

Abstract
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is an increasingly recognized adult-onset autosomal dominant vascular dementia, caused by highly stereotyped mutations in the Notch3 receptor. CADASIL is a widespread angiopathy characterized by a degeneration of vascular smooth muscle cells (VSMCs) and the abnormal accumulation of electron-dense granular material called GOM and Notch3 protein, because of an impaired clearance. Evidence that VSMCs are the primary target of the pathogenic process is supported by the restricted expression of Notch3 in these cells but mechanisms of their degeneration remain essentially unknown. We generated transgenic mice in which the SM22α promoter drove, in VSMCs, the expression of a full-length human Notch3 carrying the Arg90Cys mutation, a CADASIL archetypal mutation. Transgenic mice showed no evidence of prominent brain parenchyma damage but demonstrated the two hallmarks of the CADASIL angiopathy, GOM deposits and Notch3 accumulation, within both the cerebral and peripheral arteries. Of interest, arteries of the tail were more severely affected with prominent signs of VSMC degeneration. Time-course analysis of vessel changes revealed that disruption of normal VSMC anchorage to adjacent extracellular matrix and cells, VSMC cytoskeleton changes as well as starting signs of VSMC degeneration, which were detected around 10 months of age, preceded Notch3 and GOM accumulation appearance, which were observed only by 14 to 16 months of age. In conclusion, we have generated transgenic mice that recapitulate the characteristic vascular lesions observed in CADASIL. Our results indicate that Notch3 or GOM accumulation are unlikely to be the prerequisites for the induction of VSMC degeneration and suggest that degeneration of VSMCs may rather be triggered by the disruption of their normal anchorage, based on the important role of adhesion for cell survival.
CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy; MIM 125310) is an increasingly recognized autosomal dominant small-artery disease of the brain. 1 The onset of symptoms occurs generally in mid-life although it can range from 25 to >60 years. The symptoms typically include recurrent ischemic strokes and/or cognitive impairment. Progression of the disease leads to dementia and premature death ∼15 to 20 years after clinical onset. Magnetic resonance imaging of the brain displays T2-weighted hyperintensities within the white matter in all patients and small deep infarcts in up to 60% of the patients. Such magnetic resonance imaging abnormalities can also be detected in asymptomatic patients, indicating that brain parenchyma damages are already present at the preclinical stage of the disease. 2-5 CADASIL is caused by highly stereotyped mutations in the Notch3 receptor. 6 Notch3 belongs to the highly conserved Notch receptor family involved in cell fate specification. 7 It contains all typical Notch motifs, including a characteristic extracellular domain exhibiting 34 tandem epidermal growth factor-like repeats. All CADASIL mutations result in the addition or the loss of a cysteine residue within 1 of the 34 epidermal growth factor-like repeats and therefore to an odd number of cysteine residues in the affected epidermal growth factor domain. 8-10 Prevalence of CADASIL is unknown but more than 400 affected families all over the world as well as sporadic cases carrying a de novo mutation have been identified. 11
On neuropathological examination, CADASIL brains show a diffuse myelin loss and multiple, small deep infarcts located within the white matter and basal ganglia. The pathological hallmark of CADASIL is a nonamyloid and nonarteriosclerotic angiopathy, which affects predominantly the small penetrating arteries. Vascular lesions are characterized by degeneration and loss of smooth muscle cells and the presence of a granular osmiophilic material (GOM) accumulating within the smooth muscle cell basement membrane and the surrounding extracellular matrix. 12-14 Examination of heart, muscle, skin, and many other peripheral organs revealed vessel changes, including the presence of GOM deposits, that were identical, although less severe, to those of cerebral arteries, providing evidence that CADASIL is indeed a systemic arteriopathy. 15-17 GOM deposits are highly specific of CADASIL and their detection in cerebral and peripheral vessels from skin or muscle biopsy material has been widely used as a diagnostic marker of this disease. 15,18,19 Importantly, VSMC alterations and GOM deposits have been detected in skin vessels of asymptomatic mutation carriers having a normal brain magnetic resonance imaging, indicating that arterial lesions are present at a very early stage of the disease before brain parenchyma damages and clinical symptoms occurrence (MMR, ETL, and AJ; unpublished data). 18
First clues to the pathogenic mechanisms underlying CADASIL came recently from the expression analysis of Notch3 in tissues from healthy individuals and from CADASIL patients. Data indicate that vascular smooth muscle cells (VSMCs) are the primary target of the pathogenic process in CADASIL. In healthy human adults, expression of Notch3 is highly restricted to the vessel wall and to VSMCs. Notch3, like the other Notch receptors, undergoes a constitutive proteolytical cleavage generating an extracellular (Notch3ECD) and a transmembrane/cytosolic (Notch3TMIC) fragment, that remain associated at the cell surface to form a heterodimeric receptor. In CADASIL patients, there is an abnormal accumulation of Notch3ECD in both brain vessels and peripheral tissue arteries. Accumulation takes place at the cell membrane of smooth muscle cells in very close vicinity but not within the GOM and results from an impaired clearance of the receptor from the cell membrane. 20 Notch3 accumulation is a specific hallmark of CADASIL and its detection by simple immunostaining of skin biopsy is now commonly used as an easy and reliable diagnostic marker of CADASIL. 21 Notch3 accumulation, like GOM deposits, can be detected in mutation carriers during the long preclinical stage of the disease (ETL and AJ, unpublished observation).
So far the mechanisms underlying pathological alterations in CADASIL remain unclear. Vascular smooth muscle cell degeneration may be caused by a defect in Notch3 signaling or a toxic effect of Notch3 or GOM accumulation. A defect in Notch3 signaling may result from a defect in ligand binding or a defect in transmitting the appropriate signal. It has also been proposed that the Notch3ECD, because of its failure to be cleared from the cell membrane, may dominantly inhibit the normal Notch3 pathway through competitive inhibition of ligand binding. 22,23 However, no evidence of such defects has been detected so far using in vitro assays (AJ, unpublished results). 24 In late-onset neurodegenerative disorders, such as Alzheimer’s or Huntington’s diseases, a toxic role of abnormal protein deposition is suspected to play a major role in pathogenesis. By analogy, one might hypothesize that smooth muscle cell degeneration in CADASIL arises from a toxic effect of Notch3ECD and/or GOM accumulation.
To investigate the mechanisms of smooth muscle cell degeneration caused by Notch3 mutations, we generated transgenic mice in which the SM22α promoter drove the expression of the full-length human Notch3 with the Arg90Cys mutation, one CADASIL archetypal mutation, in VSMCs. We report that transgenic mice demonstrated vascular changes, including GOM deposits, Notch3 accumulation, and evidence of smooth muscle cell degeneration, strikingly similar to those observed in CADASIL patients. Vessel changes were detected in cerebral and peripheral vessels, including the tail arteries where alterations were the most prominent. Time-course analysis of vessel changes revealed that early signs of VSMC damages were present before Notch3 and GOM accumulation occurred, and that the first ultrastructural defects included the disruption of normal VSMC anchorage to adjacent extracellular matrix and cells as well as VSMC cytoskeleton changes.
