: Overexpression of the V3 variant of versican alters arterial smooth muscle cell adhesion, migration, and proliferation in vitro. J Cell Physiol 2002, 190:38-45 [DOI] [PubMed] [Google Scholar]
- 21.Pauly RR, Passaniti A, Bilato C, Monticone R, Cheng L, Papadopoulos N, Gluzband YA, Smith L, Weinstein C, Lakatta EG: Migration of cultured vascular smooth muscle cells through a basement membrane barrier requires type IV collagenase activity and is inhibited by cellular differentiation. Circ Res 1994, 75:41-54 [DOI] [PubMed] [Google Scholar]
- 22.Zempo N, Koyama N, Kenagy RD, Lea HJ, Clowes AW: Regulation of vascular smooth muscle cell migration and proliferation in vitro and in injured rat arteries by a synthetic matrix metalloproteinase inhibitor. Arterioscler Thromb Vasc Biol 1996, 16:28-33 [DOI] [PubMed] [Google Scholar]
- 23.Palumbo R, Gaetano C, Melillo G, Toschi E, Remuzzi A, Capogrossi MC: Shear stress downregulation of platelet-derived growth factor receptor-beta and matrix metalloprotease-2 is associated with inhibition of smooth muscle cell invasion and migration. Circulation 2000, 102:225-230 [DOI] [PubMed] [Google Scholar]
- 24.Ellis PD, Chen Q, Barker PJ, Metcalfe JC, Kemp PR: Nov gene encodes adhesion factor for vascular smooth muscle cells and is dynamically regulated in response to vascular injury. Arterioscler Thromb Vasc Biol 2000, 20:1912-1919 [DOI] [PubMed] [Google Scholar]
- 25.Champagne MJ, Dumas P, Orlov SN, Bennett MR, Hamet P, Tremblay J: Protection against necrosis but not apoptosis by heat-stress proteins in vascular smooth muscle cells: evidence for distinct modes of cell death. Hypertension 1999, 33:906-913 [DOI] [PubMed] [Google Scholar]
- 26.Zhang R, Averboukh L, Zhu W, Zhang H, Jo H, Dempsey PJ, Coffey RJ, Pardee AB, Liang P: Identification of rCop-1, a new member of the CCN protein family, as a negative regulator for cell transformation. Mol Cell Biol 1998, 18:6131-6141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Castellot JJ, Jr, Addonizio ML, Rosenberg R, Karnovsky MJ: Cultured endothelial cells produce a heparinlike inhibitor of smooth muscle cell growth. J Cell Biol 1981, 90:372-379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kireeva ML, Mo F-E, Yang GP, Lau LF: Cyr61, a product of a growth factor-inducible immediate-early gene, promotes cell proliferation, migration, and adhesion. Mol Cell Biol 1996, 16:1326-1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar S, Hand AT, Connor JR, Dodds RA, Ryan PJ, Trill JJ, Fisher SM, Nuttall ME, Lipshutz DB, Zou C, Hwang SM, Votta BJ, James IE, Rieman DJ, Gowen M, Lee JC: Identification and cloning of a connective tissue growth factor-like cDNA from human osteoblasts encoding a novel regulator of osteoblast functions. J Biol Chem 1999, 274:17123-17131 [DOI] [PubMed] [Google Scholar]
- 30.Babic AM, Chen CC, Lau LF: Fisp12/mouse connective tissue growth factor mediates endothelial cell adhesion and migration through integrin alphavbeta3, promotes endothelial cell survival, and induces angiogenesis in vivo. Mol Cell Biol 1999, 19:2958-2966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oemar BS, Werner A, Garnier JM, Do DD, Godoy N, Nauk M, Marz W, Rupp J, Pech M, Luscher TF: Human connective tissue growth factor is expressed in advanced atherosclerotic lesions. Circulation 1997, 95:831-839 [DOI] [PubMed] [Google Scholar]
- 32.Bradham DM, Igarashi A, Potter RL, Grotendorst GR: Connective tissue growth factor: a cysteine-rich mitogen secreted by human vascular endothelial cells is related to the SRC-induced immediate early gene product CEF-10. J Cell Biol 1991, 14:1285-1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Igarashi A, Okochi H, Bradham DM, Grotendorst GR: Regulation of connective tissue growth factor gene expression in human skin fibroblasts and during wound repair. Mol Biol Cell 1993, 4:637-645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwartz SM: Smooth muscle migration in vascular development and pathogenesis. Transpl Immunol 1997, 5:255-260 [DOI] [PubMed] [Google Scholar]
- 35.Zoubine MN, Banerjee S, Saxena NK, Campbell DR: Banerjee SK WISP-2: a serum-inducible gene differentially expressed in human normal breast epithelial cells and in MCF-7 breast tumor cells. Biochem Biophys Res Commun 2001, 282:421-425 [DOI] [PubMed] [Google Scholar]
- 36.Frazier K, Williams S, Kothapalli D, Klapper H, Grotendorst GR: Stimulation of fibroblast cell growth, matrix production, and granulation tissue formation by connective tissue growth factor. J Invest Dermatol 1996, 107:404-411 [DOI] [PubMed] [Google Scholar]
- 37.Kireeva ML, Latinkic BV, Kolesnikova TV, Chen CC, Yang GP, Abler AS, Lau LF: Cyr61 and Fisp12 are both ECM-associated signaling molecules: activities, metabolism, and localization during development. Exp Cell Res 1997, 233:63-77 [DOI] [PubMed] [Google Scholar]
- 38.Ball DK, Surveyor GA, Diehl JR, Steffen CL, Uzumcu M, Mirando MA, Brigstock DR: Characterization of 16- to 20-kilodalton (kDa) connective tissue growth factors (CTGFs) and demonstration of proteolytic activity for 38-kDa CTGF in pig uterine luminal flushings. Biol Reprod 1998, 59:828-835 [DOI] [PubMed] [Google Scholar]
- 39.Jedsadayanmata A, Chen CC, Kireeva ML, Lau LF, Lam SC: Activation-dependent adhesion of human platelets to Cyr61 and Fisp12/mouse connective tissue growth factor is mediated through integrin alpha(IIb)beta(3). J Biol Chem 1999, 274:24321-24327 [DOI] [PubMed] [Google Scholar]
- 40.Hishikawa K, Oemar BS, Tanner FC, Nakaki T, Fujii T, Luscher TF: Overexpression of connective tissue growth factor gene induces apoptosis in human aortic smooth muscle cells. Circulation 1999, 100:2108-2112 [DOI] [PubMed] [Google Scholar]
- 41.Hishikawa K, Oemar BS, Tanner FC, Nakaki T, Luscher TF, Fujii T: Connective tissue growth factor induces apoptosis in human breast cancer cell line MCF-7. J Biol Chem 1999, 274:37461-37466 [DOI] [PubMed] [Google Scholar]