

Abstract
Aβ is the major component of amyloid plaques characterizing Alzheimer’s disease (AD). Aβ accumulation can be affected by numerous factors including increased rates of production and/or impaired clearance. Insulin-degrading enzyme (IDE) has been implicated as a candidate enzyme responsible for the degradation and clearance of Aβ in the brain. We have previously shown that AD patients exhibit abnormalities in insulin metabolism that are associated with apoliprotein E (APOE) status. The possible association of IDE with AD, as well as the link between APOE status and insulin metabolism, led us to examine the expression of IDE in AD. We report that hippocampal IDE protein is reduced by approximately 50% in ε4+ AD patients compared to ε4− patients and controls. The allele-specific decrease of IDE in ε4+ AD patients is not associated with neuronal loss since neuron-specific enolase levels were comparable between the AD groups, regardless of APOE status. Hippocampal IDE mRNA levels were also reduced in AD patients with the ε4 allele compared to AD and normal subjects without the ε4 allele. These findings show that reduced IDE expression is associated with a significant risk factor for AD and suggest that IDE may interact with APOE status to affect Aβ metabolism.
The florid deposition of the β amyloid protein (Aβ) into senile plaques is a hallmark feature of Alzheimer’s disease (AD). Aβ, which is derived from the γ-secretase cleavage of the β-amyloid precursor protein (APP), is a product of normal cellular activity. 1,2 However, chronic accumulation due to enhanced production and/or decreased clearance gradually leads to its deposition into plaques. While the relatively rare familial early-onset AD is associated with increased Aβ generation, defective Aβ clearance may be involved in late-onset AD (LOAD), the form of the disease that constitutes approximately 90% of all cases. 3 Insulin-degrading enzyme (IDE), which catabolizes Aβ, has been implicated as a candidate enzyme responsible for the degradation and clearance of Aβ in the brain. 4-8 IDE has also recently been shown to degrade the APP intracellular domain (AICD), 9 a product of γ-secretase cleaved APP that may function in nuclear signaling. 10-14 IDE is a metalloprotease with a molecular weight of 110 kd that is expressed in many tissue types, with high concentrations noted in brain. 15 In addition to Aβ and AICD, IDE degrades a variety of peptides, including insulin, atrial natriuretic peptide, and amylin. While IDE substrates share little sequence homology, many can form amyloid fibrils, suggesting IDE may have specificity for amyloid-forming peptides. 3,16,17 Interestingly, the gene encoding IDE is located within a region of chromosome 10 where recent reports have identified a locus associated with LOAD. 18-20
IDE has a preferential affinity for insulin such that the presence of insulin will inhibit IDE-mediated degradation of other substrates, including Aβ. 7,9,21 The ability of insulin to inhibit IDE degradation of Aβ has particular relevance for recent work suggesting that patients with late-onset AD (LOAD) have elevated insulin levels (hyperinsulinemia) and abnormalities of insulin metabolism that are associated with apoliprotein E (APOE) status. 22,23 Previous work has demonstrated that as a group, LOAD patients without the ε4 allele, the isoform known to be a risk factor for LOAD, have post-glucose challenge hyperinsulinemia in conjunction with reduced insulin action (insulin resistance). For these patients, insulin abnormalities may represent a novel mechanism contributing to the development of AD. In contrast, LOAD patients with the ε4 allele have normal peripheral levels of insulin. Given the possible association of IDE with LOAD as well as the link between APOE status and insulin metabolism, we hypothesized that IDE expression may be altered in LOAD. To investigate this possibility we measured IDE protein and mRNA expression in hippocampal tissue from 26 patients with LOAD and 15 normal adults and examined the effect of the APOE-ε4 allele.
