

Abstract
The mannose 6-phosphate/insulin-like growth factor 2 receptor (M6P/IGF2R) encodes a multifunctional protein involved in lysosomal enzyme trafficking, fetal organogenesis, tumor suppression, and T cell- mediated immunity. M6P/IGF2R is an imprinted gene in mice with expression only from the maternal allele. Complete knockout of this gene causes neonatal lethality, thus preventing analysis of its multifunctional role postnatally. To help elucidate the biological functions of M6P/IGF2R in adulthood, we generated both complete and tissue-specific M6P/IGF2R knockout mice using the Cre/loxP system. We confirm that complete M6P/IGF2R knockout results in fetal overgrowth and neonatal lethality. In contrast, tissue-specific inactivation of this gene in either the liver or skeletal and cardiac muscle gives rise to viable animals with no obvious phenotype. The successful creation of viable tissue-specific M6P/IGF2R knockout mouse models will now allow for detailed analysis of receptor function in a number of cellular processes including brain development, carcinogenesis, lysosomal trafficking, and T cell-mediated immunity.
Both the 275-kd cation-independent mannose 6-phosphate/insulin-like growth factor II receptor (M6P/IGF2R) and the 46- kd cation-dependent mannose 6-phosphate receptor (M6PR) function in the intracellular trafficking of lysosomal enzymes. 1,2 Homozygous M6PR null mice are both phenotypically normal and fertile, demonstrating that M6PR is not essential for either egg fertilization or organogenesis. 3,4 However, these animals exhibited defects in the targeting of multiple lysosomal enzymes, and increased levels of phosphorylated lysosomal enzymes are present in the body fluids. Thus, a physiological level of M6P/IGF2R does not compensate for M6PR loss, indicating that these two receptors target distinct subsets of lysosomal enzymes.
M6P/IGF2R encodes a multifunctional receptor that interacts with a diverse group of ligands not only intracellularly, but also at the cell surface. 1,5 Many of these ligands are glycoproteins that contain a mannose 6-phosphate (M6P) residue as a component of their N-linked oligosaccharide side chains. They include numerous lysosomal enzymes, the latent complex of transforming growth factor-β (TGF-β), and granzyme B. 1,2,6 In addition to binding M6P-modified glycoproteins, M6P/IGF2R also interacts specifically with several molecules through M6P-independent mechanisms. The best characterized of these is the mitogen insulin-like growth factor 2 (IGF2). It binds to a M6P-independent, high-affinity receptor binding site 7 that evolved with the appearance of Therian mammals. 8 Bound IGF2 is then internalized and transported to the lysosomes where it is degraded; M6P/IGF2R is subsequently recycled back to the membrane. 9 Thus, M6P/IGF2R controls the extracellular bioavailability of IGF2 and TGFβ, thereby regulating cell proliferation and apoptosis.
M6P/IGF2R function is critical for normal mammalian development. Mice that are deficient in M6P/IGF2R throughout fetal development die around the time of birth from a somatic overgrowth phenotype that is accompanied by severe skeletal, cardiac muscle, and lung abnormalities. 10-12 M6P/IGF2R-deficient mice can be rescued by a concomitant deficiency in IGF2 or the IGF1 receptor. In view of the diversity of ligands for M6P/IGF2R, it is surprising that this complex phenotype appears to result solely from the loss of the receptor’s IGF2-binding function. This indicates that it is the failure to target IGF2 to the lysosomes, and the subsequent excess signaling through the IGF1 receptor that gives rise to the lethal phenotype. Furthermore, large offspring syndrome frequently observed in cloned animals is highly associated with epigenetic changes in gene regulation that decrease M6P/IGF2R expression. 13
The variety and pleiotropic activities of the M6P/IGF2R ligand demonstrate that this receptor plays a key regulatory role in mammalian embryonic development, and indicate that altered receptor function could contribute to pathophysiology such as cancer. This postulate is supported by the finding that M6P/IGF2R loss of heterozygosity, coupled with intragenic loss-of-function mutations in the remaining allele, is a common early event in a number of human cancers. 14-18 Tumor cell growth is also inhibited when M6P/IGF2R expression is restored to normal and increased when gene expression is reduced. 19-21 The results of these mutational and functional studies clearly show that the M6P/IGF2R possesses the characteristics necessary to be classified as a tumor suppressor gene. 22
M6P/IGF2R is also an imprinted gene in most viviparous mammals, and is expressed only from the maternal allele. 23 M6P/IGF2R imprinting evolved approximately 150 million years ago in a common ancestor to marsupials and Eutherian mammals, 8,24 but was subsequently lost about 75 million years ago in an ancestral progenitor to primates. 25 Thus, in contrast to mice, both copies of M6P/IGF2R are functional in humans.
It is important, therefore, to clarify the function of M6P/IGF2R in postnatal life since alteration of receptor activity may affect not only cancer formation and treatment, 26,27 but also wound healing, 28 autoimmune disease, 29,30 cognitive function, 31 and organ transplantation. 6 Although perinatal lethality in M6P/IGF2R knockout mice can be overcome by a deficiency in either IGF2 or IGF1 receptors, these animals are severely growth inhibited. 11 Therefore, M6P/IGF2R knockout mice are preferable for studying receptor function in postnatal animals.
In this report, we describe the generation of mice carrying a modified M6P/IGF2R, in which exon 10 is flanked by loxP sites (floxed). Exposure of the floxed M6P/IGF2R to Cre-recombinase expressed from either albumin (Alb) or muscle creatine kinase (Ckmm) promoters resulted in viable mice deficient in liver or skeletal and cardiac muscle M6P/IGF2R, respectively. The availability of a M6P/IGF2R conditional knockout mouse will finally enable the complex biological functions of this multifunctional receptor to be thoroughly investigated in vivo.
Materials and Methods
To circumvent the neonatal lethality observed in M6P/IGF2R-deficient mice generated using classical embryonic stem cell targeting methods, 10-12 we used the Cre-loxP system to create M6P/IGF2R null alleles in a tissue- and time-dependent manner. 32 Animals in this study were maintained in facilities approved by the American Association for Accreditation of Laboratory Animal Care in accordance with current regulations and standards of the United States Department of Agriculture, the Department of Health and Human Services, and the National Institutes of Health. All mice were fed ad libitum on a diet of Purina Laboratory Chow and were maintained in an animal isolation facility on a standard 12-hour light/dark cycle. The study protocol was approved by the Administrative Panel on Laboratory Animal Care of Duke University.
