

Abstract
Neovascularization in the retina and iris of diabetic patients is a major cause of severe visual loss. However, study of these lesions is compromised by the lack of a comparable diabetic rodent model. Because the vasoactive and angiogenic agent, angiotensin II, is involved in diabetic microvascular disease, we aimed to determine whether endothelial cell proliferation could be induced in the retinae and irides of hypertensive transgenic (mRen-2)27 rats that display an enhanced extra-renal renin-angiotensin system (RAS), including the eye. Six-week-old Ren-2, spontaneously hypertensive, and Sprague-Dawley rats received either streptozotocin or control vehicle and were studied for 36 weeks. Additional nondiabetic and diabetic Ren-2 rats were treated throughout with the angiotensin-converting enzyme inhibitor lisinopril (LIS) (10 mg/kg/day in drinking water). Endothelial cell proliferation was only observed in retinae and irides of diabetic Ren-2 rats and was reduced with LIS. In diabetic Ren-2, vascular endothelial growth factor (VEGF) and VEGFR-2 mRNA were increased in retinae and irides and reduced with LIS. Diabetes activated ocular renin in Ren-2 but not Sprague-Dawley rats. The diabetic Ren-2 rat is a model of intraocular endothelial cell proliferation that can be attenuated by RAS blockade via VEGF-dependent pathways. RAS blockade is a potential treatment for vision-threatening diabetic microvascular complications.
Longstanding diabetes mellitus is associated with alterations to the retinal and iris vasculature that eventually may progress to neovascularization, the hallmark feature of proliferative diabetic retinopathy (PDR), and rubeosis iridis. 1 These intraocular lesions are a major cause of visual loss in the Western world, and can progress despite advances in laser photocoagulation and vitrectomy and medical approaches including intensified glycemic control. 1,2
Recent studies have highlighted the importance of hypertension and the renin-angiotensin system (RAS) in the pathogenesis of diabetic ocular microvascular complications. 3-5 There is also evidence that angiotensin II (Ang II), in addition to its vasoactive effects, is a proangiogenic cytokine that is synthesized within the eye. 6-9 Importantly, a therapeutic role for blockade of the RAS has been suggested in the EUCLID study (EURODIAB Controlled Trial of Lisinopril in Insulin-Dependent Diabetes Mellitus), in which the angiotensin-converting enzyme (ACE) inhibitor lisinopril (LIS) slowed the progression of retinopathy including the development of PDR in diabetic patients. 10
It is well documented that the potent angiogenic and permeability factor, vascular endothelial growth factor (VEGF), has a pivotal role in the development of retinal and iris neovascularization in diabetes. 11,12 Of interest are recent reports that VEGF and Ang II interact to elicit an angiogenic response in the eye. 13-15 However, investigation into the role of hypertension, Ang II, VEGF, and other pathogenetic factors in PDR and rubeosis iridis is to a large extent compromised by the absence of diabetic rodent models that progress to retinal and iris neovascularization. 16 The reasons for rodent resistance to the development of diabetic ocular neovascularization is unknown, but could relate to the duration of diabetes and the absence of systemic hypertension, two major factors linked to the development of retinopathy. 3-5 With this in mind, the transgenic Ren-2 rat, which is both hypertensive and exhibits enhanced extra-renal renin and angiotensin was investigated. 17 We have previously reported the Ren-2 rat to develop advanced diabetic renal disease after 3 months of streptozotocin (STZ) diabetes and the rat has many similarities to human diabetic nephropathy including renal impairment and advanced glomerulosclerosis and tubulointerstitial injury. 18-20
In the present study, we examined the possibility that intraocular endothelial cell proliferation may occur in the transgenic Ren-2 rat after a longer duration of diabetes and whether the lesion is associated with up-regulation of ocular VEGF and its second receptor, VEGFR-2. The effect of ACE inhibition on endothelial cell proliferation in the transgenic Ren-2 rat was also examined. To distinguish between the effects of the local RAS and systemic hypertension in the pathogenesis of the ocular lesion in the diabetic transgenic Ren-2 rat, comparisons were made with two strains of age-matched diabetic rats. The Sprague-Dawley (SD) rat is normotensive and exhibits a suppressed tissue RAS, whereas the spontaneously hypertensive rat (SHR) has elevated blood pressure and low tissue renin. 18,21
Materials and Methods
Animals
Six-week-old female SD, SHR, and heterozygous Ren-2 rats were randomized to receive either 55 mg/kg of STZ (diabetic; Sigma, St. Louis, MO) diluted in 0.1 mol/L of citrate buffer, pH 4.5, or citrate buffer alone (nondiabetic) by tail vein injection after an overnight fast. Two days after STZ or control vehicle a separate group of nondiabetic and diabetic Ren-2 rats were administered the ACE inhibitor LIS (10 mg/kg/day in drinking water), and this treatment continued throughout the study. All rats were studied for 36 weeks. Six to eight animals per group were examined.
All rats were housed in a stable environment (maintained at 20 ± 2°C with a 12 hour light-dark cycle) and allowed free access to tap water and standard rat chow (GR2; Clark-King and Co., NSW, Australia). Each week, rats were weighed and blood glucose (nondiabetic, 4 to 8 mmol/L; diabetic, 18 to 27 mmol/L) estimated using an Accutrend α glucometer (Mannheim Boehringer, NSW, Australia). Diabetic animals received a daily injection of insulin (4 to 8 U i.p.; Ultratard, Novo Nordisk, Bagsraerd, Denmark) to promote weight gain and prevent ketonuria. Glycated hemoglobin (HbA1c) was measured by an automated high-performance liquid chromatography technique (Biorad Diamat, Richmond, CA) as previously described. 22 Every 2 weeks, systolic blood pressure was recorded in prewarmed conscious rats by tail cuff plethysmography. 23 Arterial pressure changes detected by a Pneumatic Pulse Transducer PE-300 (Narco Biosystems Inc., Houston, TX) were recorded using a Chart program (version 3.5) on a Maclab/2E System (AD Instruments Pty. Ltd., Castle Hill, NSW, Australia). Systolic blood pressure was taken at the same time of the day (1400 to 1700 hours) to minimize circadian influences (5 to 6 hours after treatment administration) from an average of at least three consecutive measurements to reduce variability. 23 All experimental procedures adhered to the guidelines of the National Health and Medical Research Council of Australia’s Code for the Care and Use of Animals for Scientific Purposes and were approved by the Bioethics Committee of the University of Melbourne.
