

Abstract
This study assesses the individual contributions of the nonalloreactive factor, cold ischemia (CI), and alloreactivity to late functional and structural renal graft changes, and examines the effect of the association of both factors on the progression of chronic allograft nephropathy. Lewis rats acted as receptors of kidneys from either Lewis or Fischer rats. For CI, kidneys were preserved for 5 hours. The rats were divided into four groups: Syn, syngeneic graft; SynI, syngeneic graft and CI; Allo, allogeneic graft; AlloI, allogeneic graft and CI. Renal function was assessed every 4 weeks for 24 weeks. Grafts were evaluated for acute inflammatory response at 1 week and for chronic histological damage at 24 weeks. Only when CI and allogenicity were combined did immediate posttransplant mortality occur, while survivors showed accelerated renal insufficiency that induced further mortality at 12 weeks after transplant. Solely ischemic rats developed renal insufficiency. Renal structural damage in ischemic rats was clearly tubulointerstitial, while significant vasculopathy and glomerulosclerosis appeared only in the allogeneic groups. There was increased infiltration of macrophages and expression of mRNA-transforming growth factor-β1 in the ischemic groups, irrespective of the allogeneic background. The joint association of CI plus allogenicity significantly increased cellular infiltration at both early and late stages, aggravating tubulointerstitial and vascular damage considerably. In summary, CI is mainly responsible for tubulointerstitial damage, whereas allogenicity leads to vascular lesion. The association of both factors accelerates and aggravates the progression of experimental chronic allograft nephropathy.
Chronic allograft nephropathy (CAN) is a multifactorial process that leads to late allograft loss in renal transplantation and is caused by both alloreactive and nonalloreactive factors. 1 Research efforts have usually focused on improving HLA mismatches and reducing acute rejection because they lead to the development and progression of CAN. 2 However, recently, nonalloreactive factors have received increasing attention as risk determinants. 2,3 One such factor is cold ischemia (CI), 2-6 although how it contributes to this late damage remains to be determined. In addition, its synergism with the allogeneic stimulus, both inevitably associated in clinical transplantation, is unclear. 3,4,6 Few studies have attempted to evaluate the influence of CI on late events that lead to chronic renal dysfunction and graft loss. 6-9
Our group 9,10 and others 7,11,12 have reported that ischemia-reperfusion injury and allogenicity induce long-term chronic lesions in the rat that mimic those found in clinical CAN. The pathogenic mechanisms of late organ dysfunction in chronic rejection are not fully understood, although it has been established that grafted organs are progressively infiltrated by T cells and macrophages, 8,11 mainly located in the tubulointerstitium, in glomeruli, 12 and around vessels. 13 The presence of these cell populations is associated with the release of biological mediators, such as interleukin-1, platelet activating factor (PAF), tumor necrosis factor-α, transforming growth factor (TGF)-β1 and endothelin-1 and the induction of nitric oxide. 8,11,14-16 Cells, cytokines, and chemoattractants initiate a cascade that may ultimately stimulate the proliferation of arterial smooth muscle cells and fibroblasts. 13
In our previous study, chronic graft nephropathy induced by CI mainly affected the tubulointerstitial compartment. 9 However, as CAN is usually associated with graft vasculopathy and glomerulosclerosis, in addition to persistent tubulointerstitial inflammation, the low level of vascular involvement in our nonalloreactive model is puzzling. Whether it is because of the lack of allogeneic stimulus or it indicates that nonalloreactive and alloreactive factors vary in their responsibility for the distinct elementary lesions of CAN is unclear.
In seeking to answer these questions, this study was designed to identify the individual contributions of CI and alloreactivity to late functional and structural graft changes, and to examine how the association of both factors affected the progression of CAN. To assess the differences between nonalloreactive and alloreactive injury, we studied syngeneic (Lewis-to-Lewis) and allogeneic (Fischer-to-Lewis) models of rat renal transplantation. To determine the importance of ischemia-reperfusion injury in the development of CAN, kidneys were stored in the cold for 5 hours before transplant. Collectively, our data show that CI is mainly responsible for tubulointerstitial changes whereas allogenicity induces vascular damage, and that the association of both factors aggravates the progression of CAN by increasing both functional and histological damage.
Materials and Methods
Animals and Surgical Technique
Male Lewis rats (250 g body weight) acted as receptors of either inbred male Lewis or Fischer kidneys for syngeneic (Syn) and allogeneic (Allo) renal transplant. All animals (Harlan UK Limited, Bicester, United Kingdom) were maintained in accordance with the guidelines of the Committee on Care and Use of Laboratory Animals and Good Laboratory Practice. For both kidney harvesting and transplantation, anesthesia was induced and maintained by intramuscular injection of droperidol (0.5 mg/100 g body weight) and fentanyl (0.01 mg/100 g body weight). After a midline incision, the left kidney was gently exposed and a cannula was introduced into the infrarenal aorta to the ostium of the renal artery. A single dose of sodium heparin (1000 UI) was administered and the kidney was immediately washed with Euro-Collins solution (EC; 4 ml, 4°C, 20 ml/hour flow rate). The renal artery was excised with an aortic patch, the renal vein was cut proximal to its junction with the cava and the ureter next to the bladder. The kidney was then either transplanted immediately or preserved for 5 hours in EC at 4°C before transplantation.
The kidney was grafted heterotopically. For this purpose, the host left kidney was gently exposed, the ureter was cut next to the kidney pelvis and nephrectomy was then performed. The recipient major abdominal vessels were occluded with vascular clamps and the donor artery and vein were anastomosed to the receptor aorta and cava respectively with a 9-0 monofilament. After the vascular clamps were removed, allowing the graft to be reperfused, the ureter was anastomosed end-to-end with four individual 11-0 monofilament stitches. Finally, the laparotomy was closed with 3-0 silk suture and a single intramuscular dose of Cyprofloxacine (5 mg) was administered. Reanastomosis took no more than 30 minutes, and total surgical time did not exceed 60 minutes. All of the animals received a single daily dose of 5 mg/kg of cyclosporin (Sandimmun Neoral, Novartis, Spain) dissolved in olive oil and administered by oral gavage from the transplant day until the 15th day after transplantation. On day 7 after transplantation, the recipient was again anesthetized by intramuscular injection of Droperidol (0.5 mg/100 g body weight) plus Fentanyl (0.01 mg/100 g body weight), right native nephrectomy was performed and the grafted kidney was examined for hydronephrosis. The wound was closed and a single intramuscular dose of Cyprofloxacine was administered. The animals were housed in a light/dark cycle chamber and allowed free access to tap water and rat chow.
Study Design and Follow-Up
The animals were monitored for 24 weeks. Before transplantation and monthly until the end of the study (at 4, 8, 12, 16, 20, and 24 weeks after transplant), rats were weighed and placed in metabolic cages for 24-hour urine collection, and a blood sample was obtained from the tail vein. At the end of the study, the grafted kidney was excised and processed for histological and molecular studies. Sixty-eight rats were transplanted and divided into four study groups: Syn group, Lewis kidneys flushed with EC solution at 4°C and immediately transplanted into a Lewis recipient (syngeneic transplant) (n = 14); SynI group, kidneys preserved in EC for 5 hours before syngeneic transplantation (n = 11); Allo group, Fischer kidneys flushed with EC and immediately transplanted into a Lewis recipient (allogeneic transplant) (n = 15); and AlloI group, kidneys preserved in EC for 5 hours before allogeneic transplantation (n = 28). The high number of animals included in the AlloI group was a result of the high mortality caused by this experimental association. Only three rats were excluded from the study because of hydronephrosis, observed at the moment of native nephrectomy (2 of 14 in the Syn group and 1 of 11 in the SynI group).