Construction of the Targeting Vector
A targeting vector was constructed to enable Cre-mediated deletion of M6P/IGF2R exon 10. The exon/intron splice junctions for exons 9–10 and exons 10–11 are categorized as type 2 (codon interrupted after the second nucleotide) and type 1 (codon interrupted after the first nucleotide), respectively. 33 Thus, deletion of exon 10 from the mouse genome will shift the coding sequence out of frame, resulting in a stop codon and truncation of the receptor 5′ of both mannose 6-phosphate binding sites, the IGF2 binding site, and the transmembrane region. Approximately 5-kb of mouse (Sv129) M6P/IGF2R genomic sequence (from intron 7 to intron 10) was cloned immediately upstream of a dual thymidine kinase, neomycin resistance cassette previously engineered to be flanked by two loxP sites (Figure 1) ▶ . Approximately 2-kb of mouse genomic sequence corresponding to the remaining portion of M6P/IGF2R intron 10, exon 11, and intron 11 was then cloned downstream of the dual selection cassette. In two further modifications a third loxP site was introduced into intron 9 and a diptheria toxin selection cassette was inserted immediately downstream of the M6P/IGF2R sequence adjacent to intron 11.
Figure 1.

LoxP targeting of M6P/IGF2R exon 10. A: Generation of the M6P/IGF2R LoxP targeting construct involved the cloning of 7-kb of the mouse genomic DNA encompassing exons 8 through 11 and flanking intronic sequence into an appropriate LoxP vector. The M6P/IGF2R LoxP targeting construct was then transfected into Sv129 ES cells. Homologous recombinants were selected for using G418 (neomycin) and confirmed by PCR. B: Cre-recombinase plasmid (1 μg) controlled from the constitutively active CMV promoter was transfected into the ES cells which were then grown in the presence of gancyclovir to select for recombinants that had lost the TK/Neo cassette; PCR was used to confirm loss of the TK/Neo cassette. Recombinant ES cells were injected into the blastocoele cavity of the 3.5-day-old C57Bl/6J embryos before implantation into pseudo-pregnant mice. C: Expression of Cre-recombinase in mice with a floxed exon 10 results in its excision generating a M6P/IGF2R protein that encodes for a truncated receptor lacking the M6P and IGF2 extracellular binding domains. DT, TK, and Neo enable both positive and negative selection.
Generation of Floxed M6P/IGF2R Mice
Mouse Sv129 embryonic stem (ES) cells were transfected with the linearized targeting vector and grown in selection medium containing G418 (neomycin). Only cells that have integrated the G418 resistance (Neo) cassette and lost the diptheria toxin (DT) cassette survive in this medium. The G418-resistant cells were then transiently transfected with a vector in which expression of Cre recombinase is directed from the constitutively active cytomegalovirus (CMV)-promoter to delete the phosphoglycerate kinase (PGK)-Neo and PGK-TK genes flanked by loxP sites (Figure 1A) ▶ . Removing the thymidine kinase (TK) gene results in cells becoming resistant to gancyclovir. Gancyclovir-resistant cells were screened by polymerase chain reaction (PCR) (data not shown). Those that had undergone partial recombination, such that they had lost the TK-Neo selection cassette but maintained the floxed exon 10 of the M6P/IGF2R, were identified (11% of total). Following karyotype analysis, three clones were selected for micro-injection, and between 2 and 10 ES cells of individual clones were micro-injected into 3.5-day-old blastocysts isolated from C57Bl/6J donating females. Approximately 25 injected blastocysts were surgically implanted into the uterus of each of three pseudo-pregnant female mice. From the resultant litters, three chimeric females were obtained which were then back-crossed with C57Bl/6J males. Germ-line transmission of the floxed exon 10 sequence by one of the three chimeras was confirmed by PCR and sequence analysis (data not shown). Individuals heterozygous for the floxed M6P/IGF2R exon 10 were then crossed to generate homozygous animals. These were born at the expected Mendelian ratios and presented no obvious abnormalities.
Mouse Genotyping
A PCR-based assay was used to determine the presence of the loxP site in M6P/IGF2R intron 9. 100 ng of mouse genomic DNA was used as template in a PCR reaction (35 cycles of 95°C for 30 seconds, 60°C for 30 seconds, and 72°C for 30 seconds) using the primers INT9F2 (5′-CCTTCCCTCCAGGCCGTTAC-3′) and INT9R1 (5′-GGTGAGGTCTCCATCTGAGTACC-3′).
Western Blot Analysis
Tissues were removed and homogenized in ice-cold dH2O using a Caframo RZR1 stirrer homogenizer (Caframo, Ltd., Wiarton, Canada). The homogenates were then sonicated with a Misonix XL2020 sonicator (Misonix Inc., Farmingdale, NY) for 3 × 15 seconds at a setting of 10; the particulate matter was removed by centrifugation. Samples were then electrophoresed in a NuPAGE 4 to 12% gradient polyacrylamide gel (Invitrogen, Carlsbad, CA) and subsequently blotted onto nitrocellulose filters. M6P/IGF2R protein was then detected using a polyclonal rabbit antibody preparation against rat M6P/IGF2R 34 (kindly provided by C. Scott, University of Sydney), and visualized using a goat anti-rabbit antibody conjugated to horseradish peroxidase (Amersham, Piscataway, NJ).