Materials and Methods
Generation of Transgenic Animals
A 7021-bp full-length human Notch3 cDNA carrying the codon 90 CGT > TGT mutation was ligated to a 2191-bp fragment of the SM22α gene, including 2126 bp upstream of the initiation transcription site and the complete noncoding first exon (a kind gift from Dr. Denise Paulin, Paris VII University). 25 A 584 bp of the bovine β-globin intron was inserted between the SM22α regulatory sequence and the Notch3 sequence. Polyadenylation signal was derived from the SV40 poly(A) DNA and cloned 3′. Final construct was confirmed by sequencing. Transgenic mice were generated by microinjection of the purified linear DNA fragment insert into fertilized eggs of C57Bl/6/DBA2 mice. Founder mice were backcrossed to wild-type C57Bl/6 mice. Presence of the transgene in the founders and their littermates was checked by polymerase chain reaction analysis of the genomic DNA using the following set of primers (5′-CGATGGAATGGGTTTCCACT -3′ and 5′-AGGCAGGAGCAGGAAAAG-3′). The approximate copy number of the transgene was estimated by densitometric comparison of hybridization intensity with known standard from Southern blot of DNA digested by HindIII restriction enzyme cutting twice in the transgene using a transgene-specific probe (EcoRI-HindIII fragment of the construct).
Mice were housed on a 12-hour light-dark cycle at constant temperature with free access to food and water, in agreement with EC guidelines for care of animal laboratories.
RNA Isolation and Reverse Transcriptase-Polymerase Chain Reaction
Total RNA was prepared from various tissues of 1-month-old mice. cDNA templates were reverse-transcribed from 2 μg of total RNA primed with random hexamers and then subjected to polymerase chain reaction using a set of primers designed from exon 1 of the murine SM22α gene and from exon 11 of the human Notch3 gene, respectively (5′-CGAAGCTACTCTCCTTCCAG-3′) and (5′-ACCTGGCTCTCGCAGCGTGT-3′).
Immunoblot Analysis
Tissues were homogenized in RIPA extraction buffer [150 mmol/L NaCl, 50 mmol/L Tris-HCl, 1% Nonidet P-40, 0.1% sodium dodecyl sulfate (SDS), 0.5% sodium desoxycholate] containing a cocktail of protease inhibitors. Protein concentrations were determined using the bicinchoninic acid protein assay reagent (Pierce, Rockford, IL). Homogenates were clarified after the addition of an equal volume of SDS-lysis buffer containing 125 mmol/L Tris-HCl, pH6.8, 4% SDS, 10% β-mercaptomethanol, and 20% glycerol, by centrifugation at 18,000 × g for 30 minutes. One hundred μg of protein per lane was subsequently resolved on 6% SDS-polyacrylamide gel and then electrophoretically transferred to nitrocellulose membrane. The following anti-Notch3 antibodies were used: murine anti-Notch3ECD (clone 5E1, 1:800 dilution; clone 11A1, 1:2500 dilution) and rabbit anti-Notch3TMIC (BC4, 1:5000 dilution). 20 Mouse anti-α-tubulin (clone DM 1A, 1:15,000 dilution) (Sigma, St. Louis, MO) and rabbit anti-smooth muscle myosin heavy chain (1:5000 dilution) (Biomedical Technologies Inc., Stoughton, MA) were used to check that an equal amount of protein extract was loaded.
Histological and Immunohistochemistry Analysis
Mice were killed by cervical dislocation. Tissues were removed, immediately fixed by immersion in formalin 10%, and routinely processed for paraffin embedding. Tissue sections were stained with hematoxylin and eosin (H&E). Brain sections were stained with H&E and luxol-Barrera method.
Immunohistochemistry was performed on 8-μm paraffin sections. The following primary antibodies were used: murine anti-Notch3ECD (clone 1E4, 1:5 dilution), 20 murineanti-smooth muscle α-actin (clone 1A4, 1:100 dilution) (DAKO, Glostrup, Denmark), rabbit anti-glial fibrillary acidic protein (1:2000 dilution) (Sigma). Immunoreactivity was visualized with the appropriate biotinylated anti-mouse or anti-rabbit antibody and avidin/biotin-horseradish peroxidase complex (Vectastain ABC-HRP kit; Vector Laboratories, Burlingame, CA) and developed with 3,3′-diaminobenzidine (Sigma). Sections were examined on a Leica DMR microscope.
Electron Microscopy
Trimmed sections (1 to 2 mm thick) of tissues were fixed in a solution of Carson pH 7.2 (3.5% formaldehyde, 110 mmol/L Na2HPO4). All samples were transferred to 2% osmium tetroxide in 0.1 mol/L of phosphate buffer pH 7.2. This was followed by rapid dehydration using a series of graded concentrations of alcohol and acetone and embedded in Epon. Semithin sections (1 μm) stained with toluidine blue were observed under a light microscope to select areas of interest. Ultrathin sections were cut and mounted on copper grids, stained with uranyl acetate and lead citrate, and observed with an electron microscope (Leo 906).
Ultrastructural Morphometry
Analysis was conducted on all available arterioles displaying one layer of smooth muscle cells (metarteriole); larger arterioles as well as veins and capillaries have been excluded from the analysis. Investigators were unaware of the genotype of mice. Photographic prints of arterioles were made, imported into an image-analysis system (AnalySIS 2.1), and stored on magnetic media (Samsung Computer Pentium Sync Master 700b). Electron micrographies at the original magnification of ×1000 to 6000 were analyzed with a modified digitizer (Soft Imaging System, Digivision Software) coupled with the computer.
The following morphometric variables were measured: internal diameter (i = lumen diameter), external diameter (e), subendothelial space, intersmooth muscle cell space, and calculated as follows. The limits of the external diameter correspond to the outermost zone of the VSMC basement membrane facing the adventitia. In arteries with an elliptical profile, measurement was taken perpendicular to the long axis; in arteries with a round-like shape profile, measurement was taken in two different axes and the average value was recorded. The subendothelial space is the space between the abluminal endothelial cell plasmalemma and the luminal plasmalemma of the first VSMC layer, an average of 10 measurements all around the circumference was recorded. The intersmooth muscle cell space corresponds to the extracellular space located between the VSMC plasmalemma, the average of 10 measurements was recorded.
Results
Generation of Transgenic Lines Expressing the Mutant Human Notch3Arg90Cys Protein
We introduced the Arg90Cys missense mutation, one of the archetypal mutations found in CADASIL patients, into the human Notch3 cDNA by site-directed mutagenesis and made a mutant Notch3 expression vector as shown in Figure 1A ▶ . Arterial smooth muscle cell-specific expression of this mutant human Notch3cDNA was driven by regulatory elements of the mouse SM22α gene. This promoter has been shown to specifically drive expression of a LacZ transgene in arterial smooth muscle cells in both murine embryo and adult mouse (AJ, personal observation). 25
Figure 1.