Histology
All rats were anesthetized with pentobarbital sodium (Nembutal, 50 mg/kg body wt i.p.; Boehringer Ingelheim, Australia) and perfused via the abdominal aorta with 0.1 mol/L of phosphate-buffered saline (PBS) (∼150 ml, pH 7.4, 180 to 220 mmHg) for 1 to 2 minutes to remove circulating blood. The eyes and kidneys were removed and immersion-fixed overnight in Bouins (BDH Laboratory Supplies, Poole, UK). After routine processing through graded alcohols and histolene, tissues were embedded in paraffin. Eyes and kidneys were serially sectioned at 3 μm, mounted on 1% gelatin-coated glass slides and stained with hematoxylin and eosin (H&E) for histopathological examination.
Immunohistochemistry for Endothelial Cell Proliferation in Retina and Iris
Six to eight pairs of adjacent sections were randomly selected from one eye per animal. Paraffin wax was removed from the tissue sections by immersion in histolene (Australia Biostain Pty. Ltd., Victoria, Australia), rehydrated in graded ethanols, and briefly washed in running tap water. Sections were then incubated for 10 minutes with normal goat serum diluted 1:10 with 0.1 mol/L of PBS at pH 7.4. In each pair, one section was labeled with a monoclonal antibody to proliferating cell nuclear antigen (PCNA) (Sigma Aldrich, Pty. Ltd., Castle Hill, NSW, Australia) while the consecutive section was labeled with the endothelial cell marker Banderia simplicifolia isolectin (Australian Laboratory Services, Sydney, NSW, Australia).
For PCNA labeling, sections were incubated with 1:75 of PCNA diluted in 0.1 mol/L of PBS at pH 7.4 for 2 hours at room temperature. Sections were washed thoroughly with PBS (3 × 5-minute changes), flooded with hydrogen peroxide diluted 1:5 with methanol for 10 minutes, rinsed with PBS (1 × 5 minutes), and then incubated with biotinylated goat anti-mouse IgG (Dakopatts, Glostrup, Denmark) diluted 1:200 with PBS for 1 hour. Sections were then rinsed with PBS (1 × 5 minutes) and incubated with the avidin-biotin-peroxidase complex (Vector, Burlingame, CA) diluted 1:200 with PBS for 1 hour at room temperature. After rinsing with PBS (1 × 5 minutes), sections were incubated with 0.05% diaminobenzidine and 0.05% hydrogen peroxide (Pierce, Rockford, IL) in PBS at pH 7.6 for 10 minutes, rinsed in tap water, counterstained in Mayer’s hematoxylin, differentiated in Scott’s tap water, dehydrated, and mounted in Depex.
For isolectin labeling, sections were incubated with 1:90 isolectin diluted in 0.1 mol/L of PBS at pH 7.4 for 3 hours at room temperature. Sections were washed thoroughly with PBS (3 × 5-minute changes), flooded with hydrogen peroxide diluted 1:4 with methanol for 10 minutes, and rinsed with PBS (3 × 5 minutes). Sections were then incubated with the avidin-biotin-peroxidase complex (Vector) diluted 1:200 with PBS for 1 hour at room temperature. After rinsing with PBS (5 minutes), sections were incubated with 0.05% diaminobenzidine and 0.05% hydrogen peroxide (Pierce) in PBS at pH 7.6 for 1 to 15 minutes, rinsed in tap water, counterstained in Mayer’s hematoxylin, differentiated in Scott’s tap water, dehydrated, and mounted in Depex. In each experiment a negative control was established by incubating sections with 1:10 normal goat serum in place of the primary antiserum.
Analysis of Endothelial Cell Proliferation
Adjacent sections of eye stained with PCNA or isolectin were photographed using a Fujix HC-2000 photomicroscope (Fuji, Tokyo, Japan) and images projected on adjacent computer monitors. Matching retinal or iris fields were identified in both sections. The number of proliferating endothelial cells in the inner retina (inner limiting membrane, ganglion cell layer, and inner plexiform layer) or the iris were counted by identifying a PCNA-stained cell and classifying it as an endothelial cell on the adjacent isolectin-labeled section. Three fields of inner retina or iris were viewed per section and six to eight pairs of sections per eye analyzed. Six to eight animals in each group were studied.
In Situ Hybridization for VEGF and VEGFR-2
Riboprobes were synthesized from cDNAs encoding mouse VEGF and VEGFR-2 (Dr. S. Stacker, Ludwig Institute, Parkville, Australia). 24 The cDNAs were cloned into pGEM 4Z (Promega, Madison, WI) and linearized with HindIII to produce anti-sense probes using SP6 RNA polymerase (Promega). Three-μm paraffin sections of eye premounted on 1% 3-aminopropyltriethoxysilane-coated slides were dewaxed, rehydrated in graded ethanol and milliQ water, equilibrated in P buffer [50 mmol/L Tris-HCl, pH 7.5, 5 nmol/L ethylenediaminetetraacetic acid (EDTA)], and incubated in 125 μg/ml of Pronase E (Bio Scientific, NSW, Australia) in P buffer for 10 minutes at 37°C. Sections were then washed in 0.1 mol/L of sodium phosphate buffer (pH 7.2), briefly refixed in 4% paraformaldehyde (Crown Scientific Pty. Ltd., Victoria, Australia) for 10 minutes, rinsed in milliQ water, dehydrated in 70% ethanol, and air-dried. Hybridization buffer containing 2 × 10 4 cpm/μl riboprobe in 300 mmol/L NaCl, 10 mmol/L Tris-HCl (pH 7.5), 10 mmol/L Na2HPO4, 5 mmol/L EDTA (pH 8.0), 1× Denhardt’s solution, 50% formamide, 17 mg/ml yeast RNA, and 10% w/v dextran sulfate was heated to 85°C for 5 minutes, and 25 μl of this solution was then added to each section. Hybridization was performed overnight at 60°C in 50% formamide-humidified chambers. Sections hybridized with sense probes for VEGF and VEGFR-2 were used as controls for nonspecific binding. After hybridization, slides were washed in 2× standard saline citrate containing 50% formamide prewarmed to 50°C to remove coverslips. Sections were then washed in the above-described solution for 1 hour at 55°C, rinsed three more times in RNase buffer (10 mmol/L Tris-HCl, pH 7.5, 1 mmol/L EDTA, pH 8.0, 0.5 mol/L NaCl), and incubated with RNase A (150 μg/ml) for 1 hour at 37°C. Sections were later washed in 2× standard saline citrate for 45 minutes at 55°C, dehydrated in graded ethanol, air-dried, and exposed to Kodak X-Omat autoradiographic film (Eastman-Kodak, Rochester, NY) for 5 days. Slides were subsequently dipped in Illford LM1 emulsion (Ilford, Cheshire, UK), stored in a light-free box with desiccant at 4°C for 4 weeks, immersed in Kodak D19 developer (Eastman-Kodak), fixed in Ilford Hypam (Ilford), and stained with H&E.