Immunohistochemistry
Immunohistochemical staining for M6P/IGF2R was conducted as described previously. 35 Briefly, tissues were fixed overnight in Omnifix (Bovie Medical, Melville, NY), paraffin-embedded, and 5-μm sections were mounted on glass slides. Duplicate serial sections were mounted on each slide to allow for a negative control. Sections were de-paraffinized in Xylenes (2 × 5 minutes) and endogenous peroxidases were quenched by H2O2 (3% in methanol for 10 minutes). After gradually rehydrating the sections, the tissues were blocked (60 minutes at room temperature) in 10% goat serum (Vector Laboratories, Burlingame, CA) and 5% milk diluent (Kirkegaard and Perry Laboratories, Gaithersburg, MD). Sections were then incubated overnight at 4°C in a humidified chamber with affinity-purified rabbit anti-bovine M6P/IGF2R antiserum (1:1800, kindly provided by P. Lobel, Rutgers University). Negative control sections were incubated with the same concentration of non-immune rabbit IgG. Tissue sections were immunoperoxidase-stained the next day using the Vectastain ABC Elite Kit per manufacturer’s instructions (Vector Laboratories).
Statistical Analysis
Population means were compared by the unpaired t-test; a P value ≤ 0.05 was considered to be statistically significant.
Results
Cre-loxP Strategy for Creating a Functionally Null M6P/IGF2R Allele
M6P/IGF2R is imprinted in mice with expression predominantly from the maternally inherited allele. 23 Therefore, heterozygous mice inheriting a null M6P/IGF2R allele from their mothers are functionally equivalent to homozygous knockout individuals and display neonatal lethality. 10-12 In contrast, heterozygous individuals inheriting a null M6P/IGF2R allele from their fathers are functionally equivalent to wild-type mice. Previous M6P/IGF2R knockout mice retained selection cassettes, and such exogenous sequences can cause phenotypic effects in mice. 36 Therefore, it was important to determine whether the phenotype observed in the previous knockout studies is reproduced when global M6P/IGF2R deficiency during development is induced using the Cre-loxP strategy.
To accomplish this, a two-stage breeding protocol was devised. The first stage involved mating males of mixed genetic background, homozygous for the floxed M6P/IGF2R exon 10, with females from a transgenic line in which Cre-recombinase was expressed constitutively from the CMV promoter. 37 The maternally inherited M6P/IGF2R allele was normal in the resulting offspring while the paternally inherited M6P/IGF2R allele was floxed. Since Cre recombinase is expressed in all cells, Cre/loxP-mediated excision of the paternally inherited exon 10 sequence occurred in every cell. Functionally, this has no effect since the paternal allele of M6P/IGF2R is normally silenced through genomic imprinting. Because Cre was expressed ubiquitously in these mice, half of their germ cells (ie, those containing the paternally inherited floxed M6P/IGF2R allele) have exon 10 deleted from the M6P/IGF2R and the other half (ie, those possessing the maternally inherited wild-type allele) retain an intact M6P/IGF2R.
Stage 2 involved breeding female progeny of the first breeding stage (in which 50% of the germ cells carry an inactivated copy of M6P/IGF2R) with wild-type male mice (C57Bl/6). This produced litters where 50% of the progeny inherited the deleted exon 10 M6P/IGF2R allele and 50% inherited a wild-type M6P/IGF2R allele. Consequently, half of the litter should be deficient in M6P/IGF2R throughout development, whereas the other half should be wild-type.
This two-stage breeding process produced small litters without mutant mice, indicating that mice inheriting a mutated M6P/IGF2R allele from their mothers were not viable. Timed pregnancies were therefore established, and embryos were harvested at day 18.5 of gestation. Genotyping and Western blot analysis showed that M6P/IGF2R protein was absent in individuals inheriting a deleted M6P/IGF2R exon 10 from their mothers (Figure 2C) ▶ . Knockout embryos and their corresponding placentas were 36% and 31% larger than wild-type, respectively (Figure 2, A and B) ▶ . Many of the knockout embryos also had an extra post-axial digit on fore and/or hind limbs, and the tails of mutant embryos were shorter and blunter than those of wild-type mice with a characteristic kink in the tip of the tail in some mutant embryos (Figure 2A) ▶ .
Figure 2.

Cre/loxP strategy successfully creates a functionally null M6P/IGF2R allele. A: Wild-type (WT) and M6P/IGF2R knockout (KO) mice at 18.5 days of gestation. Embryos inheriting a deleted M6P/IGF2R exon 10 allele from their mothers (KO) exhibited an overgrowth phenotype. They also frequently displayed extra post-axial digits on the fore and/or hind limbs (arrowhead); shorter, blunter tails often with a characteristic kink in the tip (*); and extravasation of overgrown abdominal organs (arrow). B: Wet weight measurements demonstrate that knockout (KO) embryos (n = 12) and their corresponding placentas are significantly (P < 0.0001) larger than those in WT mice (n = 20); standard errors of the mean are shown. C: Western blot analysis performed on tail tips from the 18.5-day-old embryos confirmed the absence of M6P/IGF2R protein in KO mice.
Histological analysis of mutant animals indicated that the lungs were poorly developed, with a very low frequency of bronchiole structures observed (Figure 3,A and B) ▶ . Immunohistochemical analysis of four knockout embryos confirmed the absence of M6P/IGF2R protein shown by Western blot. However, in two animals, intense heterogeneous immunostaining was observed in the lungs that co-localized with developing bronchiolar structures (Figure 3C) ▶ . This staining may represent incomplete silencing of the wild-type paternally inherited M6P/IGF2R allele, as it is expressed at significant levels in early development. 38 Alternatively, it could result from re-expression of the paternal allele later in development. In summary, the phenotype of these mice is essentially identical to those of previous models of complete M6P/IGF2R deficiency. 10-12
Figure 3.