Materials and Methods
Human Tissue from AD Subjects and Controls
Tissue was obtained from the University of Washington Alzheimer’s Disease Research Center Neuropathology Core and from the Alzheimer’s Disease Brain Bank of Vanderbilt University. LOAD cases were diagnosed clinically with dementia and fulfilled National Institute on Aging-Reagan criteria for AD at autopsy. Control subjects had no clinical history of dementia and no significant neuropathological changes indicative of AD or other clinically relevant disease. AD and non-AD cases were Braak-staged using previously published criteria. 24 Braak staging provides a multilevel quantification of the presence of neurofibrillary tangles, senile plaques, and neuronal loss, and is a widely accepted measure of the extent of AD neuropathology. In addition, neuron-specific enolase (NSE) levels were measured in AD subjects to provide an index of neuronal loss. LOAD and normal subjects were divided into four groups: LOAD with or without the ε4 allele (ε4+, n = 19; ε4−, n = 7), and normal adults with or without the ε4 allele (ε4+, n = 6; ε4−, n = 9). These groups were comparable with respect to gender distribution (LOAD ε4+, 9 females, 10 males; ε4−, 6 females, 1 male; normal ε4+, 3 females, 3 males; ε4− 6 females, 3 males). Post-mortem interval was shorter for the LOAD patients than for the normal group (LOAD time, mean hours = 6.2, SE = 1.0; normal mean = 9.6, SE = 1.2, P = 0.04), but this effect was due to a single LOAD outlier (P = 0.11 without this subject). No difference in post-mortem interval was observed between LOAD patients with or without the ε4 allele (P = 0.96). On average, LOAD patients were older at death than were normal subjects (LOAD age, mean = 78.6, SE = 2.2; normal age, mean = 70.7, SE = 2.9); however, no interaction for age at death was observed between diagnostic group and ε4 status (P = 0.53), and no differences in age at death were observed between LOAD patients with or without the ε4 allele (P = 0.37). At autopsy, fresh tissue blocks from a single hemisphere, including the medial temporal lobe, were snap-frozen in liquid nitrogen-cooled isopentane (Sigma, St. Louis, MO). Serial frozen sections (20-μm thickness) were cut on a cryostat, thaw-mounted onto Superfrost/Plus-charged slides (Fisher Scientific, Pittsburgh, PA) and stored at −70°C until use.
Western Blot Analysis
To assess IDE protein levels, 10 consecutive frozen hippocampal sections, at the level of the lateral geniculate nucleus, were used for each case. To accurately assess IDE protein levels without contamination from surrounding white matter and parahippocampal/temporal cortices, the hippocampal formation (including subiculum, hippocampus, and dentate) was dissected from each section on dry ice using a frozen single-edged razor. Hippocampal tissue lysates were prepared by sonication followed by solubilization in lysis buffer (0.1 mol/L Tris-HCl, 1% Triton X-100, 5 μg/ml aprotinin, 2 μg/ml pepstatin A, 2 μg/ml leupeptin). Total protein content was determined using a BCA protein assay kit (Pierce, Rockford, IL) and subsequently confirmed by Coomassie staining of the immunoblots at the conclusion of the experiments. Solubilized proteins were boiled in sample loading buffer (40 mmol/L Tris/HCl, pH 6.8, 2% SDS, 2% glycine, 5% 2-mercarptoethanol) and resolved by SDS-PAGE. IDE levels were determined by standard Western blot procedures using the well-characterized anti-IDE monoclonal antibody 9B12. 25 To standardize IDE levels between gels, a master standard was prepared from human frontal cortex and frozen in aliquots. Throughout the study, aliquots of this standard were processed in graded amounts (25 μg to 100 μg) in parallel with the subject samples. In this manner it was possible to control for small differences in the processing and blotting of samples over the course of the study and to obtain standard IDE band intensities on each gel that were both less and greater than the experimental sample values. IDE band intensities were quantified using a Kodak Digital Science Image Station 440 CF and Kodak Digital Science 1D Image Analysis Software. Standard values for 100 μl, 50 μl, and 25 μl concentrations were entered into a regression analysis to ensure linearity. Only samples with intensity values falling within the linear range of the assay were analyzed. Subject samples were run in duplicate or triplicate. Standard and sample values were averaged and then subjected to analysis of covariance with mean IDE sample densitometry values as the dependent variable, mean IDE standard values and age at death as covariates, and group (AD or control) and APOE status (ε4 or no ε4) as independent variables. Between-group comparisons were conducted with Bonferroni-corrected t-tests. To assess potential neuronal loss in AD patients, NSE protein levels were also measured as described above using the anti-NSE monoclonal antibody MAB324 (Chemicon International, Temecula, CA).