Four additional animals were included in each group for evaluation of the early effect of CI and alloreactivity on graft histology at 7 days after transplantation. These animals conserved the right native kidney and were not subjected to renal functional studies.
Biochemistry Data
Serum creatinine (sCr, μmol/L) and urine creatinine were determined before transplantation and at 4, 8, 12, 16, 20, and 24 weeks after transplant by Jaffe’s reaction on an autoanalyzer (Beckman Instruments, Palo Alto, CA), and creatinine clearance (CrCl, μl/min/100 g body weight) was calculated by the standard formula. Proteinuria (Prot, mg/24 hours) was determined at the same intervals using a commercial kit based on the Ponceau method (Bayer Diagnósticos, Madrid, Spain).
Quantification of Renal TGF-β1 by Real-Time Polymerase Chain Reaction (PCR)
For molecular studies the kidney was immediately frozen in liquid nitrogen and stored at −80°C. Total RNA was extracted from 50 to 100 mg of tissue by the TriPure isolation reagent (Boehringer Mannheim, Mannheim, Germany), an improvement on the Chomczynski and Sacchi method. 17 Afterward, the RNA was size-fractionated on 1% agarose gel, and stained with ethidium bromide for verification of RNA integrity and loading equivalency. Before reverse transcription, total RNA (1 μg) was incubated (30 minutes at 37°C plus 15 minutes at 95°C) with 10 U of DNase (Roche Diagnostics SL, Spain) and used for first strain cDNA synthesis. Reverse transcription mix consisted of 10 μl of RNA sample, 0.05 μmol of dNTPs (Ecogen, Spain), 2 U of RNAsin ribonuclease inhibitor (Promega, Spain), 8 μl of 5× First-Strand buffer, 2 μl of 0.1 mol/L dithiothreitol, and 400 U of RT (SuperScript II RT; Gibco BRL, Life Technologies, Spain) in a total volume of 40 μl. RNA was reverse-transcribed at 42°C for 40 minutes and finally inactivated at 95°C for 5 minutes. Reverse-transcribed negative controls were performed using distilled water.
Tissue cDNA TGF-β1 was amplified and quantified by real-time PCR (ABI Prism 7700; Applied Biosystems, Spain) using the comparative CT (threshold cycle) method and the PDAR (predeveloped TaqMan assay reagents) methodology (Applied Biosystems, Spain). The comparative CT method is a valid process when the amplifying efficiencies of both amplicons are similar (User Bulletin 2, ABI Prism 7700 Sequence Detection System, Relative Quantification of Gene Expression) and all PDARs are designed to have similar amplifying efficiencies (predeveloped TaqMan assay reagents, gene expression quantification, protocol). Moreover, we validated the method for our pair of amplicons (rat TGF-β1 and rat rRNA 18S as the reference calibrator) (results not shown) to ensure that their amplifying efficiencies were similar, so the comparative CT method could be used.
For the PCR reaction, 1 μl of each cDNA sample was mixed with 2× TaqMan Universal PCR Master Mix (12.5 μl) and 20× target primers and probe (1.25 μl) in a total reaction volume of 25 μl. Amplification was performed following the universal amplification program proposed by Applied Biosystems: 10 minutes at 95°C and 40 cycles of (15 seconds at 95°C and 1 minute at 60°C). Syngeneic values were pooled and used as the reference value. Results were expressed as many fold of the unknown sample respect to the Syn reference. No PCR products were detected using reverse-transcribed negative controls as template.
Histological Studies
For histological studies, 1- to 2-mm-thick coronal slices of the kidney were fixed in 4% formaldehyde and embedded in paraffin. For light microscopy 3- to 4-μm-thick tissue sections were stained with hematoxylin and eosin (H&E), periodic acid-Schiff, and silver methenamine. A pathologist who was blind to the treatment groups reviewed all sections.
In 1-week kidneys, tubulitis, vasculitis, interstitial infiltration, and glomerulitis were assessed in sections stained with H&E and periodic acid-Schiff, and semiquantitatively graded from 0 to + 3 following the Banff 97 criteria for acute/active lesion scoring. 18
In 24-week kidneys, sections stained with H&E and periodic acid-Schiff were examined for chronic glomerular, vascular, and tubulointerstitial damage. For glomerulosclerosis quantification (%), the global sclerosed glomeruli of each kidney section were counted and divided by the total number of glomeruli. Tubular atrophy, interstitial cellular infiltrate, interstitial fibrosis, and vasculopathy were graded as follows: 0, no abnormalities; 1+, abnormalities affecting less than one third of the sample; 2+, abnormalities affecting between one and two thirds of the sample; 3+, abnormalities affecting more than two thirds of the sample. A total tubulointerstitial score (Tub-Int) was obtained by the addition of the three individual tubulointerstitial parameters.
The interstitial area (Int Area), defined as the space of the cortex comprised between tubular membranes, glomeruli, and vessels, was quantified by morphometric analysis on silver methenamine-stained slices examined by light microscopy at ×200 in at least five fields of the cortex per kidney. Briefly, color video images were recorded from a Leitz DMRB, Leica light microscope equipped with a Sony CCD-IRIS video camera. All of the reticule intersections of a 17 × 25 point-reticule lying on the structures of interest were counted. Results were expressed as the percentage of interstitium with respect to the total surface of the kidney cortex (%).
Immunohistochemistry
Representative tissue sections were immunostained by the immunoperoxidase technique (Vectastain ABC kit anti-rabbit IgG; Vector Laboratories, Burlingame, CA). As primary antibodies, we used a monoclonal mouse anti-rat ED1 antibody for monocyte/macrophage (Oxford Biomarketing, Oxford, UK), a monoclonal mouse anti-rat MHC-II (RT1B, clone OX-6; BD Pharmingen, BD Biosciences, Europe) and a rabbit polyclonal IgG for TGF-β1 (this antibody is specific for TGF-β1 and does not cross-react with TGF-β2 or TGF-β3) (Santa Cruz Biotechnology Inc., Santa Cruz, CA). Briefly, 3- to 4-μm-thick paraffin-embedded sections were cleared and rehydrated. After washing with phosphate-buffered saline (PBS), endogenous peroxidases were inactivated by 3% H2O2. To optimize antigen expression, samples were boiled for 25 minutes in pH 6 citrate buffer. After three 5-minute washes in PBS, samples were incubated for 2 hours in 20% goat serum to block unspecific antibody binding. Samples were incubated overnight with the 1:100 diluted anti-ED1, 1:50 anti-MHC-II, and 1:20 anti-TGF-β1 primary antibodies. Then, after washing with PBS, purified rat adsorbed anti-mouse IgG (1:200; Vector Laboratories) for ED1 and MHC-II and anti-rabbit IgG (1:200; Vectastain ABC kit, Vector Laboratories) for TGF-β1, were added and incubated for 1 hour at room temperature. Then, samples were incubated in avidin-biotin complex 1:100 for 1 hour at room temperature, revealed with diaminobenzidine, washed in water, counterstained with hematoxylin, dehydrated, and mounted in DPX (BDH, Merck). Negative controls were performed by immunostaining matched serial sections without the addition of the primary antibodies. Positive ED1- and MHC-II-stained cells infiltrating kidneys were counted and expressed as mean ± SEM of cells per field of view (ED1+ cells/FV, ×400, ≥20 counted fields/kidney). Immunostaining for TGF-β1 was used to localize this growth factor in renal structures.