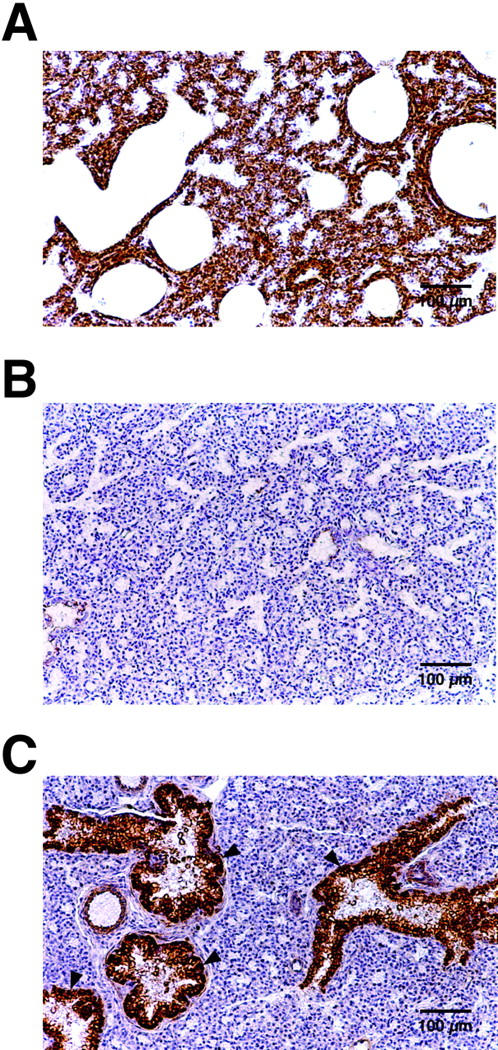
M6P/IGF2R immunohistochemical staining of lung at 18.5 days of gestation. A: Wild-type mouse lung, B: M6P/IGF2R knockout mouse lung, and C: M6P/IGF2R knockout mouse lung with reactivated receptor expression. Arrowheads; bronchial alveolar structures with expression of the normally silenced paternal M6P/IGF2R allele.
Liver-Specific Knockout
To create a liver-specific M6P/IGF2R knockout, a transgenic line was used in which the promoter and upstream enhancer of the rat albumin gene directs liver-specific expression of Cre recombinase (Alb-Cre). 39 Female mice of mixed genetic background, heterozygous for the floxed M6P/IGF2R exon 10, were crossed with Alb-Cre males to create litters in which 50% of animals inherited a floxed allele maternally and 50% inherited a wild-type allele maternally. Since the paternal allele is normally silenced through imprinting, this strategy should produce liver-specific knockout animals and wild-type littermates at equal ratios. Western blot analysis was performed on protein extracts from liver, heart, skeletal muscle, kidney, and spleen of both genotypes. M6P/IGF2R protein was undetectable in the liver of the knockout animals at 3 (n = 5) and 12 (n = 5) months of age in comparison with wild-type littermates (n = 5) (Figure 4A) ▶ . M6P/IGF2R levels in the other tissues of these knockout animals were at normal levels, confirming the tissue specificity of the M6P/IGF2R inactivation.
Figure 4.

Western blot analysis of tissue-specific M6P/IGF2R knockout mice. A: Protein extracts were prepared from tissues of M6P/IGF2R liver-specific knockout (KO) (n = 5) and wild-type (WT) (n = 5) animals; representative samples are shown. 100 μg of total protein from each tissue was electrophoresed in a NuPAGE 4 to 12% gradient polyacrylamide gel, blotted onto nitrocellulose filters, and probed with a polyclonal M6P/IGF2R antibody. B: Protein extracts were prepared from tissues removed from M6P/IGF2R skeletal/cardiac KO (n = 5) and WT (n = 5) animals; representative samples are shown. 100 μg of total protein from each tissue was electrophoresed in a NuPAGE 4 to 12% gradient polyacrylamide gel, blotted onto nitrocellulose filters, and probed with a polyclonal M6P/IGF2R antibody. The blot labeled “Muscle” was exposed longer than the other tissues to reveal the faint band in the KO lane.
Knockout and wild-type mice were also examined for changes in body and organ weights and for any evidence of morphological changes at the cellular level. There were no significant differences in either average body weight or organ weight (liver, kidney, heart, and spleen) between knockout and wild-type animals (data not shown). Similarly, histological analysis of liver sections from knockout and wild-type animals did not appear significantly different (data not shown).
Despite our inability to detect significant M6P/IGF2R in the liver of knockout animals by Western analysis, it was possible that reactivation of the normally silent paternal M6P/IGF2R allele, as occasionally observed in the lung (Figure 3, A and B) ▶ , could mask an abnormal liver phenotype. We addressed this possibility in a separate set of experiments. Breeding strategies were designed to produce mice that inherited floxed M6P/IGF2R alleles from both parents, together with the Alb-Cre transgene. These mice should not possess a potentially functional paternal M6P/IGF2R allele. Body and organ weights and liver sections of individuals inheriting two floxed M6P/IGF2R alleles were indistinguishable from the maternally inherited heterozygous mice at 3 and 12 months of age (data not shown). These findings indicate that the lack of abnormal liver phenotype in the heterozygous liver knockout animals was not due to reactivation of the paternal M6P/IGF2R.
Muscle-Specific Knockout
A transgenic line with Cre-recombinase under the control of the muscle creatine kinase promoter (Ckmm-Cre) was used to create mice in which floxed M6P/IGF2R exon 10 was deleted only in skeletal and cardiac muscle. 40 Female mice of mixed genetic background and heterozygous for the floxed M6P/IGF2R were crossed with Ckmm-Cre males. This created litters in which 50% of the animals inherited a floxed allele maternally and 50% inherited a wild-type allele maternally. This strategy was intended to produce functionally homozygous cardiac and skeletal muscle-specific M6P/IGF2R knockouts paired with wild-type littermates. M6P/IGF2R protein levels were reduced by approximately 90 to 95% in the cardiac and skeletal muscle of the floxed animals compared to their wild-type littermates (Figure 4B) ▶ . Other tissues examined (liver, kidney, and spleen) showed normal levels of M6P/IGF2R in these knockout animals (Figure 4B) ▶ , again demonstrating the tissue specificity of the knockout.
The presence of a small but significant level of M6P/IGF2R protein in the cardiac and skeletal muscle of knockout individuals (Figure 4B) ▶ may be due to either expression of M6P/IGF2R protein in non-muscle cell types or reactivation of the silent paternal allele. To address the latter possibility, mice were bred such that both the maternal and paternal alleles were floxed. A low level of M6P/IGF2R protein, similar to that shown in Figure 4B ▶ , was still detected in the cardiac and skeletal muscle of homozygous knockout individuals (data not shown). This indicates that the residual receptor protein is derived either from cardiac cells in which Cre-recombinase was not expressed and/or in cells associated with the muscle tissue such as fibroblasts and vascular endothelial cells.