Immunohistochemistry
The pattern of IDE protein expression within specific hippocampal regions was assessed by immunohistochemistry. Briefly, hippocampal paraffin sections were deparaffinized in xylene, hydrated through graded alcohols and water, and then pretreated by microwaving in AR solution (Dako, Carpinteria, CA). Slides were treated for endogenous peroxidases with 3% hydrogen peroxide in phosphate-buffered saline (PBS) followed by incubation with the primary IDE antibody 9B12 for one hour at room temperature and with the secondary antibody (anti-mouse IgG) for 45 minutes. Finally, sections were incubated in an avidin-biotin complex (Vectastain Elite ABC kit, Vector, Burlingame, CA) and the reaction product was visualized with 0.05% diaminobenzidine/0.01% hydrogen peroxide in PBS. To demonstrate the specificity of 9B12, a hippocampal section was incubated with secondary antibody in the absence of primary antibody.
In Situ Hybridization
The IDE probe used in this study is a 400-bp fragment corresponding to nucleotides 62 to 461 of the human IDE cDNA subcloned into pGEM7Z vector (Promega, Madison, WI). The IDE antisense (complementary) and sense RNA probes were in vitro transcribed using the Riboprobe Gemini System (Promega) according to the manufacturer’s instructions in the presence of 12 μmol/L 35S UTP. IDE mRNA expression levels were determined by quantitative in situ hybridization histochemistry as described previously. 26 Briefly, frozen hippocampal sections were post-fixed in 4% paraformaldehyde, treated with acetic anhydride, and delipidated and dehydrated through a graded series of ethanol and chloroform. Slides were hybridized overnight under saturating conditions with the 35S-labeled IDE antisense or sense RNA probe (0.1 pmol/slide) at 60°C. Following hybridization, slides were washed in 1X SSC (150 mmol/L NaCl and 15 mmol/L Na-citrate) at room temperature, treated with RNase A (10 mmol/L Tris, pH 8.0; 1 mmol/L EDTA, pH 8.0; 20 μg/ml RNase A) and washed in 0.1X SSC at 65°C. Slides were then dehydrated through a graded series of ethanols containing ammonium acetate, air-dried and exposed to Kodak Hyperfilm (Eastman Kodak, Rochester, NY) for 3 weeks. Hybridization signal from film autoradiograms was analyzed using a microcomputer-based image analysis system (MCID, Imaging Research, St. Catherines, Ontario, Canada). Optical density measurements of IDE mRNA were obtained from the CA-2/3 and CA1 of the cornu ammonis as well as the dentate and hilus. Data were analyzed by repeated analysis of variance measures to test for differences in IDE mRNA levels across the designated hippocampal regions among normal and AD subjects with and without an APOE-ε4 allele.
Results
IDE protein levels differed for AD patients and controls according to APOE status, as indicated by a significant interaction between diagnostic group and APOE, F(1,40) = 8.52, P = 0.0061. As shown in Figure 1A ▶ , hippocampal IDE protein levels were reduced in AD patients with an APOE-ε4 allele compared to patients without the ε4 allele (P = 0.0008), to normal adults with the ε4 allele (P = 0.0004), and to normal adults without the ε4 allele (P = 0.0011). IDE protein levels did not differ for AD patients with a single ε4 allele compared to patients with two ε4 alleles (P = 0.68, data not shown). In addition, APOE-related differences in IDE protein levels in AD patients were not due to variations in disease severity as measured by Braak staging. Braak neurofibrillary tanglevalues were virtually identical for LOAD patients with and without an ε4 allele (ε4−, mean = 5.3, SE = 0.4; ε4+, mean = 5.3, SE = 0.2, P = 0.99), as were senile plaque values (ε4−, mean = 2.9, SE = 0.3; ε4+, mean = 2.9, SE = 0.13, P = 0.99). Figure 1B ▶ demonstrates that AD patients with and without the ε4 allele also had comparable levels of NSE (P = 0.89), indicating that the marked loss of IDE in the AD ε4+ subjects cannot be accounted for by neuronal loss. Finally, IDE protein expression was not related to age at death (P = 0.91) or to post-mortem interval (P = 0.69).