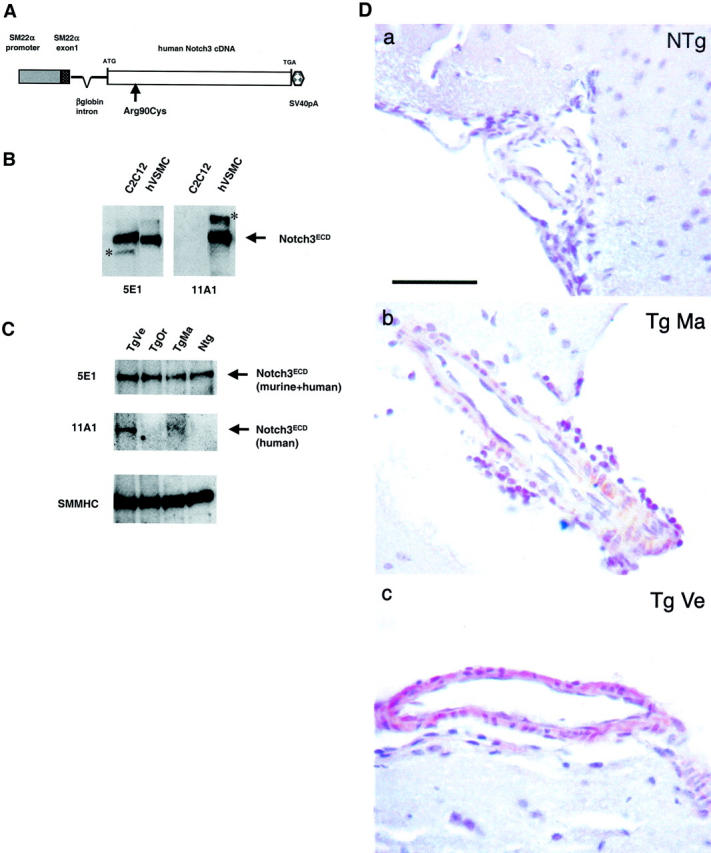
Transgene construct and analysis of transgene expression in brain tissues. A: Schematic view of the Notch3 transgene. The construct contains the SM22α promoter, a fragment of the SM22α gene including 2126 bp upstream of the initiation transcription site, and the complete noncoding first exon, the full-length human Notch3 cDNA with the Arg-to-Cys mutation at codon 90 (Arg90Cys). The bovine β-globin intron was inserted between the SM22α regulatory sequence and the Notch3 sequence; the SV40 polyadenylation (SV40pA) 3′ of the Notch3 cDNA. B: Specificity of the 5E1 and 11A1 anti-Notch3 monoclonal antibodies against the human and murine Notch3 proteins. Protein extracts prepared from murine C2C12 cells and human vascular smooth muscle cells (hVSMCs) were separated on a 6% SDS-polyacrylamide gel electrophoresis and immunoblotted with the 5E1 anti-Notch3ECD monoclonal antibody (left) and with the 11A1 anti-Notch3ECD monoclonal antibody (right). A 210-kd band, corresponding to the Notch3ECD, is detected in both murine and human cell extracts with the 5E1 antibody, while the 210-kd band is detected only in the human cell extract with the 11A1 antibody. The asterisk marks bands of unknown significance (cross-reacting material or postlysis degradation product). C: Immunoblot analysis of total and mutant Notch3 in transgenic mice and nontransgenic littermate brain tissue. Protein extracts were prepared from half a brain including the meningeal vessels of 1-month-old transgenic mice (Tg) (lines Ma, Ve, and Or) and of nontransgenic littermate (Ntg) separated on a 6% SDS-polyacrylamide gel electrophoresis and immunoblotted with the 5E1 and the 11A1 antibodies. Equal loading of protein extracts (∼100 μg) was assessed by Ponceau staining and anti-smooth muscle myosin heavy chain (SMMHC) antibody further confirmed that protein extracts contained a similar amount of vessel. Notice that the blot incubated with the 11A1 antibody required at least a fivefold longer exposure than the one incubated with the 5E1 antibody. D: Immunohistochemistry analysis of Notch3 expression in transgenic mice and nontransgenic littermate brain tissue. Paraffin brain sections from a 1-month-old nontransgenic littermate control (Ntg) (a) and from 1-month-old transgenic mice (Tg) (b, line Ma; c, line Ve) were immunolabeled with the 1E4 anti-Notch3ECD monoclonal antibody (hematoxylin counterstaining). No staining was detected in the vessel of the NTg mouse and vessels from both Tg lines exhibited a moderate staining. Scale bar, 50 μm.
We established three lines from three distinct founders that integrated and transmitted the transgene. Normal Mendelian ratios were observed in the subsequent breeding, indicating that no embryonic lethality occurred. Genomic Southern blotting showed that the approximate transgene copy number ranged from 5 to 15 across the three lines (Ve, Ma, Or) (data not shown). Reverse transcriptase-polymerase chain reaction analysis, using a set of primers specific for the noncoding exon 1 of SM22α and for the exon 11 of human Notch3, confirmed the expression of the transgene mRNA in all three transgenic lines (data not shown).
The total amount of mutant and endogenous Notch3 proteins was then assessed by immunoblot analysis with the 5E1 monoclonal antibody, which recognizes both human and murine Notch3ECD proteins and was found to be almost identical in various tissues from 1-month-old transgenic and wild-type littermates (Figure 1, B and C ▶ , and data not shown). Similar results were obtained when the blots were probed with the BC4 polyclonal antibody, which recognizes both human and murine Notch3TMIC proteins (data not shown). To further specifically study the expression of the transgene by Western blot, we used the 11A1 monoclonal anti-Notch3 antibody that proved to specifically recognize the human but not the murine Notch3ECD protein (Figure 1B) ▶ . The human mutant protein was detected in two of the three lines, with the highest expression in line Ve, although its absolute level was very low (Figure 1C) ▶ . We also examined the expression of mutant and endogenous Notch3 proteins by immunohistochemistry analysis using the 1E4 monoclonal anti-Notch3ECD antibody. No signal was detected in the brain vessels from nontransgenic mice, suggesting that the 1E4 clone, like the 11A1, recognizes the human but not the murine Notch3 protein. A weak signal was detected in the brain vessels from lines Ve and Ma, with line Ve displaying a stronger signal than line Ma (Figure 1D) ▶ . Together these data indicate that the two transgenic lines, Ve and Ma, expressed the human Notch3 transgene in the vessels, at a very low level, which can be estimated to be approximately <25% of the endogenous Notch3 protein; expression level of the murine endogenous protein in these two lines being almost identical to the one in nontransgenic mice.
Old Transgenic Mice Develop Notch3 Accumulation and GOM Deposits within the Cerebral Vessels
Transgenic lines Ma and Ve were used for further analysis. No significant difference in body weight, appearance, home cage behavior, or social interaction was observed between the transgenic and the wild-type littermates. Moreover, the survival of transgenic mice was not significantly different from that of wild-type mice, with >85% of mice surviving until 18 months of age.
Because there is a long preclinical stage in CADASIL patients during which vessel changes and brain parenchyma damages can be detected, we conducted a detailed histopathological analysis in these mice. Brain sections were stained using H&E and luxol-Barrera methods searching for lacunar infarcts and myelin loss, respectively, they were immunostained with an antibody specific to glial fibrillary acidic protein to search for reactive gliosis. We also extensively examined brain vessels searching for the pathological hallmarks of the CADASIL vasculopathy, including the accumulation of Notch3ECD and the presence of GOM deposits in the smooth muscle cells. Brains sections were examined using immunohistochemistry with antibodies specific to smooth muscle α-actin and Notch3ECD (clone 1E4), and we performed electron microscopy analysis.
Transgenic animals and age-matched wild-type littermates were first analyzed at 10 months of age (four transgenic and two nontransgenic). Transgenic animals showed no evidence of brain parenchyma damage or of vessel changes, including neither Notch3 accumulation or GOM deposits (data not shown).