At 3 and 12 months of age, no gross phenotypic changes were observed in mice deficient in cardiac and skeletal muscle M6P/IGF2R, whether mice heterozygous or homozygous for the exon 10 deletion were examined. Additionally, body and organ weights of knockout individuals did not differ significantly from wild-type littermates while histological comparison of cardiac and skeletal muscle sections revealed no significant differences between knockout and wild-type mice (data not shown).
Discussion
The critical function of M6P/IGF2R in regulating IGF2 availability has been clearly shown by the lethal phenotype for mice deficient in receptor function during embryonic development, and their rescue from lethality by concomitant IGF2- or IGF1-receptor deficiency. 10-12 Unfortunately, because of this lethal phenotype, these mice are not useful for studying the role of this multifunctional receptor in postnatal life. The Cre/loxP recombinase system exploits the ability of Cre recombinase to direct excision of a DNA sequence that has been flanked by directly repeated copies of the loxP recombination site. Expression of Cre by an inducible or tissue-specific promoter therefore allows excision of the loxP flanked DNA.
We used the Cre/loxP technology in this study to create a transgenic line amenable to inactivating the M6P/IGF2R gene in a tissue-specific manner. To validate the ability of the Cre/loxP system to inactivate M6P/IGF2R, we mated wild-type male mice with female mice heterozygous for the Cre/loxP-mediated exon 10 deletion. Embryos that inherit a mutated M6P/IGF2R allele from their mother have a global deficiency of receptor throughout embryonic development, and should therefore reproduce the phenotype observed in previous reports of receptor deficiency. 10-12 As predicted, litters studied at embryonic day 18.5 contained wild-type embryos and embryos deficient in M6P/IGF2R. The latter embryos had the somatic overgrowth phenotype and skeletal abnormalities previously associated with maternal inheritance of a mutated M6P/IGF2R allele. 10-12 These results confirm the null status of the floxed M6P/IGF2R, and reaffirm its critical role in fetal development. Having confirmed that Cre-mediated excision of M6P/IGF2R exon 10 efficiently ablates receptor function, subsequent transgenic lines were then created in which M6P/IGF2R was knocked out in a tissue-specific manner.
We have previously reported genetic evidence that M6P/IGF2R behaves as a tumor-suppressor gene in a number of tissues including the liver. 14,41 To further elucidate the tumor suppressor role of M6P/IGF2R in the liver, we created liver-specific M6P/IGF2R knockouts by breeding M6P/IGF2R floxed mice with animals in which the liver-specific albumin promoter controls Cre recombinase expression. 39 Recombination induced by the expressed Cre is reported to be approximately 40% complete immediately after birth and increases steadily until about 6 weeks of age when recombination is complete. 42 In keeping with the known albumin promoter specificity and expression kinetics, M6P/IGF2R levels were 95 to 100% reduced in the liver of adult mice (> 3 months) inheriting the floxed allele maternally. All other tissues examined, including kidney, spleen, heart, and skeletal muscle, had similar levels of receptor in the knockout animals compared to wild-type controls.
Human M6P/IGF2R is implicated in tumor suppression in the breast, gastrointestinal tract, liver, and lung. 14-18 The M6P/IGF2R is thought to contribute to tumor suppression by degrading IGF2, activating TGF-β, and mediating the action of cytotoxic T cells. 6,29 Therefore, the absence of these activities in M6P/IGF2R-deficient liver is predicted to increase susceptibility to hepatocellular carcinomas. We did not observe any gross histological abnormalities or any evidence of hyperplasia in the mutant livers at 3 and 12 months of age; however, the frequency of liver tumors in mice overexpressing IGF2 was not greater than that in control animals until they were over 18 months old. 43 Long-term carcinogenesis studies in mice with selective M6P/IGF2R deficiency in the liver, and other tissues, can now be conducted with the animals described in this report. They will allow, for the first time, a definitive assessment of the tumor suppressor activity of the M6P/IGF2R.
Viable mice deficient in M6P/IGF2R also represent an important model system for optimization of therapeutic approaches to inherited disorders of lysosomal metabolism. Numerous lysosomal storage disorders have been characterized in humans and, in most cases, are due to deficiency of a single acid hydrolase. 44 For example, Pompe disease (ie, glycogen storage disease II) is characterized by mutations in the gene coding for acid α-1,4-glucosidase (GAA) and abnormal accumulation of glycogen in skeletal muscle and heart (OMIM entry: no. 232300). 45 With a number of the recombinant enzymes now available for treatment purposes, a major challenge is to efficiently target the enzymes to the appropriate tissue in vivo. Because M6P residues are incorporated into most acid hydrolases during their biosynthesis, it may be feasible to exploit the endocytic activity of the M6P/IGF2R for tissue targeting.
To provide a model system for evaluating enzyme delivery in vivo, we generated mice deficient in skeletal muscle and heart M6P/IGF2R using the muscle creatine kinase promoter to drive expression of Cre recombinase. M6P/IGF2R levels in adult heart and skeletal muscle of mutant mice were reduced by up to 95% compared to control tissues, while receptor levels in other tissues of these animals remained normal. Despite the low levels of muscle M6P/IGF2R, there was no evidence of gross histological abnormalities in either heart or skeletal muscle. There were also no obvious signs of muscular dystrophy, breathing or feeding difficulties, or muscular hyperplasia and/or hypertrophy. Therefore, these mice may represent a valuable model system for analysis of in vivo targeting of GAA and other lysosomal enzymes. Mice deficient in M6P/IGF2R in numerous other tissues may similarly be useful for evaluating enzyme therapy in other lysosomal disorders with different tissue involvement.
Cardiac M6P/IGF2R expression, which is high during embryogenesis, also persists at an elevated level in the adult rodent. 46 This indicates that this receptor plays a significant role in the heart postnatally. Recently, cultured cardiac cells were shown to bind, internalize, and activate prorenin in a M6P-dependent manner. 47 The relevance of these observations to the in vivo situation is currently unknown, but the renin-angiotensin system plays an important role in the regulation of blood pressure and salt and fluid homeostasis. Agents that interfere with angiotensin II formation, the ACE inhibitors in particular, are now widely used for the treatment of hypertension and heart failure. 48,49 If M6P/IGF2R is shown to contribute to angiotensin II formation in vivo, this knowledge may help in the development of alternative therapeutic strategies. Mice with cardiac deficiency of M6P/IGF2R, such as those described in this study should facilitate experiments that test the physiological relevance of M6P-dependent prorenin activation.