Statistical Analysis
Mortality ratios between groups were compared using the chi-square test, and overall survival was analyzed using the Kaplan-Meier and log-rank methods. To compare more than two groups for proteinuria, serum creatinine, and creatinine clearance throughout the follow-up, the one-way analysis of variance for two-factor repeated measures was performed. To determine the origin of the differences at any time point for these parameters, the one-way analysis of variance followed by Scheffé’s test was used. To compare histological data, the nonparametric Kruskall-Wallis test and subsequent Connover’s test were used. All P values were two-tailed, and a P value <0.05 was considered statistically significant. Data are presented as mean ± SEM.
Results
Animal Survival
Early posttransplant mortality because of renal failure was observed only when CI and allogenicity were combined (AlloI group), occurring between day 2 and 6 after nephrectomy (13 of the 28 rats, 46%) (Figure 1) ▶ . To prevent mortality we delayed nephrectomy to day 15 or 21 in additional animals in the AlloI group, but without success (data not included). Moreover, at the time of nephrectomy, macroscopic examination revealed a much smaller graft with a rough surface, while in the rest of the groups grafts were unchanged. Survivors from the AlloI group showed accelerated renal insufficiency and 8 of 15 (53%) were killed at 12 weeks after transplant. One of 10 rats (10%) in the SynI group and 1 of 15 (7%) in the Allo group died of late renal insufficiency in weeks 16 and 20, respectively. All of the animals in the Syn group survived the study period (Figure 1) ▶ .
Figure 1.

Animal survival. In AlloI group (▪), 13 of 28 rats died from early renal insufficiency between days 2 and 6 after nephrectomy, and 8 of the 15 survivors had to be killed during week 12 because of renal insufficiency. One of 10 rats in the SynI group (•) and 1 of 15 in the Allo group (□) died from late renal insufficiency during weeks 16 and 20, respectively. The 12 rats in the Syn group (○) survived the study period. a: P < 0.05 AlloI versus all; Kaplan-Meier, log rank.
Renal Function
Serum creatinine (μmol/L) and proteinuria (mg/24 hours) levels before transplantation were similar in all groups (results not shown). No change in serum creatinine levels were noted in Syn group rats throughout the 24-week follow-up (Figure 2) ▶ , while a slightly higher serum creatinine level was recorded in the Allo group rats. This difference was maintained throughout the entire follow-up. Syngeneic ischemic animals displayed a similar increase in sCr level to that shown by the Allo group until week 16 (Figure 2) ▶ . However, after week 20 they developed renal insufficiency, presenting higher creatinine levels than those of the Syn group. Renal failure was so severe in some SynI animals that their health deteriorated and one animal died at week 16. Serum creatinine levels in the allogeneic ischemic group were high throughout the entire follow-up, and markedly higher than the levels recorded in the other groups (Figure 2) ▶ . The animals in this group developed such severe renal failure that almost half of them died in the early period after transplant. The weight of the survivors fell progressively, as they showed evident symptoms of illness half of them had to be killed at week 12 after transplantation. Creatinine clearance (CrCl) showed an inverse profile to serum creatinine in all four groups (Table 1) ▶ .
Figure 2.

Serum creatinine (μmol/L). Rats from Syn group (○) showed no changes in serum creatinine levels throughout the follow-up. Creatinine levels in the Allo group (□) paralleled the profile of the Syn group but with slightly higher values. In contrast, both ischemic groups developed renal insufficiency, which appeared during week 12 in the SynI group (•) while in the AlloI group (▪) this was evident from month 1 until the end of the experiment. The gray line (-x-) corresponds to AlloI animals that had to be killed during the 12 weeks. a: P < 0.05 AlloI versus all; b: P < 0.05 SynI versus Syn; c: P < 0.05 AlloI versus Syn and Allo; analysis of variance, Scheffé’s test.
Table 1.
Body Weight, Urinary Volume, and Creatinine Clearance
| Syn | SynI | Allo | AlloI | P | ||
|---|---|---|---|---|---|---|
| BW (g) | 4 weeks | 301 ± 8 | 276 ± 12 | 290 ± 8 | 280 ± 10* | ns |
| 12 weeks | 391 ± 9 | 348 ± 11* | 382 ± 11† | 348 ± 21* | 0.024 | |
| 24 weeks | 441 ± 9 | 373 ± 23* | 444 ± 15† | 389 ± 33 | 0.018 | |
| CrCl (μmol/min/100 g) | 4 weeks | 344 ± 23 | 256 ± 39* | 235 ± 8* | 123 ± 22*†‡ | 0.0001 |
| 12 weeks | 323 ± 28 | 224 ± 30* | 224 ± 17* | 138 ± 29*†‡ | 0.0012 | |
| 24 weeks | 223 ± 10 | 156 ± 26* | 190 ± 17* | 111 ± 36*‡ | 0.0067 | |
| UrVol (ml) | 4 weeks | 21 ± 2 | 29 ± 3* | 31 ± 3* | 32 ± 4* | 0.014 |
| 12 weeks | 14 ± 1 | 21 ± 2* | 21 ± 2* | 31 ± 3*†‡ | 0.0001 | |
| 24 weeks | 10 ± 1 | 18 ± 2* | 20 ± 2* | 24 ± 2*† | 0.0001 |
Body weight (BW) and urinary volume (UrVol) were measured every 4 weeks. Creatinine clearance (ClCr) was calculated according to the standard formula. Animals from the ischemic groups (SynI, AlloI) recorded a lower gain in BW than the nonischemic animals (Syn, Allo). Urine volume in SynI, Allo, and AlloI groups was significantly higher than in Syn group. Creatinine clearance was lowered by both initial cold ischemia (SynI) and allogenicity (Allo). When the two factors were combined, the decline in creatinine clearance was further aggravated (AlloI).
*, P < 0.05 versus Syn;
†, P < 0.05 versus SynI;
‡, P < 0.05 versus Allo; analysis of variance, Scheffe’s test.
Proteinuria levels in the syngeneic nonischemic group remained unchanged throughout the follow-up (Figure 3) ▶ . Animals from the syngeneic ischemic group developed progressive proteinuria and by the end of the study these levels were higher than those in the Syn group rats. Importantly, the increase in proteinuria in syngeneic ischemic animals was similar to that observed in the allogeneic group. Both allogeneic groups displayed progressive proteinuria, although the increase was greater than that in the syngeneic group. As with the syngeneic animals, ischemia also increased proteinuria at the end of the follow-up in the allogeneic group. Those animals killed at week 12 because of renal insufficiency presented with a severe increase in proteinuria (Figure 3) ▶ .
Figure 3.

Proteinuria (mg/24 hours). The Syn group (○) had no proteinuria. Initial CI (SynI group, •) and allogenicity, with or without ischemia (Allo, □; AlloI, ▪), induced proteinuria that increased progressively. Those animals sacrificed during week 12 because of renal insufficiency (AlloI, -x-) presented a severe increase in proteinuria. a: P < 0.05 Allo versus Syn; b: P < 0.05 Syn versus SynI and AlloI; analysis of variance, Scheffé’s test.