Figure 1.

Quantitative Western blot analysis of hippocampal IDE (A) and NSE (B) protein expression. Representative immunoblots contain 100 μg (IDE) or 4 μg (NSE) of protein per lane from three normal and/or three Alzheimer’s (AD) subjects without (ε4−) and with (ε4+) the APOε4 allele. Graphs represent quantitative differences in expression of IDE protein in normal and AD subjects (A) and of NSE protein in AD subjects (B) with and without the ε4 allele. Each bar represents the mean +/− SEM. IDE protein is reduced in AD patients with an APOE-ε4 allele compared to patients without the ε4 allele (***, P = 0.0008), to normal adults with the ε4 allele (***, P = 0.0004), and to normal adults without the ε4 allele (**, P = 0.0011). No difference in NSE levels were observed for AD groups (P = 0.89).
To examine the pattern of expression of IDE protein and mRNA within specific regions of the hippocampus, immunohistochemistry and in situ hybridization analyses were performed. Figure 2 ▶ depicts the pattern of expression of IDE protein and mRNA in specific hippocampal sub-fields of a normal subject. IDE protein is expressed at high levels in the dentate granule layer as well as in the CA-2/3 field and hilus (Figure 2, A and C) ▶ . In contrast, IDE protein levels are very low in the CA-1 field of the hippocampus (Figure 2, A and D) ▶ . IDE protein is highly expressed in neuronal cells, consistent with other reports demonstrating localization of IDE to neurons. 5,27 In situ hybridization analysis demonstrated that the pattern of IDE mRNA expression is similar to that of IDE protein (Figure 2E) ▶ .
Figure 2.

Pattern of IDE protein (A–D) and mRNA (E–F) expression in the hippocampus of a normal adult. A: Representative immunostained section of whole hippocampus (magnification, × 1) depicting high levels of IDE protein in the dentate granular layer (DGL), hilus, and CA-2/3, and low levels in the CA-1. No immunostaining is detected when the primary IDE antibody is omitted (B). Increased magnification (× 20) depicting high neuronal expression in the hilus (C) compared to the CA-1 (D). Representative film autoradiogram of in situ hybridization with the IDE antisense RNA probe (E) demonstrates that the pattern of IDE mRNA expression is similar to that of protein. No hybridization signal is observed with the IDE sense RNA probe (F).
Given the decrease in IDE protein levels in APOE-ε4+ AD patients described in Figure 1 ▶ , quantitative in situ hybridization was performed to determine whether there was a corresponding decrease in IDE mRNA expression and to determine whether specific regions of the hippocampus were differentially affected. Our results indicate that IDE mRNA levels differed for both ε4+ and ε4− normal and AD subjects according to the hippocampal region analyzed, as indicated by a significant interaction between hippocampal region and diagnostic group F(3,93) = 7.15, P = 0.0002, and hippocampal region and APOE status F(3,93) = 8.75, P = 0.0059. These results are demonstrated in Figure 3 ▶ . As observed for protein levels, IDE mRNA levels were reduced in AD patients with the APOE-ε4 allele compared to AD patients without the ε4 allele. Statistically significant decreases in IDE mRNA were observed in the dentate (P = 0.026) and hilar regions (P = 0.035), and a trend for a decrease was observed in the CA-2/3 (P = 0.082). IDE mRNA levels were also reduced in ε4+ AD patients compared to normal subjects without the ε4 allele (dentate, P = 0.0002; CA-2/3, P = 0.009; hilus, P = 0.002). Surprisingly, and in contrast to IDE protein expression, we also observed reduced IDE mRNA levels in normal subjects who were ε4+ compared to normal subjects without the ε4 allele. This down-regulation of IDE expression in ε4+ normal subjects was observed in the hilus (P = 0.025). In fact, independent of diagnostic group, decreased IDE mRNA expression was associated with the ε4 allele across hippocampal regions, F(1,31) = 7.07, P = 0.0123. IDE mRNA levels did not differ for subjects with a single ε4 allele compared to subjects with two ε4 alleles (P = 0.46, data not shown). We also observed significantly more IDE mRNA expression in the dentate compared to the other hippocampal regions analyzed in both normal and AD subjects independent of APOE status (all P values <0.0001). Finally, in contrast to the dentate, CA-2/3, and hilus, IDE mRNA levels were very low in the CA-1 and were unaffected by APOE status and diagnostic group.