Interestingly, M6P/IGF2R in mice is imprinted in all tissues except for the brain where both alleles are expressed. 50 It is highly expressed in neurons of the forebrain, with the highest expression in the pyramidal cells, the polymorphic layers of the hippocampus, and the granule cell layer of the dentate gyrus; regions involved in emotional behavior, information processing, and memory formation. 51 These findings indicate that M6P/IGF2R may assist in the development of these brain functions. This postulate is reinforced by the identification of M6P/IGF2R as the first putative “IQ gene.” 31 By comparing children with an IQ of 160 or higher to those with an average IQ, M6P/IGF2R was shown to be linked with general cognitive ability (“g”). The role of this receptor in the development of cognitive function can now be systematically assessed with M6P/IGF2R conditional knockout mice.
M6P/IGF2R functions in T cell activation and T cell-mediated apoptosis. Polyclonal antibodies to M6P/IGF2R block T cell differentiation at the CD4− and CD8 − stage, indicating that this receptor is actively involved in the early steps of T cell differentiation. 52 A common feature of autoimmune diseases is the formation of autoantibodies targeted against a wide spectrum of cellular molecules. Interestingly, M6P/IGF2R has been identified as a novel target of autoantibodies in patients with autoimmune diseases. 30 Furthermore, M6P/IGF2R facilitates T cell activation by internalizing CD26/DPPIV (dipeptidyl peptidase IV), a cell surface T cell activation antigen. 29 Since a large number of CD26+ T cells are found in inflamed tissues of patients with autoimmune disease, and M6P/IGF2R is a target for autoantibodies, studying the interaction between CD26 and the M6P/IGF2R may help better understand the pathogenesis of autoimmune diseases.
T cells and natural killer (NK) cells kill tumor cells by two major mechanisms involving the cross-linking of death receptors on the tumor cell surface and granule exocytosis. The major serine proteinase released from these granules is granzyme B. It enters a cell targeted for death through M6P/IGF2R-mediated endocytosis, and induces apoptosis on being released from the endosomes by the pore-forming ability of perforin. 6 Thus, M6P/IGF2R is not only essential for allogeneic cell rejection in vivo, but cancer cells may also avoid T cell-mediated immune surveillance by inactivating M6P/IGF2R and/or perforin function. 53 Thus, the efficacy of cancer immunotherapy may partly depend on the M6P/IGF2R mutation status in cancer. The M6P/IGF2R conditional knockout mouse provides a unique model in which to investigate the function of this receptor in both T cell activation and T cell-mediated apoptosis in cancer.
In summary, we have generated a transgenic mouse possessing a M6P/IGF2R modified by the presence of loxP sites. We have used the Cre/loxP system to generate mice deficient in M6P/IGF2R in a tissue-specific manner. The increasing availability of transgenic mice in which Cre recombinase expression is exquisitely controlled in a tissue- and/or age-dependent manner will allow for the elucidation of the normal physiological roles of this multifunctional receptor in postnatal life. Understanding the contribution of aberrant M6P/IGF2R expression to the pathophysiology of human diseases may ultimately lead to the development of novel therapeutic approaches.
Acknowledgments
We thank M. Magnuson for the Alb-Cre mice, R. Kahn for the Ckmm-Cre mice, and K. Rajewsky for the CMV-Cre mice. We also thank J. Dai for her excellent technical assistance and C. Scott and P. Lobel for M6P/IGF2R antibodies.
Footnotes
Address reprint requests to Dr. Randy L. Jirtle, Box 3433, Duke University Medical Center, Durham, NC 27710. E-mail: jirtle@radonc.duke.edu.
Supported by the National Institutes of Health grants CA25951 and ES08823, and AstraZeneca Pharmaceuticals, Inc.
Further information on genomic imprinting is available at http://www.geneimprint.com.
References
- 1.Kornfeld S: Structure and function of the mannose 6-phosphate/insulin-like growth factor II receptors. Annu Rev Biochem 1992, 61:307-330 [DOI] [PubMed] [Google Scholar]
- 2.Jirtle RL: Mannose 6-phosphate receptors. Creidton TE eds. Encyclopedia of Molecular Biology. 1999, :pp 1441-1447 Wiley-Liss, Inc., New York [Google Scholar]
- 3.Köster A, Saftig P, Matzner U, von Figura K, Peters C, Pohlmann R: Targeted disruption of the M(r) 46,000 mannose 6-phosphate receptor gene in mice results in misrouting of lysosomal proteins. EMBO J 1993, 12:5219-5223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ludwig T, Ovitt CE, Bauer U, Hollinshead M, Remmler J, Lobel P, Ruther U, Hoflack B: Targeted disruption of the mouse cation-dependent mannose 6-phosphate receptor results in partial missorting of multiple lysosomal enzymes. EMBO J 1993, 12:5225-5235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morgan DO, Edman JC, Standring DN, Fried VA, Smith MC, Roth RA, Rutter WJ: Insulin-like growth factor II receptor as a multifunctional binding protein. Nature 1987, 329:301-307 [DOI] [PubMed] [Google Scholar]