Quantitative in Situ Hybridization
Dark-field images were captured using light microscopy and a Fujix HC-2000 digital camera (Fuji, Tokyo, Japan). The outline of the inner retina (inner limiting membrane, ganglion cell layer, and inner plexiform layer) and iris were defined by interactive tracing. Gene expression was then quantitatively measured to determine the proportion of the area occupied by autoradiographic grains as previously described 24 using computerized image analysis (AIS; Imaging Research, Ontario, Canada). All sections were hybridized to their respective probes in the same experiment and analyzed in duplicate (n = 8 sections per rat, six to eight rats per group).
Ocular Renin Radioimmunoassay
Thirty-six weeks after STZ or control vehicle, a separate group of nondiabetic and diabetic SD and Ren-2 rats (n = 6 rats per group) were anesthetized with pentobarbital sodium (Nembutal, 50 mg/kg body wt i.p.; Boehringer Ingelheim, Australia) and perfused via the abdominal aorta with 0.1 mol/L PBS (∼150 ml, pH 7.4, 180 to 220 mmHg) for 1 to 2 minutes to remove circulating blood. The eyes were enucleated and the vitreous removed. For estimations of total renin, the left eye was placed in 150 μl of 0.1 ml of PBS at pH 7.4. For estimations of active renin, the right eye was snap-frozen in 150 μl of 0.1 mol/L PBS containing 0.1% bovine serum albumin, 0.01% thimerosol, and the following protease inhibitors: 7.5 mmol/L N-ethylmaleimide, 6 mmol/L EDTA-Na2, and 30 mmol/L benzamidine, pH 7.4. All samples were thawed, homogenized, and refrozen twice and assayed for renin by an enzyme kinetic method with hog renin (National Standards Laboratory, London, UK) as the reference standard and 24-hour nephrectomized rat plasma as the angiotensinogen substrate. 9,25 Duplicate samples were incubated for 1 hour at 37°C, pH 7.4, and the renin present in the samples was estimated by immunoassay of generated ANG I referenced against the amount of ANG I generated by 2 × 10−6 Goldblatt units (GU) of hog renin. Total renin was assayed using tissue extracted without inhibitors after activation of prorenin by trypsin treatment (2 mg/ml for 10 minutes on ice). Prorenin was derived as total renin minus active renin.
Statistical Analysis
Data were analyzed by analysis of variance followed by a Tukey’s posthoc comparison with P < 0.05 considered statistically significant. Diabetic and nondiabetic data within each strain were analyzed with a t-test for individual samples.
Results
Body Weight, HbA1c, Systolic Blood Pressure, and Ocular Renin Content
At 36 weeks, all diabetic rats had a lower body weight than their respective nondiabetic controls (Table 1) ▶ . HbA1c levels throughout the experimental period were consistently higher (P < 0.01) in diabetic Ren-2 rats compared to nondiabetic Ren-2 rats (8, 16, and 36 weeks, respectively; nondiabetic, 3.3 ± 0.1, 3.4 ± 0.2, and 3.3 ± 0.2; diabetic, 15.4 ± 0.3, 15.8 ± 0.4, 1and 5.9 ± 0.5). In diabetic Ren-2 rats treated with LIS, HbA1c levels were similar to untreated diabetic Ren-2 rats (8, 16, and 36 weeks, respectively, 15.5 ± 0.4, 16 ± 0.2, 15.8 ± 0.3) and higher than nondiabetic Ren-2 rats (P < 0.01). Throughout the study, SHR and Ren-2 rats had higher systolic blood pressure than SD rats (Table 1) ▶ . Diabetes had no significant effect on systolic blood pressure. Nondiabetic and diabetic SD rats had similar amounts of ocular active renin and prorenin (nondiabetic SD: active renin, 2.3 ± 2.0 μGU; prorenin, 0.72 ± 0.25 μGU and diabetic SD: active renin, 5.1 ± 2.3 μGU; prorenin, 5.0 ± 2.7 μGU). Ocular renin levels were significantly higher in Ren-2 rats compared with SD rats (P < 0.001) and in nondiabetic Ren-2 rats active renin and prorenin levels were similar (nondiabetic Ren-2: active renin, 28.8 ± 2.7 μGU; prorenin, 16.6 ± 6.0 μGU). In diabetic Ren-2 rats, ocular renin was mainly in an active form (diabetic Ren-2: active renin, 25.5 ± 9.5 μGU; prorenin, 8.6 ± 5.3 μGU; P < 0.05).
Table 1.
Body Weight and Systolic Blood Pressure of SD Rats, SHR, and Transgenic (mRen-2)27 Rats, 36 Weeks after STZ Diabetes or Control Vehicle
| Body weight, g | Systolic blood pressure, mmHg | |
|---|---|---|
| Nondiabetic SD | 587.9 ± 16.4 | 129.2 ± 12.1 |
| Diabetic SD | 291.8 ± 13.9* | 128.8 ± 12.1 |
| Nondiabetic SHR | 411.5 ± 3.3 | 182.6 ± 3.3† |
| Diabetic SHR | 380.7 ± 6.7* | 201.1 ± 5.8† |
| Nondiabetic Ren-2 | 390.0 ± 15.4 | 172.3 ± 6.0† |
| Diabetic Ren-2 | 304.6 ± 10.9* | 176.3 ± 6.0† |
| Nondiabetic Ren-2+LIS | 350.7 ± 8.9 | 119.2 ± 2.9 |
| Diabetic Ren-2+LIS | 265.0 ± 21.1* | 110.5 ± 4.8 |
Values expressed as means ± SEM. n = 6 to 8 rats per group.
*P < 0.001 versus respective nondiabetic control at 36 weeks.
†P < 0.01 versus all SD rats and Ren-2 rats treated with LIS.