Because the CADASIL phenotype become apparent on aging, we suspected that older transgenic mice might show histological lesions. Brains of 17- to 20-month-old mice (six transgenic and three nontransgenic) were examined by the same protocol used above. Brain parenchyma of 17- to 20-month-old transgenic mice showed no significant difference from those of age-matched nontransgenic littermates, apart from one infarct and some glial scars observed in one 20-month-old transgenic mouse. However, brain vessels of all transgenic animals examined (n = 6) exhibited a positive granular immunostaining with the 1E4 anti-Notch3ECD antibody that was not detected in age-matched nontransgenic littermates (n = 3) (Figure 2) ▶ . In addition, electron microscopy analysis revealed in all transgenic animals examined (n = 4), GOM deposits within the meningeal and the intracerebral arteries that were never seen in age-matched nontransgenic littermates (n = 2) (Figure 3) ▶ . Approximately one to three GOM patches around one smooth muscle cell were observed per vessel section. GOM deposits were located close to the smooth muscle cells, often within an infolding of the cell membrane and their electron microscopic structure was very similar to the one of GOM from CADASIL patients (Figure 3 ▶ , insets). GOM deposits were made of 10- to 15-nm electron dense granules, without filament-like profiles, their size ranged from very small, barely detectable to large deposits (0.2 to 0.6 μm). Importantly, both Notch3 accumulation and GOM deposits were detected in the two transgenic lines Ma and Ve analyzed.
Figure 2.

Notch3 accumulation in cerebral blood vessels from 17- to 20-month-old transgenic mice. Paraffin brain sections from a 20-month-old nontransgenic littermate control (Ntg) (A), 20-month-old transgenic mice (Tg) (B, line Ma; C, line Ve), and a 60-year-old CADASIL patient carrying the Arg153Cys mutation (D, hum CADASIL) were immunolabeled with the 1E4 anti-Notch3ECD monoclonal antibody. Vessels of the transgenic mice and of the CADASIL patients showed the same characteristic granular immunostaining that was not detected in the nontransgenic mouse. Scale bar, 20 μm.
Figure 3.
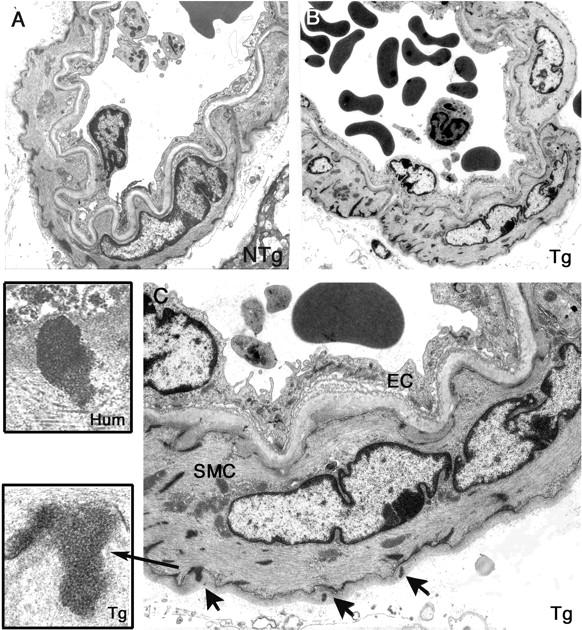
GOM deposits in cerebral blood vessels from 17- to 20-month-old transgenic mice. Ultrathin brain vessel sections from a 19-month-old nontransgenic littermate control (NTg) (A) and from a 17-month-old transgenic mouse (line Ma) (Tg) (B, C) were examined by electron microscopy. Vessel from the transgenic mouse showed no prominent changes but exhibited electron-dense granular deposits corresponding to GOM (arrows) within the basement membrane that are better seen on the higher magnification (C). Higher magnification of a GOM in a transgenic mouse brain vessel (inset, Tg) and of a GOM in a CADASIL patient brain vessel (inset, Hum) showing that GOM deposits in mouse and human had a similar structure. SMC, smooth muscle cell; EC, endothelial cell. Original magnifications: ×1293 (A, B); ×3597 (C); ×27,800 (inset, Tg); ×19,500 (inset, Hum).
Old Transgenic Mice Develop Notch3 Accumulation and GOM Deposits within the Peripheral Arteries
Because CADASIL is a systemic arteriopathy, we wondered whether transgenic mice would also display smooth muscle cell abnormalities within the extracerebral vessels. We essentially focused on the tail, because the tail in rodents contains a dense vascular network composed of four vascular bundles, each having at least one muscular artery and one vein (Figure 4) ▶ . In addition, the tail offered the advantage of being easily sampled in alive mice. Analysis was conducted on 17- to 20-month-old mice and included light microscopy analysis with H&E staining, α-actin and 1E4 anti-Notch3ECD immunostaining, and electron microscopy analysis. Because of the extreme heterogeneity in the nature of the tissues composing the tail, ranging from extremely hard, such as the bone, to very soft tissues, sectioning of paraffin blocks proved to be quite difficult. As a result only a limited number of samples could be really analyzed by immunohistochemistry. Nevertheless, we observed the characteristic granular 1E4 anti-Notch3ECD immunostaining in tail arteries from transgenic mice (n = 3) that was never detected in the wild-type littermates (n = 3) (Figure 5, A and D) ▶ . Tail samples embedded in Epon were easily processed for electron microscopy and analysis revealed the presence of GOM deposits in the tail arteries from all transgenic mice examined (n = 4) (Figure 6 ▶ , insets 1 and 2). Of interest GOM deposits were more numerous than in the brain vessels, with at least two to five GOM deposits around one smooth muscle cell per vessel section.
Figure 4.

Dense vascular network in the mouse tail. Paraffin tail section from a wild-type mouse stained with H&E. The tail contains four vascular bundles (encircled by black squares), each being located between skeletal muscle bundles (m) (A). High magnification of one vascular bundle (B), depicted by an asterisk on A, shows the high density of vessels. A, arteries; V, veins. Original magnifications: ×4 (A); ×20 (B).
Figure 5.
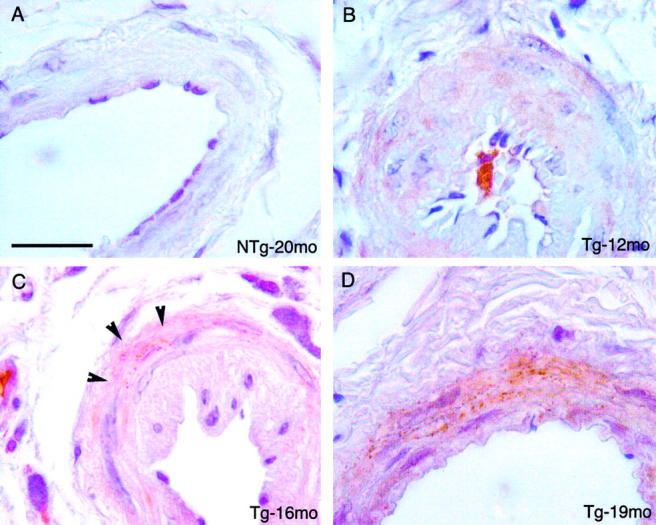
Age-dependent granular Notch3 immunoreactivity in the tail arteries from transgenic mice. Paraffin tail artery sections from a 20-month-old nontransgenic littermate (Ntg) (A) and from transgenic mice (B, 12 months old; C, 16 months old; and D, 19 months old) were immunostained with the 1E4 anti-Notch3ECD antibody. No staining was detected in the nontransgenic mouse, a homogeneous and nongranular staining was observed in the 12-month-old mouse. A granular staining was observed in the tail arteries from the 16-month-old and the 19-month-old transgenic mice, being discrete and present around a few smooth muscle cells in the 16-month-old mouse (arrowheads) and more prominent in the 19-month-old mouse. Scale bar, 20 μm.
Figure 6.