Early Effect of Cold Ischemia and Alloreactivity on Inflammatory Response
At 1 week after transplantation, allogeneic ischemic kidneys appeared moderately enlarged and edematous, in contrast to the other groups. At light microscopy, three of the AlloI kidneys (75%) displayed a striking histological picture with diffuse inflammatory cell infiltration that was moderate in the interstitium and tubuli, and moderate-to-severe in the arterial vessels. Both Allo and SynI kidneys presented mild-to-moderate tubulitis and mononuclear cell interstitial infiltration, but no vascular affectation was found (Figure 4 ▶ and Table 2 ▶ ).
Figure 4.
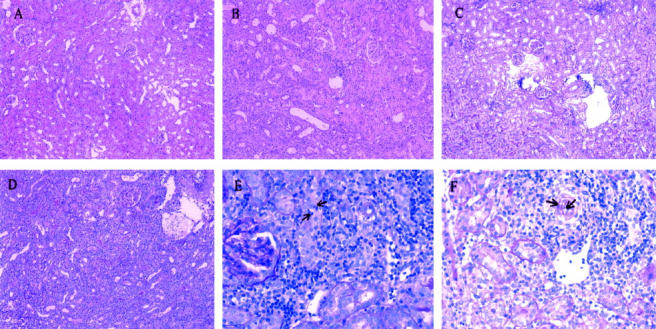
One-week histology. A: A representative kidney slice from the syngeneic nonischemic group showing a quite well-conserved histological architecture. B: A syngeneic ischemic kidney with mild-to-moderate cellular infiltration. C: An allogeneic nonischemic kidney also showing moderate cellular infiltration. D: General view of an allogeneic ischemic kidney showing severe and diffuse cellular infiltration. E: Tubulitis was clearly apparent in allogeneic ischemic kidneys (arrows). F: Detail of an arteriole infiltrated by inflammatory cells (arrows). PAS stain; original magnifications: ×100 (A–D); ×400 (E, F).
Table 2.
Kidney Graft Acute/Active Lesion Scoring at 1 Week
| Tubulitis | Interstitial infiltrate | Vasculitis | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 0 | 1 | 2 | 3 | 0 | 1 | 2 | 3 | |
| Syn | 4 | 4 | 4 | |||||||||
| SynI | 2 | 2 | 2 | 2 | 4 | |||||||
| Allo | 1 | 3 | 2 | 2 | 4 | |||||||
| AlloI | 1 | 2 | 1 | 1 | 1 | 2 | 2 | 1 | 1 | |||
On H&E and PAS specimens from 1-week kidney grafts, tubulitis, vasculitis, interstitial inflammation, and glomerulitis were semiquantitatively scored according to the Banff 97 working classification of renal allografts. There was no glomerulitis in any of the cases. Different degree of tubulitis and interstitial infiltration was seen in all groups except for syngeneic kidneys. Arterirtis was only found in the allogeneic ischemic grafts.
We measured monocyte-macrophage-positive cells, as part of the mononuclear cell infiltration responsible for the inflammatory-immune response, and MHC-II-positive cells to assess the activation of immune mechanisms. Both ED1+ and OX-6+ cells were significantly higher in the AlloI group than in the other groups, whereas they were similarly attracted and activated in both the Allo and SynI groups (Table 3) ▶ .
Table 3.
Interstitial Area, Macrophages, MHC-II Cells, and TGF-β1 mRNA Quantification
| Syn | SynI | Allo | AlloI | P | |
|---|---|---|---|---|---|
| One week | |||||
| ED1+ (cells/FW) | 4.68 ± 2.27 | 31.41 ± 7.74 | 33.37 ± 6.71 | 71.07 ± 18.28*†‡ | 0.0148 |
| OX-6+ (cells/FW) | 0.87 ± 0.14 | 6.98 ± 1.01 | 19.85 ± 6.05 | 48.77 ± 11.38*†‡ | 0.0048 |
| Twenty-four weeks | |||||
| Interstitial area (%) | 12.15 ± 0.82 | 24.45 ± 3.15* | 24.31 ± 2.87* | 36.59 ± 6.91*†‡ | 0.0050 |
| ED1+ (cells/FW) | 2.73 ± 0.98 | 27.06 ± 5.06* | 13.08 ± 2.01† | 29.37 ± 7.35*‡ | 0.0011 |
| TGF-β1 (fold/Syn) | 1.00 ± 0.16 | 6.74 ± 1.16* | 5.04 ± 1.2* | 11.31 ± 1.13*†‡ | 0.0003 |
For interstitial area, silver methenamine-stained slices were evaluated with light microscopy and quantified by the morphometric point-counting method at ×200 in at least five fields per kidney and expressed as a percentage (%). For immunohistochemical evaluation of infiltrating macrophages and MHC-II activation, ED1+ and OX-6 cells were counted at ×400 in at least 20 fields per kidney and expressed as the mean number of cells per field of view. The TGF-β1 mRNA expression was quantified by RT-real-time PCR. Syngeneic values (Syn group) were pooled and used as the reference value. All results were expressed as many fold of the unknown sample respect to the Syn reference.
In 1-week grafts, Syn kidneys appeared free of cells; moderate infiltration was seen in Allo and SynI grafts, and severe cell infiltration was found in the AlloI group. In 24-week grafts, the number of infiltrating macrophages in the ischemic groups (SynI and AlloI) was clearly greater in their corresponding nonischemic peer groups (Syn and Allo). The expansion of the interstitial area was similar when initial cold ischemia (SynI) or allogenicity (Allo) acted separately. In the AlloI group the interstitial area occupied a substantial percentage of the total cortex. Both SynI and Allo groups showed significantly more TGF-β1 expression than the Syn group. The highest TGF-β1 expression was found when cold ischemia and allogenicity were combined in the AlloI group.
*, P < 0.05 versus Syn;
†, P < 0.05 versus SynI;
‡, P < 0.05 versus Allo; analysis of variance, Scheffe’s test.
Histopathological Long-Term Consequences of Cold Ischemia and Alloreactivity
At 24 weeks after transplantation, light microscopy revealed few lesions in syngeneic nonischemic kidneys (Table 4) ▶ with only a slight interstitial cell infiltrate and no vascular or glomerular lesion (Figure 5A) ▶ . In syngeneic ischemic kidneys, however, while vascular and glomerular lesions were barely appreciable (Figure 5B) ▶ , severe tubular atrophy, interstitial fibrosis, and mononuclear cell infiltrates were observed. In allogeneic nonischemic kidneys (Figure 5C) ▶ , vascular and glomerular lesions were more evident than they were in either of the syngeneic groups, with a significantly higher glomerulosclerosis rate. However, tubular atrophy and interstitial cell infiltrate were lower than in the syngeneic ischemic group. In allogeneic ischemic kidneys (Figure 5D) ▶ histological lesions were severe and vascular, glomerular, and all tubulointerstitial parameters were significantly higher than those in the other groups. Renal architecture in allogeneic ischemic kidneys was difficult to recognize, being characterized by completely sclerosed glomeruli and a scarcity of tubuli. Furthermore, most arterioles presented severe wall thickening, with a virtually obliterated arteriolar lumen. Conserved vessels were found in both syngeneic groups (Figure 6) ▶ , while the arterioles in both allogeneic groups presented a significant increase in the media with a concomitant narrowing of the luminal space. Media hyperplasia and thickening were responsible for most of the arteriolar narrowing rather than myointimal proliferation or the combination of both lesions.
Table 4.