Retinal and Iris Histopathology
Retinal histology appeared similar in all groups (not shown). In irides of all nondiabetic rats (Figure 1A) ▶ , the architecture was similar and consisted of a single layer of nonpigmented epithelium on which two layers of vessels were superimposed. Periodically a large dilated vessel was observed. Fibroblasts were located in the iris stroma. The irides of diabetic SD and SHR rats (not shown) were similar to nondiabetic controls. In contrast, the irides of untreated diabetic Ren-2 rats were thicker and often contained three to four layers of large dilated vessels extending over the iris surface (Figure 1B) ▶ . These vessels contained numerous red and white blood cells. These irides from diabetic Ren-2 rats also had posterior synechiae (Figure 1C) ▶ . In diabetic Ren-2 rats treated with LIS, the iris appeared similar to nondiabetic animals (Figure 1D) ▶ .
Figure 1.

Histopathology of irides from nondiabetic and diabetic Ren-2 rats. A: Nondiabetic Ren-2 rat iris showing normal distribution and density of iris vessels (arrow). B: Diabetic Ren-2 rat iris contains numerous dilated vessels (arrow) with red blood cells (asterisk). C: Diabetic Ren-2 rat iris has vessels adhering to the lens capsule (arrow). D: Diabetic Ren-2 rat treated with LIS showing density and size of iris vessels (arrow) is similar to nondiabetic Ren-2 control. LC, lens capsule; L, lens. Stain, H&E. Original magnifications, ×200.
Endothelial Cell Proliferation in Retina and Iris
Representative histological sections showing endothelial cell proliferation in inner retina and iris are illustrated in Figure 2 ▶ (retina, A to J; iris, K to T). Analysis of endothelial cell proliferation in inner retina and iris is shown in Figure 3 ▶ . In nondiabetic SD, SHR, and Ren-2 rats a small number of proliferating endothelial cells were observed in blood vessels in the inner retina. LIS reduced the amount of endothelial cell proliferation in the inner retina of nondiabetic Ren-2 rats to zero. Diabetes was associated with an increase in endothelial cell proliferation in blood vessels in the inner retina of Ren-2 rats but not SD or SHR. Endothelial cell proliferation was observed in the inner retina of diabetic Ren-2 rats treated with LIS.
Figure 2.

Consecutive 3-μm paraffin sections of inner retina and iris from nondiabetic and diabetic rats labeled with PCNA or the endothelial cell marker isolectin. Retina: A and B: Diabetic SD. C and D: Diabetic SHR. E and F: Nondiabetic Ren-2. G and H: Diabetic Ren-2. I and J: Diabetic Ren-2 treated with LIS. Arrows demonstrate endothelial cells labeled with either PCNA or isolectin. Iris: K and L: Diabetic SD. M and N: Diabetic SHR. O and P: Nondiabetic Ren-2. Q and R: Diabetic Ren-2. S and T: Diabetic Ren-2 treated with LIS. Arrows demonstrate endothelial cells labeled with either PCNA or isolectin. Only in diabetic Ren-2 inner retina (G and H) and iris (Q and R) were endothelial cells positive for PCNA. Endothelial cell proliferation in both inner retina (I and J) and iris (S and T) of diabetic Ren-2 rats was reduced with LIS. Counterstain, H&E. Original magnifications, ×500.
Figure 3.

Number of proliferating endothelial cells in the inner retina (A) and iris (B) of nondiabetic and diabetic SD rats, SHRs, and transgenic (mRen-2)27 rats. Values expressed as means ± SEM; n = 6 to 8 rats per group. Ren-2, transgenic (mRen-2)27 rat. A: Retina. *, P < 0.001 versus nondiabetic Ren-2 + LIS. †, P < 0.05 versus all diabetic groups and nondiabetic Ren-2 + LIS. B: Iris. ‡, P < 0.01 versus all diabetic groups and nondiabetic Ren-2 + LIS.
Overall, in all rat groups irides contained more proliferating endothelial cells than in the inner retina. In all nondiabetic animals, endothelial cell proliferation was localized to blood vessels within the iris stroma. In diabetic Ren-2 rats, irides appeared thicker and numerous blood vessels were observed on the iris surface. Increased endothelial cell proliferation was found in diabetic Ren-2 rat irides compared to all other groups. In diabetic SD and SHR, the appearance of irides and the amount of endothelial cell proliferation was unchanged compared to nondiabetic SD and SHR. LIS reduced the amount of endothelial cell proliferation in irides of diabetic Ren-2 rats.
In Situ Hybridization for VEGF and VEGFR-2 mRNA
In retinae of all nondiabetic rats, a similar distribution of labeling for both VEGF (not shown) and VEGFR-2 mRNA (Figure 4) ▶ was evident with hybridization signal in inner limiting membrane, ganglion cell layer, inner plexiform layer, and inner nuclear layer (INL). In nondiabetic rats, the hybridization signal for VEGF and VEGFR-2 mRNA was most intense in SHR and Ren-2 animals. Diabetes did not alter the distribution of VEGF and VEGFR-2 mRNA in retinae, however intensity was increased in the diabetic Ren-2 rat but not SD and SHR (Figure 4) ▶ . Both VEGF and VEGFR-2 mRNA were reduced in the retinae of diabetic Ren-2 rats treated with LIS (Figure 4) ▶ .
Figure 4.

Cellular expression of VEGF (open bars) and VEGFR-2 (closed bars) mRNA in inner retina (A) and iris (B) of nondiabetic and diabetic SD rats, SHRs, and transgenic (mRen-2)27 rats. Values are represented as mean ± SEM. Ren-2, transgenic (mRen-2)27 rat; ND, nondiabetic; D, diabetic. A: Retina. *, P < 0.01 versus all nondiabetic groups labeled for VEGF. **, P < 0.01 versus all nondiabetic groups labeled for VEGFR-2. †, P < 0.001 versus all diabetic groups and nondiabetic Ren-2 groups labeled for VEGF. †, P < 0.001 versus all diabetic groups and nondiabetic Ren-2 groups labeled for VEGFR-2. B: Iris. †, P < 0.01 versus respective nondiabetic group labeled for VEGF. ‡, P < 0.01 versus diabetic Ren-2 + LIS labeled for VEGF. *, P < 0.001 versus all nondiabetic groups labeled for VEGFR-2. #, P < 0.05 versus respective nondiabetic group labeled for VEGFR-2. §, P < 0.05 versus all diabetic groups labeled for VEGFR-2.