Figure 3.
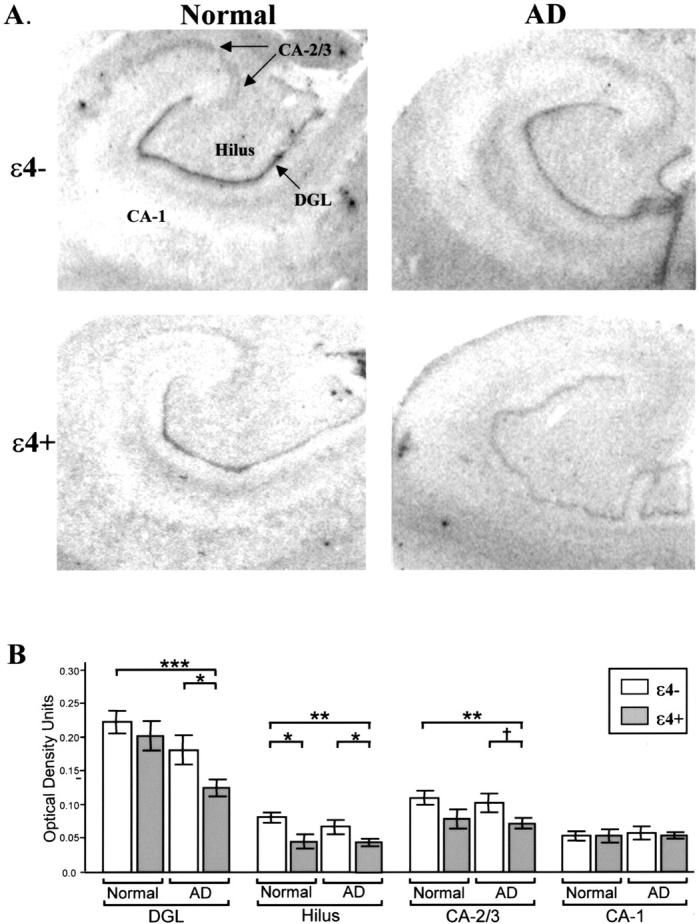
Quantitative in situ hybridization analysis of hippocampal IDE mRNA expression in normal and LOAD subjects. A: Representative film autoradiograms depicting the differences in expression of hippocampal IDE mRNA in normal and Alzheimer’s (AD) subjects with (ε4+) and without (ε4−) the APOE-ε4 allele. B: Graphic representation of the quantitative differences in IDE mRNA expression. Each bar represents the mean +/− SEM. IDE mRNA is reduced in LOAD subjects with the ε4 allele (gray bar) compared to LOAD and normal subjects without the ε4 allele (white bar) in the dentate granular layer (DGL), hilus, and CA-2/3. In addition, IDE mRNA is decreased in the hilus of normal ε4+ subjects compared to normal ε4− subjects. (*, P < 0.05, **, P < 0.01, ***, P < 0.001, †, P = 0.082).
Discussion
These findings are the first demonstration of reduced IDE levels linked to the presence of the APOE-ε4 allele in AD and suggest a novel mechanism through which APOE-ε4 may promote the development of AD. Reduced IDE protein levels presumably would diminish the capacity for degradation of Aβ in the brains of individuals with the ε4 allele, leading to chronic Aβ elevations. Because IDE degrades the APP intracellular domain as well, 9 it is possible that reduced IDE expression could also influence the putative nuclear signaling of this γ-secretase-generated APP fragment, although the consequences of such influence remain to be determined. Given that IDE levels were similar for subjects with one or two ε4 alleles, the effect of ε4 on IDE expression may not be dose-dependent. It is also unclear whether APOE-ε4 status plays a causal role in reduced IDE expression in AD or whether IDE levels are reduced through an independent process that interacts with the presence of the ε4 allele during the development of the disease. In the present study, quantitative in situ hybridization analysis demonstrated a decrease in IDE mRNA expression in the hilus in both normal and AD subjects with the ε4 allele, suggesting that reduced IDE expression may be an immutable consequence of the ε4 genotype. However, quantitative Western blot analysis of whole brain lysate indicated reduced IDE protein levels only in AD patients with the ε4 allele. This result suggests that down-regulation may be either a disease-associated process or an independently occurring risk factor. It is possible that in situ hybridization analysis examining specific regions of the hippocampus is sensitive enough to detect the difference in IDE expression between normal subjects with and without the ε4 allele while detection by Western blot analysis using the entire hippocampus may mask this difference. Alternatively, IDE protein may not be down-regulated in the hilus of normal ε4+ subjects despite the decrease in mRNA. This result would suggest that factors involved in IDE protein synthesis or degradation may be differentially regulated such that normal individuals are able to compensate for decreased IDE mRNA levels associated with the ε4 allele, whereas AD subjects are not.