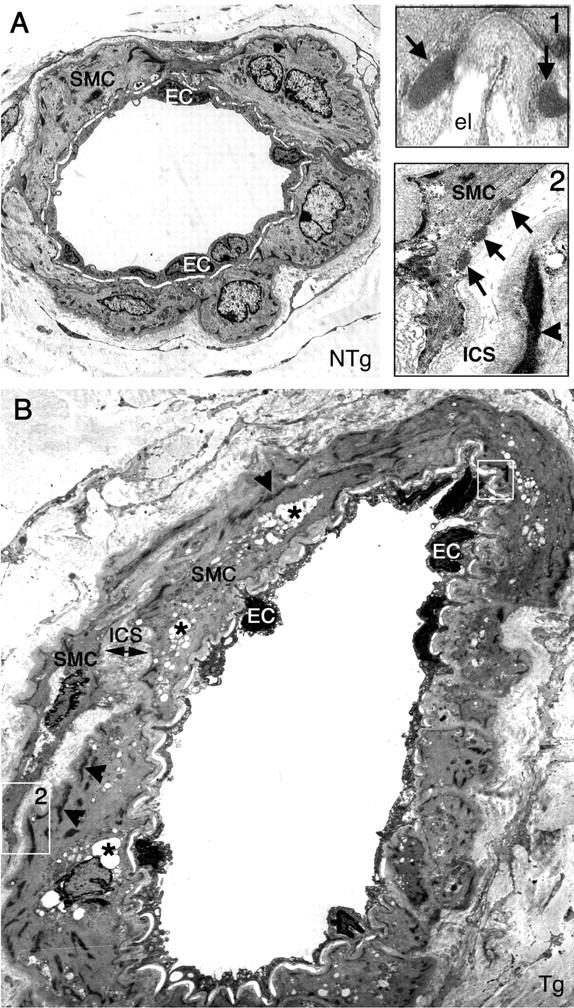
GOM deposits and prominent morphological alterations in the tail arteries from 17- to 20-month-old transgenic mice. Ultrathin tail artery sections from a 19-month-old nontransgenic littermate control (NTg) (A) and a 19-month-old transgenic mouse (Tg) (B and insets) were examined by electron microscopy. Artery from the transgenic mouse showed GOM deposits (arrows), which are better seen in the two insets (top right) (high magnification of the two areas encircled by white squares in B). Smooth muscle cells and endothelial cells in the tail artery of the transgenic mouse also exhibited prominent alterations. Endothelial cells had a thinner profile with pyknotic nuclei. Smooth muscle cells were filled with numerous electron-lucent vacuoles (asterisk) indicative of degeneration, and some cells contained an increased number as well as thicker dense plaques (arrowheads). They had an irregular shape and appeared separated from their neighboring cells, leading to an abnormal enlargement of the intersmooth muscle space. el, elastical lamina; SMC, smooth muscle cell; EC, endothelial cell; ICS, intersmooth muscle cell space. Original magnifications: ×1293 (A, B); ×12,930 (insets).
We also examined other peripheral arteries including renal, carotid, and femoral arteries. Electron microscopy analysis demonstrated the presence of GOM deposits in the transgenic mice (n = 3) but not in control age-matched mice (n = 2) (data not shown).
Tail Arteries from Transgenic Mice Show, in Addition to Notch3 Accumulation and GOM Deposits, Prominent Morphological Alterations
Importantly, electron microscopy analysis of tail arteries from transgenic mice demonstrated, in addition to GOM deposits, prominent morphological alterations with evidence of dramatic changes of both VSMCs and endothelial cells that were not seen in littermate controls (Figure 6) ▶ .
Degenerating cells and cellular debris were frequently seen within the media. Smooth muscle cells were filled with numerous electron-lucent vacuoles and swollen mitochondriae (Figures 6B and 7E) ▶ ▶ . Plasma membrane of smooth muscle cells, instead of being smooth and clearly distinguishable, appeared fuzzy and like moth-eaten (Figure 6 ▶ , inset 2; Figure 7G ▶ ). Of interest, neither the presence or the number of GOM deposits were found to be correlated with the severity of cell damage. Indeed GOM deposits could be absent on degenerating smooth muscle cells or, on the contrary, be present on less damaged cells. Smooth muscle cells showed cytoskeleton changes. Cytoplasm of some cells had an abnormal homogeneous and granular appearance. Other cells exhibited an increased number of dense bodies and more and thicker dense plaques giving the cell an irregular and indented shape (Figure 6B ▶ and inset 2). In addition, smooth muscle cells were much less closely packed, appearing separated from their neighbors and from the elastica lamina. As a result, the intersmooth muscle cell space and the subendothelial space appeared enlarged (Figures 6B and 7C ▶ ▶ ; Table 1 ▶ ). Endothelial cells also had a thinner profile, with their nuclei and their cytoplasm appearing more osmiophilic (Figure 6B) ▶ . In contrast, tail arteries from age-matched nontransgenic littermates (n = 4) showed only minor age-related ultrastructural changes, including a slight thickening of the extracellular matrix and the presence of very few electron-lucent vacuoles, but that were rarely observed and only in a very few smooth muscle cells (Figure 6A) ▶ .
Figure 7.
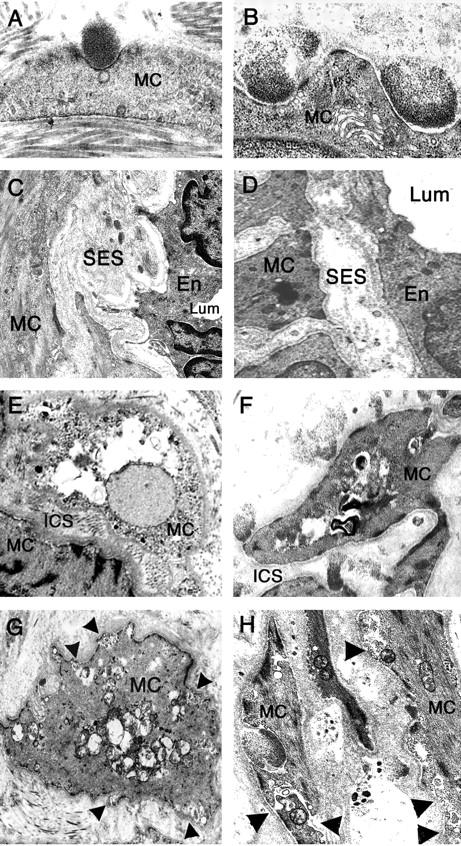
Ultrastructural vessel changes in transgenic mice are identical to those observed in human CADASIL patients. Electron micrographs of ultrathin tail artery sections from 16- to 20-month-old transgenic mice (left column: A, C, E, G) and skin vessel sections from CADASIL patients (right column: B, D, F, H) showed strikingly similar vessel changes in transgenic mice and CADASIL patients including GOM deposits located in smooth muscle cell infolding (A, B), an enlargement of the subendothelial space (C, D), prominent vacuolization of smooth muscle cells and enlargement of the intersmooth muscle cell space (E, F), and an irregular shape of smooth muscle cells with their plasma membrane having a blurred and moth-eaten appearance (arrowheads) (G, H). Lum, lumen; MC, smooth muscle cell; En, endothelial cell; SES, subendothelial space; ICS, intersmooth muscle cell space. Original magnifications: ×13,400 (A); ×15,000 (B); ×3750 (C); ×3800 (D); ×4646 (E); ×6000 (F); ×7750 (G); ×12,000 (H).
Table 1.