In irides of all nondiabetic rats, labeling for both VEGF (Figures 4 and 5) ▶ ▶ and VEGFR-2 (Figures 4 and 5) ▶ ▶ was localized to blood vessels and stromal cells. The intensity of VEGF and VEGFR-2 labeling was similar in treated and untreated nondiabetic SHR and Ren-2, however VEGFR-2 expression was lower in nondiabetic SD rats. Diabetes was associated with an increase VEGF and VEGFR-2 mRNA in irides of SD and particularly untreated Ren-2 rats (Figures 4 and 5) ▶ ▶ . In SHR irides, diabetes was associated with an increase in VEGF mRNA but not VEGFR-2. Overall, VEGF and VEGFR-2 hybridization signal appeared to be more intense in iris than retina of diabetic Ren-2 rats. LIS treatment reduced VEGF and VEGFR-2 mRNA in irides of diabetic Ren-2 rats. In all groups, diabetes did not alter the distribution of VEGF and VEGFR-2 mRNA.
Figure 5.
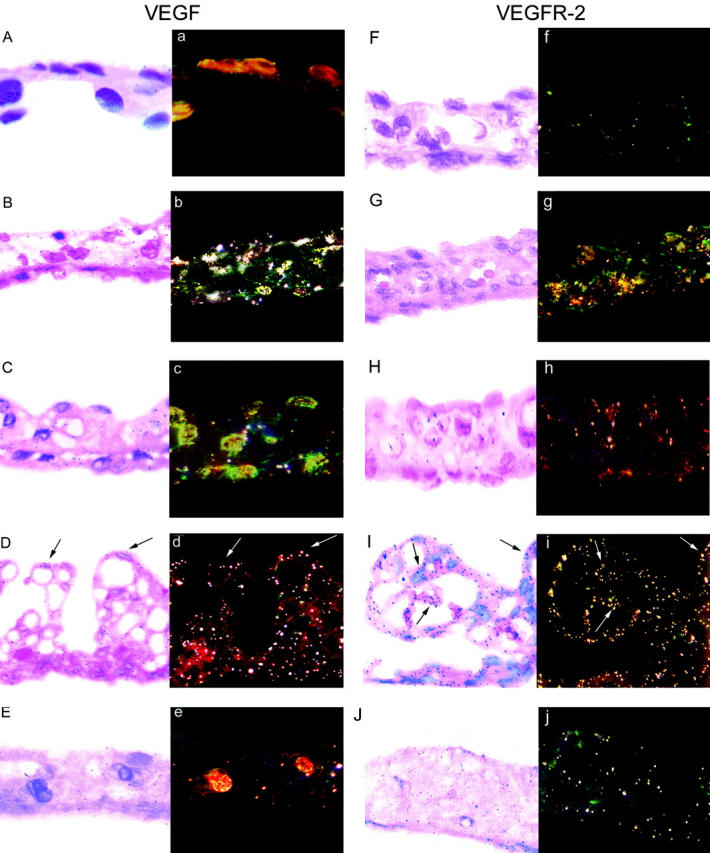
Paired bright- and dark-field photomicrographs showing VEGF and VEGFR-2 gene expression in 3-μm paraffin sections of iris from nondiabetic and diabetic SDs, SHRs, and transgenic (mRen-2)27 rats. VEGF: A and a: Diabetic SD. B and b: Diabetic SHR. C and c: Nondiabetic Ren-2. D and d: Diabetic Ren-2 showing hybridization signal on blood vessels extending from the anterior iris surface. E and e: Diabetic Ren-2 treated with LIS. VEGFR-2: F and f: Diabetic SD. G and g: Diabetic SHR. H and h: Nondiabetic Ren-2. I and i: Diabetic Ren-2 showing hybridization signal on blood vessels extending from the anterior iris surface. J and j: Diabetic Ren-2 treated with LIS. Counterstain, H&E. Original magnifications, ×380.
Discussion
The present study reports a rodent model of intraocular endothelial cell proliferation in long-term diabetic transgenic Ren-2 rats. Iris neovascularization was confirmed by the presence of proliferating endothelial cells and discrete new vessels on the iris surface. Proliferating endothelial cells in the retina of diabetic Ren-2 rats were also observed. The pathogenesis of the iris and retinal lesions in the diabetic Ren-2 rat could not be explained by systemic hypertension because endothelial cell proliferation did not occur in the diabetic SHR despite similar levels of blood pressure to the Ren-2 rat. Instead, an interaction between the local RAS and VEGF is indicated as evidenced by increased site-specific VEGF and VEGFR-2 gene expression and a rise in ocular active renin. This is consistent with previous findings in which glucose has been shown to sensitize cells to the actions of Ang II and VEGF. 26 Importantly, in both retina and iris, the ACE inhibitor LIS reduced endothelial cell proliferation and VEGF and VEGFR-2 expression, a finding that has implications for the development of new therapies for the treatment of PDR and rubeosis iridis.
Investigation into the pathogenetic mechanisms involved in the progression of diabetic ocular microvascular complications has been hindered by the lack of a diabetic rodent model that develops PDR and rubeosis iridis. Although retinal neovascularization is observed in large animals such as the galactosemic dog, these experiments are not always practical because of the duration of study (excess of 5 years) and financial and ethical considerations. 27,28 In rodents, diabetes whether induced by chemical, dietary, or genetic means only results in early retinal pathology such as pericyte loss, capillary dilatation and leakage, and basement membrane thickening, even after 1 to 2 years of diabetes. 29,30 With regard to rubeosis iridis, this is rarely observed in diabetic animals, 31 although it does occur after a combination of vitrectomy, lensectomy, and retinal detachment, or exposure of the eye to VEGF. 32-34 It could be argued that the endothelial cell proliferation observed in the diabetic Ren-2 rat does not represent distinct neovascularization but rather endothelial cell repair in response to hyperglycemia. This may be true for the retina because new blood vessels in the inner retina were not observed, and retinal endothelial cell proliferation was minimal. However, irides of diabetic Ren-2 rats displayed not only increased endothelial cell proliferation, but the appearance of vessels on the anterior surface and lens, events that typify rubeosis iridis and precede neovascular glaucoma in diabetic patients.