Histopathological Long-Term Consequences of Cold Ischemia and Alloreactivity
| Syn | SynI | Allo | AlloI | P | |
|---|---|---|---|---|---|
| Tubular atrophy | 0.0 ± 0.0 | 1.6 ± 0.3* | 1.2 ± 0.2* | 2.4 ± 0.2*†‡ | 0.0001 |
| Interstitial fibrosis | 0.0 ± 0.0 | 1.3 ± 0.1* | 0.4 ± 0.2† | 2.3 ± 0.2*†‡ | 0.0001 |
| Cell infiltrate | 0.8 ± 0.2 | 1.9 ± 0.2* | 0.9 ± 0.1† | 3.0 ± 0.0*†‡ | 0.0001 |
| Tubulointerstitial score | 0.8 ± 0.2 | 4.8 ± 0.6* | 2.6 ± 0.4*† | 7.7 ± 0.4*†‡ | 0.0001 |
| Vasculopathy | 0.0 ± 0.0 | 0.1 ± 0.1 | 0.4 ± 0.2*† | 2.3 ± 0.4*†‡ | 0.0001 |
| Glomerulosclerosis (%) | 0.0 ± 0.0 | 3.5 ± 1.5 | 17.2 ± 6.8*† | 28.6 ± 6.7*†‡ | 0.0002 |
Tubular atrophy, interstitial fibrosis, and interstitial cellular infiltrate were semi-quantitatively graded from 0 to +3. A total score was obtained by adding the three single tubulointerstitial parameters. Vasculopathy was semi-quantitatively graded in a similar form. For glomerulosclerosis the global sclerosed glomeruli of each kidney section were counted and divided by the total number of glomeruli and expressed as a percentage. Syn animals presented minimal structural changes. SynI animals had minor vascular and glomerular changes while tubulointerstitial damage was notably pronounced. Both allogeneic groups presented vasculopathy and glomerulosclerosis, but these conditions were particularly manifest in the ischemic group (AlloI). The AlloI group also revealed a significantly greater tubulointerstitial damage than was apparent in the allogeneic nonischemic group (Allo).
*, P < 0.05 versus Syn;
†, P < 0.05 versus SynI;
‡, P < 0.05 versus Allo; Kruskall-Wallis, Connover’s test.
Figure 5.
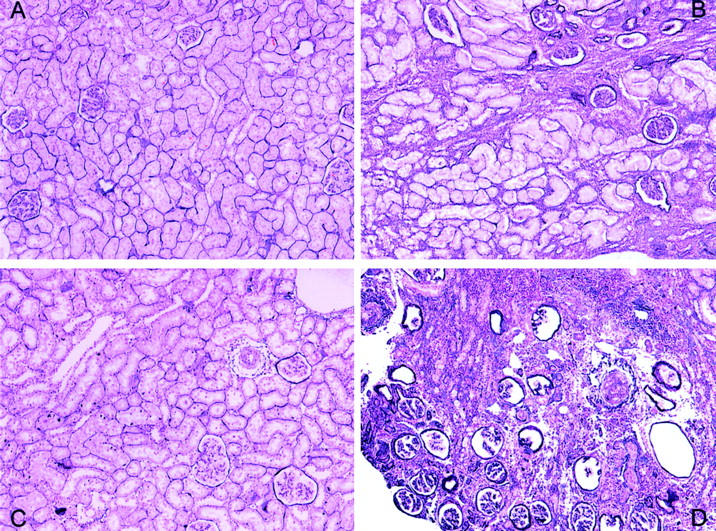
Twenty-four week histology. A: Syngeneic nonischemic group showing minimal histological affectation. B: A syngeneic ischemic kidney with marked interstitial fibrosis and tubular atrophy. C: An allogeneic nonischemic kidney showing little interstitial damage but with glomerulosclerosis and some vasculopathy. D: Histological lesions in this allogeneic ischemic kidney are clearly apparent with loss of renal architecture. PAS stain; original magnifications, ×100.
Figure 6.
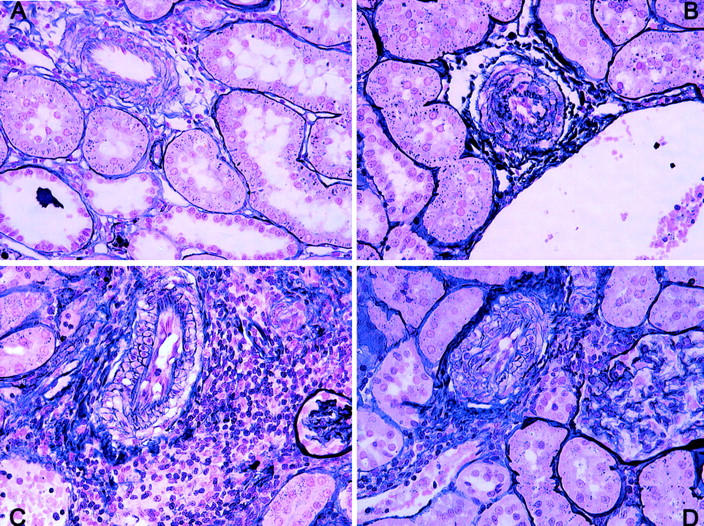
Vascular damage at 24 weeks. A: Normal conserved vessels were found in both syngeneic groups with or without ischemia. In the allogeneic groups, especially in the allogeneic ischemic kidneys, there was a clear reduction in the luminal area because of vessel wall hyperplasia (B), intimal proliferation (C), or a combination of both types (D). Periodic acid methenamine; original magnifications, ×400.
The percentage of interstitial area in syngeneic kidneys was low (Table 3) ▶ . Ischemia doubled the interstitial area in syngeneic animals, and interestingly, allogenicity increased this percentage to a similar degree. Finally, when both factors were combined in the AlloI group, the interstitial area expanded to occupy more than one third of the cortex.
Very few ED1+ cells were observed in the interstitium of syngeneic kidneys (Table 3) ▶ . In contrast, in allogeneic nonischemic kidneys, ED1+ cells were found in greater numbers. Interestingly, independent of allogenicity, ischemia significantly increased the number of infiltrating ED1+ cells in the interstitium of both the SynI and AlloI groups. Globally, the cellularity seen at 24 weeks was less evident than at 1 week when the acute process is more exudative.
TGF-β1 Assessment
Syngeneic nonischemic kidneys showed the lowest expression of TGF-β1 mRNA (Table 3) ▶ , while the association of allogenicity and ischemia clearly increased TGF-β1 expression. Interestingly, allogenicity and ischemia separately induced a similar increase in TGF-β1 expression, although this increase was not as high as when both factors were combined.
In all groups, the diffuse presence of TGF-β1 protein was identified in the epithelial tubular cells, but it was most apparent in the SynI, Allo, and AlloI groups (Figure 7) ▶ . The kidneys from both allogeneic groups also showed intense TGF-β1 immunostaining inside their glomeruli and a more moderate staining in the media of arteriolar vessels. No vascular TGF-β1 staining was found in either of the syngeneic groups, although it was occasionally observed in the glomeruli of SynI kidneys.
Figure 7.