Although little is known about the regulation of IDE gene expression, there are several possible mechanisms through which IDE levels and/or activity may be modulated. IDE may be affected by metabolic conditions that affect insulin action, such as type 2 diabetes mellitus and the hyperinsulinemia observed in LOAD patients without the ε4 allele. 23,28 In this case, inhibition of IDE activity rather than altered IDE expression levels is likely to be a factor, a possibility consistent with findings of disrupted insulin metabolism in this group. In theory, genetic mutations in IDE may also affect its expression or function, and linkage has been demonstrated for a late-onset familial AD locus on chromosome 10q in the region of the IDE gene. 18-20 At present, no such mutations or polymorphisms have been identified within the IDE gene and one report suggests that coding sequence variants in IDE probably do not account for this linkage. 29 However, variations in the regulatory region of the IDE gene could also play a role in susceptibility to LOAD.
Multiple mechanisms may thus affect IDE expression levels or activity and differentially determine IDE’s role in AD pathophysiology. The normal degradation of Aβ by IDE may be compromised by reduced expression of the protease due to independent genetic factors or some ε4-related mechanism that has yet to be identified. In addition, Aβ degradation could also be compromised by conditions that affect IDE’s activity, such as the chronic hyperinsulinemia observed in non-ε4 LOAD. Such compromise could occur in the context of normal or even elevated IDE levels. In addition, it has been hypothesized that type 2 diabetes may be a risk factor for dementia 30,31 and a recent report suggests that adults with both type 2 diabetes and the APOE-ε4 allele have an increased risk for developing AD compared to subjects with either risk factor alone. 32 Presumably, the combination of reduced IDE activity due to diabetes-associated hyperinsulinemia as well as reduced IDE expression levels associated with possession of the ε4 allele could synergistically enhance the risk for developing AD. In all of the above scenarios, reduced clearance of Aβ would result, setting the stage for its subsequent aggregation into senile plaques. In addition to IDE, other proteases, such as neprilysin, may also play a role in removal of Aβ from the brain. 33,34 NEP-deficient mice show a modest but significant increase in Aβ in the brain 35 and reduced levels of neprilysin have been associated with AD. 36 However, it is not known if decreased neprilysin is associated with the presence of the ε4 allele. In addition, while both IDE and neprilysin degrade extracellular Aβ, IDE appears to be more effective at degrading intracellular Aβ. 8 It has been postulated that deposition of Aβ may begin intracellularly; thus inhibition of intracellular Aβ degradation by IDE may be an important step in the pathology of AD.
Two previous studies examining IDE expression in AD brain produced ambiguous results. One study by Bernstein et al 27 reported increased IDE immunostaining in AD neurons compared with controls, whereas a second study by Perez et al 4 found decreased IDE activity and protein levels in AD brain. Both studies used small numbers of AD patients, however, which did not permit these authors to investigate the potential modulating effect of genetic factors such as APOE genotype. Furthermore, neither study examined the hippocampus using quantified immunoblotting or in situ hybridization techniques. These studies also were unable to address whether IDE abnormalities were due to generic disease-related factors such as neuronal loss. Our study demonstrating reduced IDE expression in AD using a larger number of samples supports the Perez et al study. 4 We also present the additional finding that reduced IDE expression characterizes ε4-related disease but not non-ε4-related disease. The fact that both groups have similar levels of AD-related neuropathology and NSE protein levels provides evidence that reduced IDE is not an artifact of the disease process.