The increased endothelial cell proliferation in the iris compared to the retina of the diabetic Ren-2 rat is unlike the sequence of events that occurs in diabetic patients in which rubeosis iridis occurs after PDR. It is generally viewed that iris neovascularization develops after the diffusion of an angiogenic agent, probably VEGF, from hypoxic areas of retinal tissue through the vitreous to the iris. 35,36 Our finding that both VEGF and VEGFR-2 gene expression were increased in the iris of diabetic Ren-2 rats, and higher than retinae from these animals, is consistent with marked local up-regulation of this angiogenic cytokine inducing blood vessel growth. Previous studies support this concept, with evidence of local iris VEGF synthesis and findings in monkeys and diabetic humans that the severity of iris neovascularization is proportional to the amount of ocular VEGF. 12,34
The appearance of ocular complications in humans is often associated with hypertension, albuminuria, and nephropathy. 3 In animal models of diabetes, hypertension may accelerate the development of renal and retinal disease but not lead to advanced nephropathy or intraocular neovascularization. 22,37 These findings are consistent with the present study in which the SHR exhibited hypertension comparable to the Ren-2 rat, but did not display ocular endothelial cell proliferation even after 36 weeks of diabetes. The difference between the Ren-2 rat and other rodents is that in this model overexpression of the RAS occurs in extra-renal tissues including the eye. 17,25 We have previously shown that the STZ diabetic Ren-2 rat develops a decline in renal function concomitant with advanced glomerulosclerosis and tubulointerstitial disease that is associated with up-regulation of the site-specific RAS. 18,19 In the present study, ocular levels of renin were elevated in the Ren-2 rat compared to SD rats and diabetes was associated with an increase in active renin. Previously we reported a similar occurrence in Ren-2 rats with retinopathy of prematurity, a condition characterized by extensive retinal neovascularization. 25 There is considerable evidence that overactivity of the RAS accompanies diabetic neovascular disorders, with increased plasma prorenin, the inactive precursor of renin, acting as a marker for PDR. 7,38 An angiogenic role for angiotensin in the eye is provided by in vitro studies of bovine retinal endothelial cells, 13 and reports that the application of angiotensin to the rabbit cornea induces an angiogenic response. 6 It is likely that the local rather than systemic RAS is involved in ocular neovascularization because all components of the RAS are synthesized in the retina and iris of a number of species including humans. 7,8 In addition, renin and VEGF have been localized to the end feet of macroglial Müller cells that are intimately associated with retinal blood vessels, hence positioning these factors at the site of retinal angiogenesis. 9,39,40
Blockade of the RAS with either ACE inhibitors or angiotensin type 1 (AT1) receptor antagonists has consistently been shown to attenuate cell damage. 18,41,42 This is particularly true for diabetic nephropathy, in which RAS blockade improves histopathology and albuminuria in both experimental models and humans. 19,43 Fewer studies have investigated the effect of RAS blockade on the progression of diabetic eye disease. Recently, we and other investigators reported that both ACE inhibitors and AT1 receptor antagonists reduced retinal neovascularization in rodent models of retinopathy of prematurity. 25,44 In the present study, LIS reduced endothelial cell proliferation in both retina and iris of diabetic Ren-2 rats. These results are consistent with previous reports that ACE inhibition reduces retinal vascular permeability and delays the progression of PDR in humans. 10,45 Overall, these findings indicate that blockade of the RAS may be an important anti-angiogenic strategy for the attenuation of PDR and rubeosis iridis.
Our observation that in the retina and iris of the diabetic Ren-2 rat, increased VEGF and VEGFR-2 mRNA are reduced by ACE inhibition is consistent with a previous report in the retina of the diabetic SD rat. 24 Most important is that in the present study, the rise in VEGF and VEGFR-2 mRNA is accompanied by endothelial cell proliferation and that both events are reduced with LIS. These findings highlight an interaction between the VEGF system and the RAS in the progression of neovascularization, 15 an association that has been reported in vascular smooth muscle, cardiac endothelial cells, renal mesangium, and retinopathy of prematurity. 25,46-48 Indeed, the increased VEGF expression that accompanies angiogenesis in these situations can be reduced with RAS blockade. 25,46-48 There is also evidence that other factors are involved because studies using cultured retinal cells have reported that Ang II potentiates VEGF-induced angiogenesis that is mediated by angiopoietin2-Tie2 and AT1 receptor protein kinase C and mitogen-activated protein kinase pathways. 13,49 Interpretation of our findings must involve a consideration of hypertension and its in vivo counterpart mechanical cell stretch, both of which have been reported to increase VEGF expression in retina. 50 Therefore, it is possible that the anti-proliferative effects of RAS blockade may be because of a reduction in systemic blood pressure rather than a specific effect on the RAS. 10,25,44 This possibility is also suggested by the United Kingdom prospective diabetes study in which β-blockers and ACE inhibitors were equally efficacious in reducing diabetic microvascular disease. 4 However, the present study showing a disparity in endothelial proliferation in diabetic SHR and Ren-2 rats, despite equivalent blood pressure, explains the likely local role of the RAS in the retina and iris.
In conclusion, the diabetic transgenic Ren-2 rat is a model of intraocular endothelial cell proliferation whose pathogenesis is linked to local actions of VEGF- and RAS-dependent pathways. It is anticipated that this model will provide an important research tool for understanding the cellular mechanisms involved in the development of diabetic ocular microvascular complications and a means of testing new noninvasive anti-angiogenic therapies. In particular, the study provides a rationale for further clinical studies to explore the potential retinoprotective effect of interrupting the RAS.
Footnotes
Address reprint requests to Dr. Jennifer Wilkinson-Berka, Department of Physiology, the University of Melbourne, Parkville, Victoria, Australia, 3010. E-mail: j.berka@physiology.unimelb.edu.au.
Supported by the National Health and Medical Research Council of Australia, Diabetes Australia, and the Juvenile Diabetes Research Foundation.