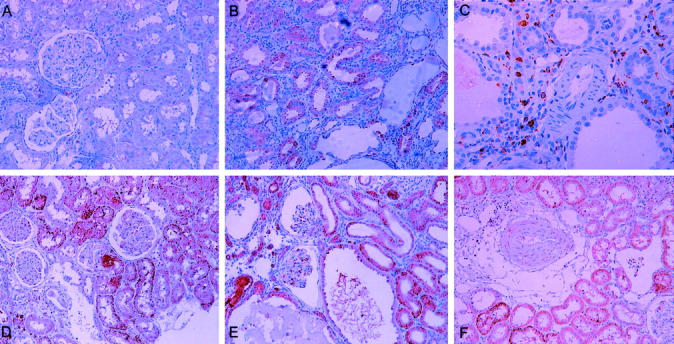
TGF-β1 immunostaining. A: TGF-β1 slightly stained in some tubular epithelial cells. B: In this syngeneic ischemic kidney, TGF-β1 was predominantly found in tubular epithelial cells. C: An artery from a syngeneic ischemic kidney negative for TGF-β1 staining. D: In this allogeneic kidney, apart from tubular epithelial cells, glomeruli were slightly stained for TGF-β1. E: Glomeruli and tubuli from an allogeneic ischemic kidney clearly positive for TGF-β1. F: A medium artery from an allogeneic ischemic kidney clearly stained for TGF-β1. Original magnifications: ×200 (A, B, D–F); ×400 (C).
Discussion
Chronic allograft nephropathy is caused by several injuries, some accumulative, to the transplanted kidney. 2 These injuries trigger inflammation and progressively deplete the tissue’s finite self-repair ability. In this study we have examined the nonalloreactive factor, CI, and alloreactivity, both separately and in association, to ascertain how they contribute to the onset and progression of CAN, and whether when combined their effects are more deleterious. Such experiments should, at least in part, show whether CAN constitutes a single pathological entity or rather whether it combines various functional and histopathological features that are each dependent on a range of causal factors.
The allogeneic Fischer-to-Lewis combination is a reproducible and worldwide accepted model of experimental chronic allograft rejection. 12,14,19-21 In a syngeneic Lewis-to-Lewis model we have shown that 5 hours of CI induced progressive chronic nephropathy in the long-term follow-up. 9 Our results, in the present study, highlighted several differences according to which factor was evaluated: ischemia, allogenicity, or the two factors in combination. Allogenicity induced progressive proteinuria associated with stable renal function, as previously described with this model. 12 In contrast, and as a consequence of CI, deterioration in renal function at late stages was concurrent with the appearance of moderate proteinuria. Finally, severe renal failure in the early and late periods after transplant was observed in the combination group, resulting in high mortality. Furthermore, those rats that reached the end of the study presented a deteriorated renal function, with severe renal failure and high levels of proteinuria. Unlike studies of the allogeneic factor, 12,14,22 studies evaluating CI in renal transplant models 23-25 have only occasionally assessed its long-term consequences. 21,26 Moreover, as in these studies serum creatinine levels are rarely considered we can only compare our results with those reported in Kouwenhoven and colleagues, 26 in which serum creatinine levels were reported as being consistently stable up to week 24. We cannot rule out the possibility that this late renal dysfunction might be related to the concurrence of cyclosporin and ischemia because cyclosporin blood levels were found within 200 and 600 μg/L (at days 7 and 15, unpublished data) in our previous study using the same model.
It has been claimed that different etiological factors trigger similar mechanisms leading to CAN, 13,14 with infiltrating macrophages, T cells, and natural killer playing a crucial role as effector cells. Our results show that, individually, CI and alloreactivity induce a similar degree of inflammatory response early in the period after transplant. Thus, during reperfusion in syngeneic ischemic animals we found a moderate macrophage infiltration and some tubulitis. These inflammatory cells derive from postischemic oxidative damage 27 and participate in regeneration or alternatively may damage tissue. 26-28 Cell infiltration and tubulitis were also moderate in the alloreactive nonischemic model. In contrast, severe macrophage infiltration and MHC-II-positive cells were observed in ischemic allografts, indicating an intense inflammatory response. This inflammation affected not only the tubulointerstitial compartment but also arterial vessels. This vascular affectation may be explained in part by the direct ischemic injury on vessels because VCAM overexpression on endothelium occurs after prolonged CI. 24 However it is conceivable that allogeneic response, formerly controlled by cyclosporin, may have been surpassed by the aggression of CI, which added to the allogeneic background turns into a strong inflammatory insult. This severe inflammatory-allogeneic response accounts for the high incidence of graft failure and animal mortality. In fact the long-term survival in the AlloI group corresponds to the early affected grafts where only one of four kidneys appeared viable.
CI produced late considerable tubulointerstitial damage because histology in syngeneic ischemic kidneys revealed higher tubular atrophy, interstitial fibrosis, and cellular infiltrate than in either syngeneic or allogeneic nonischemic kidneys. As expected, when both factors were combined, the tubulointerstitial damage was severe, supporting the functional findings. Moreover, when the interstitial area was quantified by morphometric analysis, allogeneic ischemic kidneys presented the highest values, thus confirming the severe damage caused by the combination of the two factors. It is well known that in fibrosis the altered extracellular matrix synthesis is under the complex control of many cytokines 29 including TGF-β1. 30 Allogeneic kidneys in this study presented greater interstitial fibrosis and TGF-β1 expression when they had been previously exposed to CI. Because syngeneic ischemic kidneys presented 34% higher expression of TGF-β1 and developed more fibrosis than allogeneic nonischemic grafts, it is therefore suggested that ischemia prevailed over allogenicity in the up-regulation of TGF-β1. It has been claimed that macrophages and tubular epithelial cells are a source of fibrogenic growth factors and chemokines, contributing to tubular atrophy and renal scarring. 31 As at later stages ischemic allografts continued to present more intense macrophage infiltration than the other groups, it is suggested that the inflammatory-immune status, put under considerable stress soon after transplantation, 6,25,32 continued injuring the graft thus leading to significant histological late damage. At this time both ischemic groups presented a higher number of interstitial macrophages, as well as severe interstitial fibrosis and tubular TGF-β1 expression, clearly substantiating that CI directs the long-term injury to the tubulointerstitial compartment as part of the CAN.
The hallmark of chronic rejection is arteriosclerosis, characterized by diffuse concentric intimal thickening resulting in narrowing and ultimate luminal occlusion of the arteries and arterioles of the graft. 33,34 Vasculopathy in our study was only present in the allogeneic groups and, more significantly, it was most clearly apparent when ischemia was added. Our syngeneic grafts, either with or without ischemia, did not develop vasculopathy. A number of authors 26 have described variable degrees of arteriopathy in kidney isografts. In the heart, which constitutes the specific scenario to explore transplant vasculopathy, it has been proved that syngeneic grafts were free of arteriopathy 34 and that CI in the absence of alloimmune insult did not induce obliterative vascular disease. 4,35 Most of the arterioles of the allogeneic ischemic kidneys were nearly obliterated by severe thickening of the wall, which was primarily because of the proliferation of smooth muscle cells and, less frequently, to hyperplasia of the intima. It has been described 36,37 that in the initial stages of vasculopathy there is smooth muscle cell proliferation with a posterior migration of myofibroblasts to the intima. Thus, the specific vascular changes that we saw may reflect either particular manifestations of the present model or, alternatively, that at 24 weeks vessel lesions were still at an early stage of development. The severe renal failure in allogeneic ischemic animals is probably associated with hypertension and thus part of the hypertrophia and hyperplasia of the media may also be explained by the mechanical stress of hypertension. Unfortunately, we did not measure arterial pressure and thus we cannot rule out the possibility that those highly injured animals might have developed hypertension and secondary proliferation of smooth muscle cells. Other authors have also shown that vascular changes in heart occurred in ischemic allografts at a faster rate than in nonischemic grafts. 4,38 Our findings confirm that ischemic insult acts in synergy with the antigenic stimulus in promoting vasculopathy in kidney. Thus, although alloreactivity triggers vasculopathy, CI added to the alloimmune background further exacerbates and aggravates vascular damage because it induces arteritis as early as at 1 week.