In summary, evidence is accruing that IDE plays a critical role in the degradation of Aβ in the human brain. 4-7 The present study is the first documentation of reduced IDE expression in the brain of patients with LOAD, and provides important evidence that such reduction characterizes patients with the APOE-ε4 allele. As such, these results provide a novel link between Aβ metabolism and the only clearly established genetic risk factor for LOAD, inheritance of the APOE-ε4 allele.
Acknowledgments
We thank Thomas J. Montine, M.D., Ph.D, for providing human brain tissue and Aaron Krohn and Lynne Greenup for their expert technical assistance.
Footnotes
Address reprint requests to Suzanne Craft, GRECC S-182, VA Puget Sound Health Care System, 1660 South Columbian Way, Seattle, WA 98108. E-mail: scraft@u.washington.edu.
Supported by Department of Veteran’s Affairs and NIA Alzheimer’s Disease Research Center grant AG05136.
References
- 1.Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, Lieberburg I, Koo EH, Schenk D, Teplow DB, Selkoe DJ: Amyloid β-peptide is produced by cultured cells during normal metabolism. Nature 1992, 359:322-325 [DOI] [PubMed] [Google Scholar]
- 2.Shoji M, Golde TE, Ghiso J, Cheung TT, Estus S, Shaffer LM, Cai XD, McKay DM, Tintner R, Frangione B, Younkin SG: Production of the Alzheimer amyloid β protein by normal proteolytic processing. Science 1992, 258:126-129 [DOI] [PubMed] [Google Scholar]
- 3.Selkoe DJ: Clearing the brain’s amyloid cobwebs. Neuron 2001, 32:177-180 [DOI] [PubMed] [Google Scholar]
- 4.Perez A, Morelli L, Cresto JC, Castano EM: Degradation of soluble amyloid β-peptides 1–40, 1–42, and the Dutch variant 1–40Q by insulin degrading enzyme from Alzheimer disease and control brains. Neurochem Res 2000, 25:247-255 [DOI] [PubMed] [Google Scholar]
- 5.Vekrellis K, Ye Z, Qiu WQ, Walsh D, Hartley D, Chesneau V, Rosner MR, Selkoe DJ: Neurons regulate extracellular levels of amyloid β-protein via proteolysis by insulin-degrading enzyme. J Neurosci 2000, 20:1657-1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mukherjee A, Song E, Kihiko-Ehmann M, Goodman P, Jr, Pyrek JS, Estus S, Hersh LB: Insulysin hydrolyzes amyloid β peptides to products that are neither neurotoxic nor deposit on amyloid plaques. J Neurosci 2000, 20:8745-8749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qiu WQ, Walsh DM, Ye Z, Vekrellis K, Zhang J, Podlisny MB, Rosner MR, Safavi A, Hersh LB, Selkoe DJ: Insulin-degrading enzyme regulates extracellular levels of amyloid β-protein by degradation. J Biol Chem 1998, 273:32730-32738 [DOI] [PubMed] [Google Scholar]
- 8.Sudoh S, Frosch MP, Wolf BA: Differential effects of proteases involved in intracellular degradation of amyloid β-protein between detergent-soluble and -insoluble pools in CHO-695 cells. Biochemistry 2002, 41:1091-1099 [DOI] [PubMed] [Google Scholar]
- 9.Edbauer D, Willem M, Lammich S, Steiner H, Haass C: Insulin-degrading enzyme rapidly removes the β-amyloid precursor protein intracellular domain (AICD). J Biol Chem 2002, 277:13389-13393 [DOI] [PubMed] [Google Scholar]