D. J. K. and R. E. G. are recipients of a Juvenile Diabetes Research Foundation Career Development Award.
References
- 1.Klein R, Klein B, Moss S: Epidemiology of proliferative diabetic retinopathy. Diabetes Care 1992, 15:1875-1891 [DOI] [PubMed] [Google Scholar]
- 2.Fryczkowski A, Hodes B, Walker J: Diabetic choroidal and iris vasculature scanning electron microscopy findings. Int Ophthalmol Clin 1989, 13:269-279 [DOI] [PubMed] [Google Scholar]
- 3.Klein B, Klein R, Moss S, Palta M: A cohort study of the relationship of diabetic retinopathy to blood pressure. Arch Ophthalmol 1995, 113:601-606 [DOI] [PubMed] [Google Scholar]
- 4.: UK Prospective Diabetes Study Group: Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38. Br Med J 1998, 317:703-713 [PMC free article] [PubMed] [Google Scholar]
- 5.Wan Nazaimoon W, Letchuman R, Noraini N, Ropilah A, Zainal M, Ismail I, Wan Mohamad W, Faridah I, Singaraveloo M, Sheriff I, Khalid B: Systolic hypertension and duration of diabetes mellitus are important determinants of retinopathy and microalbuminuria in young diabetics. Diabetes Res Clin Pract 1999, 46:213-221 [DOI] [PubMed] [Google Scholar]
- 6.Fernandez L, Twickler J, Mead A: Neovascularization produced by angiotensin II. J Lab Clin Med 1985, 105:141-145 [PubMed] [Google Scholar]
- 7.Danser AHJ, Van den Dorpel MA, Deinum J, Derkx FHM, Franken AAM, Peperkamp E, de Jong PTVM, Schalekamp MADH: Renin, prorenin and immunoreactive renin in vitreous fluid from eyes with and without diabetic retinopathy. J Clin Endocrinol Metab 1989, 68:160-167 [DOI] [PubMed] [Google Scholar]
- 8.Danser AHJ, Derkx FHM, Admiraal PJJ, Deinum J, Dejong PTVM, Schalekamp MADH: Angiotensin levels in the eye. Invest Ophthalmol Vis Sci 1994, 35:1008-1018 [PubMed] [Google Scholar]
- 9.Berka JL, Stubbs AJ, Wang DZ-M, Di Nicolantonio R, Alcorn D, Campbell DJ, Skinner SL: Renin-containing Müller cells of the retina display endocrine features. Invest Ophthalmol Vis Sci 1995, 36:1450-1458 [PubMed] [Google Scholar]
- 10.Chaturvedi M, Sjolie AK, Stephenson JM, Abrahamian H, Keipes M: Effect of lisinopril on progression of retinopathy in normotensive people with type 1 diabetes. Lancet 1998, 351:28-31 [DOI] [PubMed] [Google Scholar]
- 11.Aiello LP, Avery RL, Arrigg PG, Keyt BA, Jampel HD, Shah ST, Pasquale LR, Thieme H, Iwamoto MA, Park JE, Nguyen HV, Aiello LM, Ferrara N, King GL: Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med 1994, 331:1480-1487 [DOI] [PubMed] [Google Scholar]
- 12.Tolentino M, Miller J, Gragoudas E, Chatzistefanou K, Ferrara N, Adamis A: Vascular endothelial growth factor is sufficient to produce iris neovascularization and neovascular glaucoma in a nonhuman primate. Arch Ophthalmol 1996, 114:964-970 [DOI] [PubMed] [Google Scholar]
- 13.Otani A, Takagi H, Oh H, Suzuma K, Matsumura M, Ikeda E, Honda Y: Angiotensin II stimulated vascular endothelial growth factor expression in bovine retinal pericytes. Invest Ophthalmol Vis Sci 2000, 41:1192-1199 [PubMed] [Google Scholar]
- 14.Gilbert RE, Kelly DJ, Cox AJ, Wilkinson-Berka JL, Rumble JR, Osicka T, Panagiotopoulos S, Lee V, Hendrich EC, Jerums G, Cooper ME: Angiotensin converting enzyme inhibition ameliorates retinal overexpression of vascular endothelial growth factor and hyperpermeability in experimental diabetes. Diabetologia 2000, 43:1360-1367 [DOI] [PubMed] [Google Scholar]
- 15.Wilkinson-Berka JL, Kelly DJ, Gilbert RE: The interaction between the renin-angiotensin system and vascular endothelial growth factor in the pathogenesis of retinal neovascularization in diabetes. J Vasc Res 2001, 38:527-610 [DOI] [PubMed] [Google Scholar]
- 16.Robinson WG, Jr, McCaleb ML, Feld LG, Michaelis IV OE, Laver N, Mercandetti M, Robinson WG, Jr: Degenerated intramural pericytes (‘ghost cells’) in the retinal capillaries of diabetic rats. Curr Eye Res 1991, 10:339-350 [DOI] [PubMed] [Google Scholar]
- 17.Mullins J, Peters J, Ganten D: Fulminant hypertension in transgenic rats harbouring the mouse Ren-2 gene. Nature 1990, 344:541-544 [DOI] [PubMed] [Google Scholar]
- 18.Kelly DJ, Wilkinson-Berka JL, Allen TA, Cooper ME, Skinner SL: A new model of diabetic nephropathy with progressive renal impairment in the transgenic (mRen-2)27 rat. Kidney Int 1998, 54:343-352 [DOI] [PubMed] [Google Scholar]
- 19.Kelly DJ, Skinner SL, Gilbert RE, Cooper ME, Wilkinson-Berka JL: Differential effects of bosentan and valsartan with respect to progressive diabetic renal pathology in the transgenic (mRen-2)27 rat model. Kidney Int 2000, 57:1882-1894 [DOI] [PubMed] [Google Scholar]
- 20.Wilkinson-Berka J, Gibbs N, Cooper M, Skinner SL, Kelly D: Renoprotective and anti-hypertensive effects of combined valsartan and perindopril in progressive diabetic nephropathy in the transgenic (mRen-2)27 rat. Nephrol Transplant Dialysis 2001, 16:1343-1349 [DOI] [PubMed] [Google Scholar]
- 21.Campbell DJ, Rong P, Kladis A, Rees B, Ganten D, Skinner SL: Angiotensin and bradykinin peptides in the TGR(mRen-2)27 rat. Hypertension 1995, 25:1014-1020 [DOI] [PubMed] [Google Scholar]
- 22.Cooper ME, Rumble JR, Allen TJ, O’Brien RC, Jerums G, Doyle AE: Antihypertensive therapy in a model combining spontaneous hypertension with diabetes. Kidney Int 1992, 41:898-903 [DOI] [PubMed] [Google Scholar]
- 23.Bunag RD: Validation in awake rats of a tail-cuff method for measuring systolic pressure. J Appl Physiol 1973, 34:279-382 [DOI] [PubMed] [Google Scholar]
- 24.Gilbert R, Vranes D, Berka JL, Cox A, Kelly DJ, Stacker S, Cooper ME: Vascular endothelial growth factor and its receptors in control and diabetic rat eyes. Lab Invest 1998, 78:1017-1027 [PubMed] [Google Scholar]