Age-Related Values of Subendothelial Space and Intersmooth Muscle Cell Space in Tail Arteries from Transgenic and Nontransgenic Mice
| Age, months | Subendothelial space | Intersmooth muscle cell space | ||
|---|---|---|---|---|
| Nontransgenic | Transgenic | Nontransgenic | Transgenic | |
| 5 | 0.43 ± 0.02 (n = 8) | 0.42 ± 0.10 (n = 11) | 0.41 ± 0.02 (n = 8) | 0.37 ± 0.04 (n = 11) |
| 10 to 12 | 0.35 ± 0.05 (n = 10) | 0.84 ± 0.16* (n = 11) | 0.31 ± 0.05 (n = 11) | 0.72 ± 0.03* (n = 11) |
| 14 to 16 | 0.58 ± 0.08 (n = 6) | 0.85 ± 0.22* (n = 14) | 0.42 ± 0.24 (n = 6) | 1.18 ± 0.14* (n = 14) |
| 17 to 20 | 0.74 ± 0.04 (n = 7) | 1.16 ± 0.30* (n = 5) | 0.75 ± 0.08 (n = 7) | 1.06 ± 0.30* (n = 5) |
Results are are mean ± SDs μm from metarterioles (n = 5 to 14) analyzed in two to eight mice per group
*P ≤ 0.01 versus age-matched nontransgenic group (unpaired, two-tailed Student’s t-test).
Importantly, all of the morphological alterations detected in the tail arteries of 17-to 20-month-old transgenic mice were strikingly and remarkably similar to those observed in the cerebral and peripheral arteries from patients (Figure 7) ▶ (PB, MMR, and AJ, unpublished data). 16-18,26,27
Morphological Alterations of Smooth Muscle Cells and Endothelial Cells Precede Notch3 Accumulation and GOM Deposits
As a next step we aimed to determine the time course of vessel changes, in particular to ascertain the earliest vessel changes in relation to the appearance of Notch3 accumulation and GOM deposits, with the hope that it will give us important clues to the mechanisms of smooth muscle cell degeneration. Analysis was conducted in tail arteries from transgenic mice, from 5 months of age up to 16 months of age and after, and included Notch3 immunostaining and electron microscopy analysis with ultrastructural morphometry.
Notch3 Accumulation and GOM Deposits
Up to 12 months of age, granular Notch3 immunostaining was never detected in transgenic mice (n = 6) (Figure 5B) ▶ . At 14 to 16 months of age, a faint 1E4 granular immunostaining was occasionally detected in a few smooth muscle cells of tail arteries, being observed in two of the four transgenic mice that were examined (Figure 5C) ▶ . Up to 14 months of age, extensive electron microscopy analysis failed to detect any GOM deposits in the arteries (Figure 8) ▶ . GOM deposits were detected in tail arteries only by 16 months of age (Figure 7A) ▶ .
Figure 8.
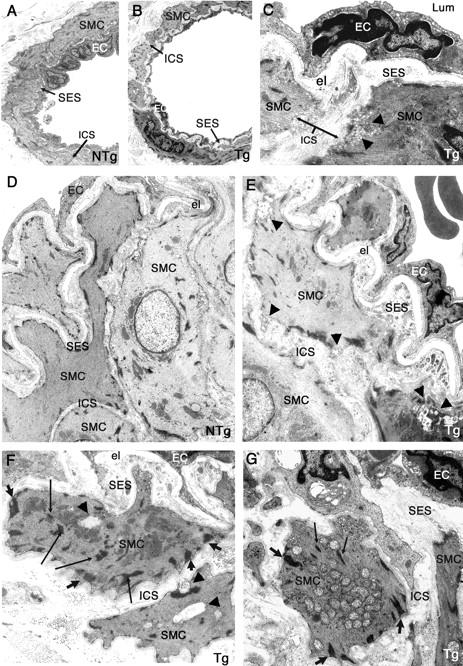
Ultrastructural changes of smooth muscle cells and of endothelial cells in tail arteries from 10- to 12 month-old transgenic mice, but no GOM deposits. Ultrathin tail artery sections of nontransgenic littermate control (NTg) (A, 10 months old; D, 12 months old) and of transgenic mice (Tg) (B and C, 10 months old; E–G, 12 months old) were examined by electron microscopy. Vessels from transgenic mice exhibited an enlarged intersmooth muscle cell space (better seen on C) and an enlarged subendothelial space filled with extracellular matrix and cellular debris (better seen on E–G). Smooth muscle cells contained more and thicker dense plaques (large arrows) having an abnormal oblique orientation and an increased number of dense bodies (thin arrows) (better seen on F and G). Starting signs of smooth muscle cell degeneration were also observed with a blurred and moth eaten appearance of the plasma membrane, the presence of some vacuoles (arrowheads) (C, E, F), or the abnormal accumulation of mitochondriae (G). Endothelial cells (ECs) with pyknotic nuclei were also detected (C, E, G). Importantly, no GOM deposit was observed. Lum, lumen; el, elastica lamina; SMC, smooth muscle cell; EC, endothelial cell; SES, subendothelial space; ICS, intersmooth muscle cell space. Original magnifications: ×1270 (A, B); ×6000 (C); ×1670 (D, E); ×4646 (G).
Morphological Alterations
In 5-month-old mice, no significant difference was detected between the transgenic mice (n = 3) and the wild-type mice (n = 2) (Table 1 ▶ and data not shown). The first visible defects were detected in 10- to 12-month-old transgenic mice. In wild-type mice (n = 6), the media was made of highly packed smooth muscle cells with an intimate contact of the innermost layer of smooth muscle cells to the internal elastica lamina (Figure 8, A and D) ▶ . In contrast, in transgenic mice (n = 6) the media showed that smooth muscle cells were less packed, and that the innermost layer did not fit any more the festoons of the internal elastic lamina (Figure 8; C, E, F, G) ▶ . As a result there was a widening of the intersmooth muscle cell space (0.72 ± 0.03 μm versus 0.31 ± 0.05 μm in the wild-type mice) and of the subendothelial space (0.84 ± 0.16 μm versus 0.35 ± 0.05 μm in the wild-type mice) (Table 1) ▶ . The enlarged subendothelial space was filled with extracellular matrix elements, cellular debris and microfibrils merged at the margins of elastica lamina (Figure 8E) ▶ . Smooth muscle cells exhibited cytoskeleton changes with the presence of more and larger dense bodies as well as more and thicker dense plaques, having sometimes a more oblique orientation (Figure 8, F and G) ▶ . Importantly, a very careful ultrastructural examination of vessels from 10- to 12-month-old transgenic mice already revealed starting signs of smooth muscle and endothelial cell damages. Some electron-lucent vacuoles, an abnormal accumulation of swollen mitochondriae, as well as a blurred and moth-eaten appearance of the plasma membrane were detected in smooth muscle cells (Figure 8; C, E, F, G) ▶ and endothelial cells having a more osmiophilic nuclei were also observed (Figure 8, E and G) ▶ .
In 14- to 16-month-old transgenic mice (n = 6), all of the alterations described above were more prominent. Both the subendothelial space and the intersmooth muscle cell space were further enlarged (subendothelial space = 0.85 ± 0.22 μm versus 0.58 ± 0.08 μm in the wild-type mice; intersmooth muscle cell space = 1.18 ± 0.14 μm versus 0.42 ± 0.24 μm in the wild-type mice) (Table 1) ▶ . Clear degenerative changes of smooth muscle cells were detected in the outermost layer of smooth muscle cells of larger arterioles and smooth muscle cell phantoms were observed (data not shown).