- 6.Motyka B, Korbutt G, Pinkoski MJ, Heibein JA, Caputo A, Hobman M, Barry M, Shostak I, Sawchuk T, Holmes CF, Gauldie J, Bleackley RC: Mannose 6-phosphate/insulin-like growth factor II receptor is a death receptor for granzyme B during cytotoxic T cell-induced apoptosis. Cell 2000, 103:491-500 [DOI] [PubMed] [Google Scholar]
- 7.Brown J, Esnouf RM, Jones MA, Linnell J, Harlos K, Hassan AB, Jones EY: Structure of a functional IGF2R fragment determined from the anomalous scattering of sulfur. EMBO J 2002, 21:1054-1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Killian JK, Byrd JC, Jirtle JV, Munday BL, Stoskopf MK, MacDonald RG, Jirtle RL: M6P/IGF2R imprinting evolution in mammals. Mol Cell 2000, 5:707-716 [DOI] [PubMed] [Google Scholar]
- 9.Kirchhausen T: Single-handed recognition of a sorting traffic motif by the GGA proteins. Nat Struct Biol 2002, 9:241-244 [DOI] [PubMed] [Google Scholar]
- 10.Lau MM, Stewart CE, Liu Z, Bhatt H, Rotwein P, Stewart CL: Loss of the imprinted IGF2/cation-independent mannose 6-phosphate receptor results in fetal overgrowth and perinatal lethality. Genes Dev 1994, 8:2953-2963 [DOI] [PubMed] [Google Scholar]
- 11.Ludwig T, Eggenschwiler J, Fisher P, D’Ercole AJ, Davenport ML, Efstratiadis A: Mouse mutants lacking the type 2 IGF receptor (IGF2R) are rescued from perinatal lethality in Igf2 and Igf1r null backgrounds. Dev Biol 1996, 177:517-535 [DOI] [PubMed] [Google Scholar]
- 12.Wang ZQ, Fung MR, Barlow DP, Wagner EF: Regulation of embryonic growth and lysosomal targeting by the imprinted Igf2/Mpr gene. Nature 1994, 372:464-467 [DOI] [PubMed] [Google Scholar]
- 13.Young LE, Fernandes K, McEvoy TG, Butterwith SC, Gutierrez CG, Carolan C, Broadbent PJ, Robinson JJ, Wilmut I, Sinclair KD: Epigenetic change in IGF2R is associated with fetal overgrowth after sheep embryo culture. Nat Genet 2001, 27:153-154 [DOI] [PubMed] [Google Scholar]
- 14.De Souza AT, Hankins GR, Washington MK, Orton TC, Jirtle RL: M6P/IGF2R gene is mutated in human hepatocellular carcinomas with loss of heterozygosity. Nat Genet 1995, 11:447-449 [DOI] [PubMed] [Google Scholar]
- 15.Hankins GR, De Souza AT, Bentley RC, Patel MR, Marks JR, Iglehart JD, Jirtle RL: M6P/IGF2 receptor: a candidate breast tumor suppressor gene. Oncogene 1996, 12:2003-2009 [PubMed] [Google Scholar]
- 16.Kong FM, Anscher MS, Washington MK, Killian JK, Jirtle RL: M6P/IGF2R is mutated in squamous cell carcinoma of the lung. Oncogene 2000, 19:1572-1578 [DOI] [PubMed] [Google Scholar]
- 17.Souza RF, Appel R, Yin J, Wang S, Smolinski KN, Abraham JM, Zou T-T, Shi Y-Q, Lei J, Cottrell J, Cymes K, Biden K, Simms L, Leggett B, Lynch PM, Frazier M, Powell SM, Harpaz N, Sugimura H, Young J, Meltzer SJ: The insulin-like growth factor II receptor gene is a target of microsatellite instability in human gastrointestinal tumours. Nat Genet 1996, 14:255-257 [DOI] [PubMed] [Google Scholar]
- 18.Ouyang H, Shiwaku HO, Hagiwara H, Miura K, Abe T, Kato Y, Ohtani H, Shiiba K, Souza RF, Meltzer SJ, Horii A: The insulin-like growth factor II receptor gene is mutated in genetically unstable cancers of the endometrium, stomach, and colorectum. Cancer Res 1997, 57:1851-1854 [PubMed] [Google Scholar]
- 19.Kang JX, Bell J, Beard RL, Chandraratna RA: Mannose 6-phosphate/insulin-like growth factor II receptor mediates the growth-inhibitory effects of retinoids. Cell Growth Differ 1999, 10:591-600 [PubMed] [Google Scholar]
- 20.O’Gorman DB, Costello M, Weiss J, Firth SM, Scott CD: Decreased insulin-like growth factor-II/mannose 6-phosphate receptor expression enhances tumorigenicity in JEG-3 cells. Cancer Res 1999, 59:5692-5694 [PubMed] [Google Scholar]
- 21.Souza RF, Wang S, Thakar M, Smolinski KN, Yin J, Zou TT, Kong D, Abraham JM, Toretsky JA, Meltzer SJ: Expression of the wild-type insulin-like growth factor II receptor gene suppresses growth and causes death in colorectal carcinoma cells. Oncogene 1999, 18:4063-4068 [DOI] [PubMed] [Google Scholar]
- 22.Clurman B, Groudine M: Tumour-suppressor genes: killer in search of a motive? Nature 1997, 389:122-123 [DOI] [PubMed] [Google Scholar]
- 23.Barlow DP, Stöger R, Herrmann BG, Saito K, Schweifer N: The mouse insulin-like growth factor type-2 receptor is imprinted and closely linked to the Tme locus. Nature 1991, 349:84-87 [DOI] [PubMed] [Google Scholar]
- 24.Nolan CM, Killian JK, Petitte JN, Jirtle RL: Imprint status of M6P/IGF2R and IGF2 in chickens. Dev Genes Evol 2001, 211:179-183 [DOI] [PubMed] [Google Scholar]
- 25.Killian JK, Nolan CM, Wylie AA, Li T, Vu TH, Hoffman AR, Jirtle RL: Divergent evolution in M6P/IGF2R imprinting from the Jurassic to the Quaternary. Hum Mol Genet 2001, 10:1721-1728 [DOI] [PubMed] [Google Scholar]
- 26.De Souza AT, Yamada T, Mills JJ, Jirtle RL: Imprinted genes in liver carcinogenesis. EMBO J 1997, 11:60-67 [DOI] [PubMed] [Google Scholar]
- 27.Jirtle RL: Genomic imprinting and cancer. Exp Cell Res 1999, 248:18-24 [DOI] [PubMed] [Google Scholar]
- 28.O’Kane S, Ferguson MW: Transforming growth factor β and wound healing. Int J Biochem Cell Biol 1997, 29:63-78 [DOI] [PubMed] [Google Scholar]