It has been stated that chronic graft deterioration represents not a unique disease but a nonspecific state induced by different etiological factors, driven by common biological mechanisms with a sole parenchyma response. 39 Our results confirm this assertion as the factors studied here induced common inflammatory mechanisms but the variation in functional and histological features suggest a different final parenchyma response. Renal stresses may act as weak or strong injuries depending on their impact on graft outcome 39,40 and vary in their effect on the compartments of the organ, 40 leading thereby to potentially different scenarios. Thus, alloreactivity in our model acts as a weak injury in contrast to CI, which by inducing severe and persistent inflammation acts as a major injury both directing the lesion to the tubulointerstitial compartment and highly aggravating overall damage. Therefore, although immune activation plays the most critical role in the etiology of CAN, ischemia clearly activates and accelerates the cellular mechanisms involved 41 providing an immunostimulatory signal that lowers the threshold for activation of the immune response. 40
Finally, in diagnostic renal biopsies, various histopathological features are usually identified, which are eventually grouped as CAN according to the Banff classification. This classification was designed primarily to group and grade the constellation of entities that represent chronic graft damage, but it is somewhat limited in defining potential etiological factors. In the light of our results, CAN should not be considered a single pathological entity and we suggest that further subclassifications be included to provide a better description of the different causal injuries seeking, at least, to identify those with stronger effects on late parenchyma changes.
In summary, the experimental renal transplant model allows allogenicity and CI to be studied independently, providing a clear description of the late consequences of the occurrence of either of these factors. From our results, it can be concluded that CI is mostly responsible for tubulointerstitial damage, whereas allogenicity induces vascular and glomerular damage. Although immune activation plays the most critical role in the etiology of CAN, ischemia clearly activates and accelerates the cellular mechanisms involved.
Footnotes
Address reprint requests to Josep M. Grinyo, Nephrology Service, Hospital of Bellvitge, Ciutat Sanitària i Universitària de Bellvitge, Feixa Llarga s/n, E-08907 L’Hospitalet de Llobregat, Barcelona, Spain. E-mail: jmgrinyo@medicina.ub.es.
Supported by grants from Fondo Europeo de Desorrollo Regional (FEDER) Plan Nacional I+D (2FD 1997-2109-C02-01 to M. R.), the Fundació Marató TV3 (00/4030); the Instituto de Salud Carlos III (ISCIII) (01/3071 to I. H. F.); and Fondo de Investigación Sanitaria (FIS) (00/0017-01 to N. L.), respectively.
References
- 1.Paul LC: Chronic renal transplant loss. Kidney Int 1995, 47:1491-1499 [DOI] [PubMed] [Google Scholar]
- 2.Paul LC: Chronic allograft nephropathy: an update. Kidney Int 1999, 56:783-793 [DOI] [PubMed] [Google Scholar]
- 3.Tilney NL, Guttmann RD: Effects of initial ischemia/reperfusion injury on the transplanted kidney. Transplantation 1997, 64:945-947 [DOI] [PubMed] [Google Scholar]
- 4.Knight RJ, Dikman S, Liu H, Martinelli GP: Cold ischemic injury accelerates the progression to chronic rejection in a rat cardiac allograft model. Transplantation 1997, 64:1102-1107 [DOI] [PubMed] [Google Scholar]
- 5.Yilmaz S, Paavonen T, Häyry P: Chronic rejection of rat renal allografts. II. The impact of prolonged ischemia time on transplant histology. Transplantation 1992, 53:823-827 [DOI] [PubMed] [Google Scholar]
- 6.Lu CY, Penfield JG, Kielar ML, Vazquez MA, Jeyarajah DR: Hypothesis: is renal allograft rejection initiated by the response to injury sustained during the transplant process? Kidney Int 1999, 55:2157-2168 [DOI] [PubMed] [Google Scholar]
- 7.Tullius SG, Heemann U, Hancock WW, Azuma H, Tilney NL: Long-term kidney isografts develop functional and morphologic changes that mimic those of chronic allograft rejection. Ann Surg 1994, 4:425-435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chandraker A, Takada M, Nadeau KC, Peach R, Tilney NL, Sayegh MH: CD28-B7 blockade in organ dysfunction secondary to cold ischemia/reperfusion injury. Kidney Int 1997, 52:1678-1684 [DOI] [PubMed] [Google Scholar]
- 9.Herrero-Fresneda I, Torras J, Lloberas N, Riera M, Cruzado JM, Condom E, Merlos M, Alsina J, Grinyó JM: Cold ischemia in the absence of alloreactivity induces chronic transplant nephropathy through a process mediated by the platelet-activating factor. Transplantation 2000, 70:1624-1631 [DOI] [PubMed] [Google Scholar]
- 10.Torras J, Cruzado JM, Riera M, Condom E, Duque N, Herrero-Fresneda J, Merlos M, Espinoza L, Lloberas N, Egido J, Grinyo JM: Long-term protective effect of UR-12670 after warm renal ischemia in uninephrectomized rats. Kidney Int 1999, 56:1798-1808 [DOI] [PubMed] [Google Scholar]
- 11.Azuma H, Nadeau K, Takada M, Mackenzie HS, Tilney NL: Cellular and molecular predictors of chronic renal dysfunction after initial ischemia-reperfusion injury of a single kidney. Transplantation 1997, 64:190-197 [DOI] [PubMed] [Google Scholar]
- 12.Diamond JR, Tilney NL, Frye J, Ding G, McElroy J, Pesek-Diamond I, Yang H: Progressive albuminuria and glomerulosclerosis in a rat model of chronic renal allograft rejection. Transplantation 1992, 54:710-716 [DOI] [PubMed] [Google Scholar]
- 13.Azuma H, Nadeau KC, Ishibashi M, Tilney NL: Prevention of functional, structural and molecular changes of chronic rejection of rat renal allografts by a specific macrophage inhibitor. Transplantation 1995, 60:1577-1582 [DOI] [PubMed] [Google Scholar]
- 14.Hancock WH, Whitley WD, Tullius SG, Heemann UW, Wasowska B, Baldwin WM, III, Tilney NL: Cytokines, adhesion molecules, and the pathogenesis of chronic rejection of rat renal allografts. Transplantation 1993, 56:643-650 [DOI] [PubMed] [Google Scholar]
- 15.Herrero I, Torras J, Riera M, Condom E, Coll O, Cruzado JM, Hueso M, Bover J, Lloberas N, Alsina J, Grinyo JM: Prevention of cold ischemia-reperfusion injury by an endothelin receptor antagonist in experimental renal transplantation. Nephrol Dial Transplant 1999, 14:872-880 [DOI] [PubMed] [Google Scholar]