Discussion
We established two lines of transgenic mice expressing the Arg90Cys mutant Notch3, at a very low level, in VSMCs. A thorough analysis clearly showed that old transgenic mice developed the specific pathological hallmarks of the CADASIL vasculopathy. As in human patients, transgenic mice demonstrated Notch3ECD accumulation and GOM deposits that were detected in both cerebral and peripheral arteries. Prominent ultrastructural alterations with evidence of degenerative changes of vascular smooth cells were also detected in the tail arteries of the transgenic mice. Vascular smooth muscle changes, which included vacuolization of the smooth muscle cells, a moth-eaten appearance of the plasma membrane, and cytoskeleton changes were strikingly similar to those observed in CADASIL patients. In addition, as in human patients, vessels in transgenic mice showed a disruption of the normal smooth muscle cell anchorage to cells and extracellular matrix (PB, MMR, and AJ; unpublished observations). 16,17,27,28 As in human patients, endothelial cells, in addition to smooth muscle cells, showed degenerative changes. 17,26 However, these transgenic mice did not show evidence of brain parenchyma damages. Altogether our data indicate that the pathological alterations seen in our old transgenic mice recapitulate the characteristic and specific aspects of the CADASIL vasculopathy, modeling the early stage of the human disease before onset of clinical symptoms and brain parenchyma damages. It is often difficult to predict the accuracy with which transgenic mouse lines will model a corresponding human disease. Because modeling the CADASIL disease in the mouse was the main purpose of this study, we did not generate as a first step mice transgenic for the wild-type Notch3 protein. Therefore, at the present time we cannot formally exclude the possibility that the phenotype observed in the Notch3Arg90Cys mice may be the result of an overexpression of Notch3 or that we can state that it is merely the result of the expression of a mutant Notch3 protein. However, this former scenario seems very unlikely because the expression level of the Notch3Arg90Cys transgene proved to be very low in both lines, being far <25% of the endogenous murine Notch3 protein. Future studies will be needed to clarify this point as well as to determine whether mice expressing much more mutant protein may develop the lesions sooner.
A surprising aspect of this study was the location of the most severe lesions in the tail arteries rather than in the cerebral arteries. This is the opposite finding of what is observed in human CADASIL patients, in which, for a still unknown reason, vessel changes are much more prominent in the brain arteries than in the peripheral tissue arteries. One possibility may be that, in transgenic mice, the expression level of the transgene and/or the ratio of the transgene to the endogenous Notch3 in the tail arteries are slightly different from the brain arteries. Another possibility may be that, depending on the vascular bed, smooth muscle cells are more or less vulnerable to the mutation. Of interest, the tail vasculature is likely to be highly solicited in terms of hemodynamic and mechanical constraints because the tail contributes to two major functions in rodents, being a major thermoregulatory organ and an important balancing and stabilizing organ.
Most pathological studies performed in human patients focused on end-stage lesions of the CADASIL arteriopathy. Modeling the pathological vascular alterations characteristic of CADASIL in the tail arteries of these transgenic mice provided the opportunity to investigate the time course of vessel changes throughout a very long period. Appearance of Notch3ECD accumulation and GOM deposits seemed clearly dependent on age, being detected by 14 to 16 months of age. More importantly, we found that morphological alterations of smooth muscle cells preceded Notch3ECD and GOM deposit appearance, at least in tail arteries. Indeed, early signs of smooth muscle cell damage were detected at 10 to 12 months of age whereas GOM deposits and Notch3 accumulation were absent. Moreover, we failed to detect any apparent correlation between the presence or the number of GOM deposits and the severity of smooth muscle cells damages. Together these data indicate that neither the development nor the distribution of Notch3ECD accumulation and GOM deposits correlate with the early changes in smooth muscle cell integrity and strongly suggest that they are not the prerequisites for the induction of smooth muscle cell damage. However we cannot rule out that Notch3ECD accumulation or GOM deposits may contribute at a later stage of the pathogenic process when smooth muscle cells are committed to degeneration.
Time-course analysis of vessel changes revealed that the first ultrastructural abnormalities included the disruption of normal VSMC anchorage to adjacent extracellular matrix and cells as well as cytoskeleton changes with an increased number of dense plaques and dense bodies, present at around 10 months of age. A detailed ultrastructural analysis of a large series of vessels from CADASIL patients previously showed that loss of VSMC anchorage was a prominent feature in CADASIL, however, this finding was interpreted as a consequence of VSMC loss. 28 Our data indicate that loss of smooth muscle cell anchorage is unlikely the result of smooth muscle cell loss but rather a very early event because there was no evidence of smooth muscle loss when it was first detected. Cell-cell and cell-matrix interactions have major effects on phenotypic features such as gene regulation and cytoskeleton structure. In addition, a growing number of studies indicate that a number of cell types are dependent on adhesion to the extracellular matrix for their continued survival and that disruption of appropriate cell-matrix contacts both in vitro and in vivo is sufficient to initiate cell death. 29-31 On this basis, we raise the hypothesis that the disruption of normal smooth muscle cell anchorage may be one of the key events initiating the cascade leading to smooth muscle cell degeneration in CADASIL.
Modeling the pathological vascular alterations characteristic of CADASIL in the tail arteries of these transgenic mice provides several new opportunities for future investigations. The emerging view is that vasculopathy in CADASIL likely leads to vascular dysfunction rather than vessel occlusion because detailed pathological analysis in human CADASIL patients failed to detect any evidence of vessel stenosis. 13,28 Because of the simple and easy accessibility of the tail arteries and its possible functional monitoring on living animals, these mice will allow the investigation of the nature of this potential vascular dysfunction as well as to determine whether it may be apparent at the early stage of the disease even before vessels exhibit prominent smooth muscle cell loss. In addition these mice could provide a preclinical model for testing therapeutic drugs.
Acknowledgments
We thank Drs. J. B. Michel and D. Paulin for their generous gifts of human vascular smooth muscle cells and SM22α construct, respectively; Pepita Masquelier for animal care; Nathalie Goetinck for technical assistance with electron microscopy; and Charles Babinet for helpful discussions and critical reading of the manuscript.
Footnotes
Address reprint requests to Anne Joutel, INSERM EMI 99-21, Faculté de Médecine Lariboisière, 10 av de Verdun, 75010 Paris, France. E-mail: joutel@paris7.jussieu.fr.
Supported by grants from INSERM, the Fondation de France and Jean Valade award, (to A.J. and E.T.L.).