- 29.Ikushima H, Munakata Y, Ishii T, Iwata S, Terashima M, Tanaka H, Schlossman SF, Morimoto C: Internalization of CD26 by mannose 6-phosphate/insulin-like growth factor II receptor contributes to T cell activation. Proc Natl Acad Sci USA 2000, 97:8439-8444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tarrago D, Aguilera I, Melero J, Wichmann I, Nunez-Roldan A, Sanchez B: Identification of cation-independent mannose 6-phosphate receptor/insulin-like growth factor type-2 receptor as a novel target of autoantibodies. Immunology 1999, 98:652-662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chorney MJ, Chorney K, Seese N, Owen MJ, Daniels J, McGuffin P, Thompson LA, Detterman DK, Benbow C, Lubinski D, Eley T, Plomin R: A quantitative trait locus associated with cognitive ability in children. Psychol Sci 1998, 9:159-166 [Google Scholar]
- 32.Kuhn R, Schwenk F, Aguet M, Rajewsky K: Inducible gene targeting in mice. Science 1995, 269:1427-1429 [DOI] [PubMed] [Google Scholar]
- 33.Killian JK, Jirtle RL: Genomic structure of the human M6P/IGF2 receptor. Mamm Genome 1999, 10:74-77 [DOI] [PubMed] [Google Scholar]
- 34.Hartshorn MA, Scott CD, Baxter RC: Immunofluorescent localization of type II insulin-like growth factor receptor in rat liver and hepatoma cells. J Endocrinol 1989, 121:221-227 [DOI] [PubMed] [Google Scholar]
- 35.Jirtle RL, Carr BI, Scott CD: Modulation of insulin-like growth factor/mannose-6-phosphate receptors and transforming growth factor-β1 during liver regeneration. J Biol Chem 1991, 266:22444-22450 [PubMed] [Google Scholar]
- 36.Scacheri PC, Crabtree JS, Novotny EA, Garrett-Beal L, Chen A, Edgemon KA, Marx SJ, Spiegel AM, Chandrasekharappa SC, Collins FS: Bi-directional transcriptional activity of PGK-neomycin and unexpected embryonic lethality in heterozygote chimeric knockout mice. Genesis 2001, 30:259-263 [DOI] [PubMed] [Google Scholar]
- 37.Schwenk F, Baron U, Rajewsky K: A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res 1995, 23:5080-5081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lerchner W, Barlow DP: Paternal repression of the imprinted mouse Igf2r locus occurs during implantation and is stable in all tissues of the post-implantation mouse embryo. Mech Dev 1997, 61:141-149 [DOI] [PubMed] [Google Scholar]
- 39.Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, Shelton KD, Lindner J, Cherrington AD, Magnuson MA: Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic β cell-specific gene knock-outs using Cre recombinase. J Biol Chem 1999, 274:305-315 [DOI] [PubMed] [Google Scholar]
- 40.Wang J, Wilhelmsson H, Graff C, Li H, Oldfors A, Rustin P, Bruning JC, Kahn CR, Clayton DA, Barsh GS, Thoren P, Larsson NG: Dilated cardiomyopathy and atrioventricular conduction blocks induced by heart-specific inactivation of mitochondrial DNA gene expression. Nat Genet 1999, 21:133-137 [DOI] [PubMed] [Google Scholar]
- 41.Yamada T, De Souza AT, Finkelstein S, Jirtle RL: Loss of the gene encoding mannose 6-phosphate/insulin-like growth factor II receptor is an early event in liver carcinogenesis. Proc Natl Acad Sci USA 1997, 94:10351-10355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Postic C, Magnuson MA: DNA excision in liver by an albumin-Cre transgene occurs progressively with age. Genesis 2000, 26:149-150 [DOI] [PubMed] [Google Scholar]
- 43.Rogler CE, Yang D, Rossetti L, Donohoe J, Alt E, Chang CJ, Rosenfeld R, Neely K, Hintz R: Altered body composition and increased frequency of diverse malignancies in insulin-like growth factor-II transgenic mice. J Biol Chem 1994, 269:13779-13784 [PubMed] [Google Scholar]
- 44.Raben N, Plotz P, Byrne BJ: Acid α-glucosidase deficiency (glycogenosis type II, Pompe disease) Curr Mol Med 2002, 2:145-166 [DOI] [PubMed] [Google Scholar]
- 45.Chen YT, Amalfitano A: Towards a molecular therapy for glycogen storage disease type II (Pompe disease). Mol Med Today 2000, 6:245-251 [DOI] [PubMed] [Google Scholar]
- 46.McCormick KM, Dahms NM, Lough J: Insulin-like growth factor-II/mannose-6-phosphate receptor expression during early heart development. Dev Dyn 1996, 207:195-203 [DOI] [PubMed] [Google Scholar]
- 47.Saris JJ, Derkx FH, Lamers JM, Saxena PR, Schalekamp MA, Danser AH: Cardiomyocytes bind and activate native human prorenin: role of soluble mannose 6-phosphate receptors. Hypertension 2001, 37:710-715 [DOI] [PubMed] [Google Scholar]
- 48.Brown NJ, Vaughan DE: Angiotensin-converting enzyme inhibitors. Circulation 1998, 97:1411-1420 [DOI] [PubMed] [Google Scholar]
- 49.Givertz MM: Manipulation of the renin-angiotensin system. Circulation 2001, 104:E14-E18 [DOI] [PubMed] [Google Scholar]
- 50.Vu TH, Hoffman AR: Comparative genomics sheds light on mechanisms of genomic imprinting. Genome Res 2000, 10:1660-1663 [DOI] [PubMed] [Google Scholar]
- 51.Couce ME, Weatherington AJ, McGinty JF: Expression of insulin-like growth factor-II (IGF-II) and IGF-II/mannose-6-phosphate receptor in the rat hippocampus: an in situ hybridization and immunocytochemical study. Endocrinology 1992, 131:1636-1642 [DOI] [PubMed] [Google Scholar]
- 52.Kecha O, Brilot F, Martens H, Franchimont N, Renard C, Greimers R, Defresne MP, Winkler R, Geenen V: Involvement of insulin-like growth factors in early T cell development: a study using fetal thymic organ cultures. Endocrinology 2000, 141:1209-1217 [DOI] [PubMed] [Google Scholar]
- 53.Igney FH, Krammer PH: Immune escape of tumors: apoptosis resistance and tumor counterattack. J Leukoc Biol 2002, 71:907-920 [PubMed] [Google Scholar]