- 16.Herrero I, Torras J, Bover J, Espinosa LI, Cruzado JM, Riera M, Hueso M, Lloberas N, Alsina J, Grinyó JM: Effect of ETA/ETB receptor antagonist administration on iNOS gene expression in a rat renal transplantation model. Transplant Proc 1999, 31:2344-2345 [DOI] [PubMed] [Google Scholar]
- 17.Chomczynski P, Sacchi N: Single step method of RNA isolation by acid guanidium thiocyanate-phenol-chloroform extraction. Anal Biochem 1987, 162:156-159 [DOI] [PubMed] [Google Scholar]
- 18.Racusen LC, Solez K, Colvin RB, Bonsib SM, Castro MC, Cavallo T, Croker BP, Demetris J, Drachenberg CB, Fogo AB, Furness P, Gaber LW, Gibson IW, Glotz D, Goldberg JC, Grande J, Halloran PF, Hansen HE, Hartley B, Hayry PJ, Hill CM, Hoffman EO, Hunsicker LG, Lindblad AS, Marcussen N, Mihatsch MJ, Nadasdy T, Nickerson P, Olsen TS, Papadimitriou JC, Randhawa PS, Rayner DC, Roberts I, Rose S, Rush D, Salinas-Madrigal L, Salomon DR, Sund S, Taskinen E, Trpkov K, Yamaguchi Y: The Banff 97 working classification of renal allograft pathology. Kidney Int 1999, 55:713-723 [DOI] [PubMed] [Google Scholar]
- 19.Benediktsson H, Chea R, Davidoff A, Paul LC: Antihypertensive drug treatment in chronic renal allograft rejection in the rat. Effect on structure and function. Transplantation 1996, 62:1634-1642 [DOI] [PubMed] [Google Scholar]
- 20.Amuchastegui SC, Azzollini N, Mister M, Pezzotta A, Perico N, Remuzzi G: Chronic allograft nephropathy in the rat is improved by angiotensin II receptor blockade but not by calcium channel antagonism. J Am Soc Nephrol 1998, 9:1948-1955 [DOI] [PubMed] [Google Scholar]
- 21.Tullius SG, Reutzel-Selke A, Egermann F, Nieminen-Kelha M, Jonas S, Bechstein WO, Volk HD, Neuhaus P: Contribution of prolonged ischemia and donor age to chronic renal allograft dysfunction. J Am Soc Nephrol 2000, 11:1317-1324 [DOI] [PubMed] [Google Scholar]
- 22.Nadeau KC, Azuma H, Tilney NL: Sequential cytokine dynamics in chronic rejection of rat renal allografts: roles for cytokines RANTES and MCP-1. Proc Natl Acad Sci USA 1995, 92:8729-8733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dragun D, Hoff U, Park JK, Qun Y, Schneider W, Luft FC, Haller H: Ischemia-repercussion injury in renal transplantation is independent of the immunologic background. Kidney Int 2000, 58:2166-2177 [DOI] [PubMed] [Google Scholar]
- 24.Dragun D, Hoff U, Park JK, Qun Y, Schneider W, Luft FC, Haller H: Prolonged cold preservation augments vascular injury independent of renal transplant immunogenicity and function. Kidney Int 2001, 60:1173-1181 [DOI] [PubMed] [Google Scholar]
- 25.Kouwenhoven EA, Bruin RWF, Bajema IM, Marquet RL, Ijzermans JNM: Cold ischemia augments allogeneic-mediated injury in rat kidney allografts. Kidney Int 2001, 59:1142-1148 [DOI] [PubMed] [Google Scholar]
- 26.Kouwenhoven EA, De Bruin RWF, Heemann UW, Marquet RL, Ijzermans JNM: Late graft dysfunction after prolonged cold ischemia of the donor kidney. Inhibition by cyclosporin. Transplantation 1999, 68:1004-1010 [DOI] [PubMed] [Google Scholar]
- 27.Lloberas N, Torras J, Herrero-Fresneda I, Cruzado JM, Hurtado I, Grinyó JM: Ischemic renal oxidative stress induces inflammatory response through PAF and oxidized phospholipids. Prevention by an antioxidant treatment. FASEB 2002, 16:908-910 [DOI] [PubMed] [Google Scholar]
- 28.Ghielli M, Verstrepen WA, Nouwen EJ, De Broe ME: Inflammatory cells in renal regeneration. Ren Fail 1996, 18:355-375 [DOI] [PubMed] [Google Scholar]
- 29.Eddy AA: Molecular insights into renal interstitial fibrosis. J Am Soc Nephrol 1996, 7:2495-2508 [DOI] [PubMed] [Google Scholar]
- 30.Stahl PJ, Felsen D: Transforming growth factor-β, basement membrane, and epithelial-mesenchymal transdifferentiation. Implications for fibrosis in kidney disease. Am J Pathol 2001, 159:1187-1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zeisber M, Bonner G, Maeshima Y, Colorado P, Müller GA, Strutz F, Kalluri R: Collagen composition and assembly regulates epithelial-mesenchymal transdifferentiation. Am J Pathol 2001, 159:1313-1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takada M, Nadeau KC, Shaw GD, Tilney NL: Prevention of late renal changes after initial ischemia/reperfusion injury by blocking early selectin binding. Transplantation 1997, 64:1520-1525 [DOI] [PubMed] [Google Scholar]
- 33.Hayry P, Isoniemi H, Yilmaz S, Mennander A, Lemström K, Räisänen-Sokolowski A, Koskinen P, Ustinov J, Lautenschlager I, Taskinen E, Krogerus L, Aho P, Paavonen T: Chronic allograft rejection. Immunol Rev 1993, 134:33-81 [DOI] [PubMed] [Google Scholar]
- 34.Russell PS, Chase CM, Sykes M, Ito H, Shaffer J, Colvin RB: Tolerance, mixed chimerism, and chronic transplant arteriopathy. J Immunol 2001, 167:5731-5740 [DOI] [PubMed] [Google Scholar]
- 35.Masetti P, DiSesa VJ, Schoen FJ, Sun SC, Byrne JG, Appleyard RF, Laurence R, Cohn LH: Ischemic injury before heart transplantation does not cause coronary arteriopathy in experimental isografts. J Heart Transplant 1991, 10:597-599 [PubMed] [Google Scholar]
- 36.Sasaguri S, Eishi Y, Tsukada T, Sunamori M, Suzuki A, Numano F, Hatakeyama S, Hosoda Y: Role of smooth-muscle cells and macrophages in cardiac allograft arteriosclerosis in rabbits. J Heart Transplant 1990, 9:18-24 [PubMed] [Google Scholar]
- 37.Shirwan H: Chronic allograft rejection. Do the TH2 cells preferentially induced by indirect alloantigen recognition play a dominant role? Transplantation 1999, 68:715-726 [DOI] [PubMed] [Google Scholar]
- 38.Schmid CS, Heemann U, Tilney NL: Factors contributing to the development of chronic rejection in heterotopic rat heart transplantation. Transplantation 1997, 64:222-228 [DOI] [PubMed] [Google Scholar]
- 39.Halloran PF, Melk A, Barth C: Rethinking chronic allograft nephropathy: the concept of accelerated senescence. J Am Soc Nephrol 1999, 10:167-181 [DOI] [PubMed] [Google Scholar]
- 40.Kirk AD: Location, location, location: regional immune mechanisms critically influence rejection. Nat Med 2002, 8:553-554 [DOI] [PubMed] [Google Scholar]
- 41.Halloran PF, Homik J, Goes N, Lui SL, Urmson J, Ramassar V, Cockfield SM: The “injury response”: a concept linking nonspecific injury, acute rejection, and long-term transplant outcomes. Transplant Proc 1997, 29:78-81 [DOI] [PubMed] [Google Scholar]