References
- 1.Tournier-Lasserve E, Joutel A, Melki J, Weissenbach J, Lathrop GM, Chabriat H, Mas JL, Cabanis EA, Baudrimont M, Maciazek J, Bach MA, Bousser MG: Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy maps to chromosome 19q12. Nat Genet 1993, 3:256-259 [DOI] [PubMed] [Google Scholar]
- 2.Chabriat H, Vahedi K, Iba-Zizen MT, Joutel A, Nibbio A, Nagy TG, Krebs MO, Julien J, Dubois B, Ducrocq X, Levasseur M, Homeyer P, Mas JL, Lyon-Caen O, Tournier-Lasserve E, Bousser MG: Clinical spectrum of CADASIL: a study of 7 families. Lancet 1995, 346:934-939 [DOI] [PubMed] [Google Scholar]
- 3.Chabriat H, Levy C, Taillia H, Iba-Zizen MT, Vahedi K, Joutel A, Tournier-Lasserve E, Bousser MG: Patterns of MRI lesions in CADASIL. Neurology 1998, 51:452-457 [DOI] [PubMed] [Google Scholar]
- 4.Dichgans M, Mayer M, Uttner I, Bruning R, Muller-Hocker J, Rungger G, Ebke M, Klockgether T, Gasser T: The phenotypic spectrum of CADASIL: clinical findings in 102 cases. Ann Neurol 1998, 44:731-739 [DOI] [PubMed] [Google Scholar]
- 5.Desmond DW, Moroney JT, Lynch T, Chan S, Chin SS, Mohr JP: The natural history of CADASIL: a pooled analysis of previously published cases. Stroke 1999, 30:1230-1233 [DOI] [PubMed] [Google Scholar]
- 6.Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, Alamowitch S, Domenga V, Cecillion M, Marechal E, Maciazek J, Vayssiere C, Cruaud C, Cabanis EA, Ruchoux MM, Weissenbach J, Bach JF, Bousser MG, Tournier-Lasserve E: Notch3 mutations in CADASIL, an hereditary adult-onset condition causing stroke and dementia. Nature 1996, 383:707-710 [DOI] [PubMed] [Google Scholar]
- 7.Artavanis-Tsakonas S, Rand MD, Lake RJ: Notch signaling: cell fate control and signal integration in development. Science 1999, 284:770-776 [DOI] [PubMed] [Google Scholar]
- 8.Joutel A, Vahedi K, Corpechot C, Troesch A, Chabriat H, Vayssiere C, Cruaud C, Maciazek J, Weissenbach J, Bousser MG, Bach JF, Tournier-Lasserve E: Strong clustering and stereotyped nature of Notch3 mutations in CADASIL patients. Lancet 1997, 350:1511-1515 [DOI] [PubMed] [Google Scholar]
- 9.Oberstein SA, Ferrari MD, Bakker E, van Gestel J, Kneppers AL, Frants RR, Breuning MH, Haan J: Diagnostic Notch3 sequence analysis in CADASIL: three new mutations in Dutch patients. Dutch CADASIL Research Group. Neurology 1999, 52:1913-1915 [DOI] [PubMed] [Google Scholar]
- 10.Dichgans M, Ludwig H, Muller-Hocker J, Messerschmidt A, Gasser T: Small in-frame deletions and missense mutations in CADASIL: 3D models predict misfolding of Notch3 EGF-like repeat domains. Eur J Hum Genet 2000, 4:280-285 [DOI] [PubMed] [Google Scholar]
- 11.Joutel A, Dodick DD, Parisi JE, Cecillon M, Tournier-Lasserve E, Bousser MG: De novo mutation in the Notch3 gene causing CADASIL. Ann Neurol 2000, 47:388-391 [PubMed] [Google Scholar]
- 12.Baudrimont M, Dubas F, Joutel A, Tournier-Lasserve E, Bousser MG: Autosomal dominant leukoencephalopathy and subcortical ischemic stroke: a clinicopathological study. Stroke 1993, 24:122-125 [DOI] [PubMed] [Google Scholar]
- 13.Ruchoux MM, Maurage CA: CADASIL: cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. J Neuropathol Exp Neurol 1997, 56:947-964 [PubMed] [Google Scholar]
- 14.Kalimo H, Vitanene M, Amberla K, Juvonene V, Martiila R, Pöyhönen M, Rinne JO, Savontaus ML, Tuisku S, Winblad B: CADASIL hereditary disease of arteries causing brain infarcts and dementia. Neuropathol Appl Neurobiol 1999, 25:257-265 [DOI] [PubMed] [Google Scholar]
- 15.Ruchoux MM, Chabriat H, Bousser MG, Baudrimont M, Tournier-Lasserve E: Presence of ultrastructural arterial lesions in muscle and skin vessels of patients with CADASIL. Stroke 1994, 25:2291-2292 [DOI] [PubMed] [Google Scholar]
- 16.Ruchoux MM, Guerouaou D, Vandenhaute B, Pruvo JP, Vermersch P, Leys D: Systemic vascular smooth muscle cell impairment in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Acta Neuropathol 1995, 89:500-512 [DOI] [PubMed] [Google Scholar]
- 17.Schröder JM, Sellhauss B, Jörg J: Identification of the characteristic vascular changes in a sural nerve biopsy of a case with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Acta Neuropathol 1995, 89:116-121 [DOI] [PubMed] [Google Scholar]
- 18.Ebke M, Dichgans M, Bergmann M, Voelter HU, Rieger P, Gasser T, Schwendemann G: Skin biopsy allows diagnosis in early stages. Acta Neurol Scand 1997, 95:351-357 [DOI] [PubMed] [Google Scholar]
- 19.Mayer M, Straube A, Bruening R, Uttner I, Pongratz D, Gasser T, Dichgans M, Muller-Hocker J: Muscle and skin biopsies are a sensitive diagnostic tool in the diagnosis of CADASIL. J Neurol 1999, 246:526-532 [DOI] [PubMed] [Google Scholar]
- 20.Joutel A, Andreux F, Gaulis S, Domenga V, Cecillon M, Battail N, Piga N, Chapon F, Godfrain C, Tournier-Lasserve E: The ectodomain of the Notch3 receptor accumulates within the cerebrovasculature of CADASIL patients. J Clin Invest 2000, 105:597-605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Joutel A, Favrole P, Labauge P, Chabriat H, Lescoat C, Andreux F, Domenga V, Cecillon M, Vahedi K, Ducros A, Cave-Riant F, Bousser MG, Tournier-Lasserve E: Skin biopsy immunostaining with a Notch3 monoclonal antibody for CADASIL diagnosis. Lancet 2001, 358:2049-2051 [DOI] [PubMed] [Google Scholar]
- 22.Spinner NB: CADASIL: Notch signaling defect or protein accumulation problem? J Clin Invest 2000, 105:561-562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fryxell KJ, Soderlund M, Jordan TV: An animal model for the molecular genetics of CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy). Stroke 2001, 32:6-11 [DOI] [PubMed] [Google Scholar]
- 24.Haritunians T, Boulter J, Hicks C, Buhrman J, DiSibio G, Shawber C, Weinmaster G, Nofziger D, Schanen C: CADASIL Notch3 mutant proteins localize to the cell surface and bind ligand. Circ Res 2002, 90:506-508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moessler H, Mericskay M, Li Z, Nagl S, Paulin D, Small JV: The SM 22 promoter directs tissue-specific expression in arterial but not in venous or visceral smooth muscle cells in transgenic mice. Development 1996, 122:2415-2425 [DOI] [PubMed] [Google Scholar]
- 26.Ruchoux MM, Maurage CA: Endothelial changes in muscle and skin biopsies in patients with CADASIL. Neuropathol Appl Neurobiol 1997, 24:60-65 [DOI] [PubMed] [Google Scholar]
- 27.Rubio A, Rifkin D, Powers JM, Patel U, Stewart J, Faust P, Goldman JE, Mohr JP, Numaguchi Y, Jensen K: Phenotypic variability of CADASIL and novel morphologic findings. Acta Neuropathol 1997, 94:247-254 [DOI] [PubMed] [Google Scholar]
- 28.Brulin P, Godfraind C, Leteurtre E, Ruchoux MM: Morphometric analysis of ultrastructural vascular changes in CADASIL. Acta Neuropathol 2002, 104:241-248 [DOI] [PubMed] [Google Scholar]
- 29.Frisch SM, Francis H: Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol 1994, 124:619-626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McGill G, Shimamura A, Bates RC, Savage RE, Fisher DE: Loss of matrix adhesion triggers rapid transformation-selective apoptosis in fibroblasts. J Cell Biol 1997, 138:901-911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ding B, Price RL, Goldsmith EC, Borg TK, Yan X, Douglas PS, Weinberg EO, Bartunek J, Thielen T, Didenko VV, Lorell BH: Left ventricular hypertrophy in ascending aortic stenosis mice: anoikis and the progression to early failure. Circulation 2000, 101:2854-2862 [DOI] [PubMed] [Google Scholar]