- 25.Moravski CJ, Kelly DJ, Cooper ME, Gilbert RE, Bertram J, Shahinfar S, Skinner SL, Wilkinson-Berka JL: Retinal neovascularization is prevented by blockade of the renin-angiotensin system. Hypertension 2000, 36:1099-1104 [DOI] [PubMed] [Google Scholar]
- 26.Sone H, Deo BK, Kumagai AK: Enhancement of glucose transport by vascular endothelial growth factor in retinal endothelial cells. Invest Ophthalmol Vis Sci 2000, 41:1876-1884 [PubMed] [Google Scholar]
- 27.Engerman R, Finkelstein D, Aguirre G, Diddie KR, Fox RR, Frank RN, Varma SD: Ocular complications. Diabetes 1982, 31:82-88 [DOI] [PubMed] [Google Scholar]
- 28.Frank RN: The galactosemic dog. A valid model for both early and late stages of diabetic retinopathy. Arch Ophthalmol 1995, 113:275-276 [DOI] [PubMed] [Google Scholar]
- 29.Kern T, Engerman R: Comparison of retinal lesions in alloxan-diabetic rats and galactose-fed rats. Curr Eye Res 1994, 13:863-867 [DOI] [PubMed] [Google Scholar]
- 30.Huang S, Khosrof S, Koletsky R, Benetz B, Ernsberger P: Characterization of retinal vascular abnormalities in lean and obese spontaneously hypertensive rats. Clin Exp Pharmacol Physiol 1995, 22(Suppl 1):S129-S131 [DOI] [PubMed] [Google Scholar]
- 31.Caspers-Velu LE, Wadhwani KC, Rapoport SI, Kador PF: Iris vasculopathy in galactose-fed rats. Exp Eye Res 1999, 68:211-221 [DOI] [PubMed] [Google Scholar]
- 32.Stefansson E, Landers MBR, Wolbarsht ML, Klintworth GK: Neovascularization of the iris: an experimental model in cats. Invest Ophthalmol Vis Sci 1984, 25:361-364 [PubMed] [Google Scholar]
- 33.Tolentino M, Miller J, Gragoudas E, Jakobiec F, Flynn E, Chatzistefanou K, Ferrara N, Adamis AP: Intravitreous injections of vascular endothelial growth factor produce retinal ischemia and microangiopathy in an adult primate. Ophthalmology 1996, 103:1820-1828 [DOI] [PubMed] [Google Scholar]
- 34.Ozaki H, Hayashi H, Vinores SA, Moromizato Y, Campochiaro PA, Oshima K: Intravitreal sustained release of VEGF causes retinal neovascularization in rabbits and breakdown of the blood-retinal barrier in rabbits and primates. Exp Eye Res 1997, 64:505-517 [DOI] [PubMed] [Google Scholar]
- 35.Michaelson I: The mode of development of the vascular system of the retina, with some observations on its significance for certain retinal diseases. Trans Ophthalmol Soc UK 1948, 68:137-180 [Google Scholar]
- 36.Ashton N, Ward B, Serpell G: Role of oxygen in the genesis of retrolental fibroplasia. Br J Ophthalmol 1954, 38:397-432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fluckiger W, Perrin I, Rossi G: Morphometric studies on retinal microangiopathy and myocardiopathy in hypertensive rats (SHR) with induced diabetes. Virchows Arch B Cell Pathol Incl Mol Pathol 1994, 47:79-94 [DOI] [PubMed] [Google Scholar]
- 38.Schalekamp MADH: Renin-angiotensin system components and endothelial proteins as markers of diabetic microvascular disease. Clin Invest 1993, 71:S3-S6 [DOI] [PubMed] [Google Scholar]
- 39.Tout S, Chan-Ling T, Hollander H, Stone J: The role of Muller cells in the formation of the blood retinal barrier. Neuroscience 1993, 55:291-301 [DOI] [PubMed] [Google Scholar]
- 40.Eichler W, Kuhrt H, Hoffmann S, Wiedemann P, Reichenbach A: VEGF release by retinal glia depends on both oxygen and glucose supply. Neuroreport 2000, 11:3533-3537 [DOI] [PubMed] [Google Scholar]
- 41.Wolf G: Angiotensin II is involved in the progression of renal disease: importance of non-hemodynamic mechanisms. Nephrologie 1998, 19:451-456 [PubMed] [Google Scholar]
- 42.Mifsud SA, Allen TJ, Bertram JF, Hulthen UK, Kelly DJ, Cooper ME, Wilkinson-Berka JL, Gilbert RE: Podocyte foot process broadening in experimental diabetic nephropathy: amelioration with renin-angiotensin blockade. Diabetologia 2001, 44:878-882 [DOI] [PubMed] [Google Scholar]
- 43.Cooper ME: Pathogenesis, prevention, and treatment of diabetic nephropathy. Lancet 1998, 352:213-219 [DOI] [PubMed] [Google Scholar]
- 44.Lonchampt M, Pennel L, Duhault J: Hyperoxia/normoxia-driven retinal angiogenesis in mice: a role for angiotensin II. Invest Ophthalmol Vis Sci 2001, 42:429-432 [PubMed] [Google Scholar]
- 45.Jackson WE, Holmes DL, Garg SK, Harris S, Chase HP: Angiotensin-converting enzyme inhibitor therapy and diabetic retinopathy. Ann Ophthalmol 1992, 24:99-103 [PubMed] [Google Scholar]
- 46.Williams B, Baker AQ, Gallacher B, Lodwick D: Angiotensin II increases vascular permeability factor gene expression by human vascular smooth muscle cells. Hypertension 1995, 25:913-917 [DOI] [PubMed] [Google Scholar]
- 47.Chua C, Hamdy R, Chua B: Upregulation of vascular endothelial growth factor by angiotensin II in rat heart endothelial cells. Biochem Biophys Acta 1998, 1401:187-194 [DOI] [PubMed] [Google Scholar]
- 48.Pupilli C, Lasagni L, Romagnani P, Bellini F, Mannelli M, Misciglia N, Mavilia C, Vellei U, Villari D, Serio M: Angiotensin II stimulates the synthesis and secretion of vascular permeability factor/vascular endothelial growth factor in human mesangial cells. J Am Soc Nephrol 1999, 10:245-255 [DOI] [PubMed] [Google Scholar]
- 49.Otani ATH, Oh H, Koyama S, Honda Y: Angiotensin II induces expression of the Tie2 receptor ligand, angiopoietin-2, in bovine retinal endothelial cells. Diabetes 2001, 50:867-875 [DOI] [PubMed] [Google Scholar]
- 50.Suzuma I, Hata Y, Clermont A, Pokras F, Rook SL, Suzuma K, Feener EP, Aiello L: Cyclic stretch and hypertension induce retinal expression of vascular endothelial growth factor and vascular endothelial growth factor receptor-2: potential mechanisms for exacerbation of diabetic retinopathy by hypertension. Diabetes 2001, 50:444-454 [DOI] [PubMed] [Google Scholar]