- 10.Sastre M, Steiner H, Fuchs K, Capell A, Multhaup G, Condron MM, Teplow DB, Haass C: Presenilin-dependent γ-secretase processing of β-amyloid precursor protein at a site corresponding to the S3 cleavage of Notch. EMBO Rep 2001, 2:835-841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu C, Kim SH, Ikeuchi T, Xu H, Gasparini L, Wang R, Sisodia SS: Characterization of a presenilin-mediated amyloid precursor protein carboxyl-terminal fragment γ: evidence for distinct mechanisms involved in γ-secretase processing of the APP and Notch1 transmembrane domains. J Biol Chem 2001, 276:43756-43760 [DOI] [PubMed] [Google Scholar]
- 12.Cao X, Sudhof TC: A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science 2001, 293:115-120 [DOI] [PubMed] [Google Scholar]
- 13.McLoughlin DM, Miller CC: The intracellular cytoplasmic domain of the Alzheimer’s disease amyloid precursor protein interacts with phosphotyrosine-binding domain proteins in the yeast two-hybrid system. FEBS Lett 1996, 397:197-200 [DOI] [PubMed] [Google Scholar]
- 14.Kimberly WT, Zheng JB, Guenette SY, Selkoe DJ: The intracellular domain of the β-amyloid precursor protein is stabilized by Fe65 and translocates to the nucleus in a notch-like manner. J Biol Chem 2001, 276:40288-40292 [DOI] [PubMed] [Google Scholar]
- 15.Kuo WL, Gehm BD, Rosner MR: Regulation of insulin degradation: expression of an evolutionarily conserved insulin-degrading enzyme increases degradation via an intracellular pathway. Mol Endocrinol 1991, 5:1467-1476 [DOI] [PubMed] [Google Scholar]
- 16.Duckworth WC, Bennett RG, Hamel FG: Insulin degradation: progress and potential. Endocr Rev 1998, 19:608-624 [DOI] [PubMed] [Google Scholar]
- 17.Authier F, Posner BI, Bergeron JJ: Insulin-degrading enzyme. Clin Invest Med 1996, 19:149-160 [PubMed] [Google Scholar]
- 18.Bertram L, Blacker D, Mullin K, Keeney D, Jones J, Basu S, Yhu S, McInnis MG, Go RC, Vekrellis K, Selkoe DJ, Saunders AJ, Tanzi RE: Evidence for genetic linkage of Alzheimer’s disease to chromosome 10q. Science 2000, 290:2302-2303 [DOI] [PubMed] [Google Scholar]
- 19.Myers A, Holmans P, Marshall H, Kwon J, Meyer D, Ramic D, Shears S, Booth J, DeVrieze FW, Crook R, Hamshere M, Abraham R, Tunstall N, Rice F, Carty S, Lillystone S, Kehoe P, Rudrasingham V, Jones L, Lovestone S, Perez-Tur J, Williams J, Owen MJ, Hardy J, Goate AM: Susceptibility locus for Alzheimer’s disease on chromosome 10. Science 2000, 290:2304-2305 [DOI] [PubMed] [Google Scholar]
- 20.Ertekin-Taner N, Graff-Radford N, Younkin LH, Eckman C, Baker M, Adamson J, Ronald J, Blangero J, Hutton M, Younkin SG: Linkage of plasma Aβ42 to a quantitative locus on chromosome 10 in late-onset Alzheimer’s disease pedigrees. Science 2000, 290:2303-2304 [DOI] [PubMed] [Google Scholar]
- 21.Bennett RG, Hamel FG, Duckworth WC: Characterization of the insulin inhibition of the peptidolytic activities of the insulin-degrading enzyme-proteasome complex. Diabetes 1997, 46:197-203 [DOI] [PubMed] [Google Scholar]
- 22.Craft S, Asthana S, Schellenberg G, Cherrier M, Baker LD, Newcomer J, Plymate S, Latendresse S, Petrova A, Raskind M, Peskind E, Lofgreen C, Grimwood K: Insulin metabolism in Alzheimer’s disease differs according to apolipoprotein E genotype and gender. Neuroendocrinology 1999, 70:146-152 [DOI] [PubMed] [Google Scholar]
- 23.Craft S, Peskind E, Schwartz MW, Schellenberg GD, Raskind M, Porte D, Jr: Cerebrospinal fluid and plasma insulin levels in Alzheimer’s disease: relationship to severity of dementia and apolipoprotein E genotype. Neurology 1998, 50:164-168 [DOI] [PubMed] [Google Scholar]
- 24.Braak H, Braak E: Neuropathological staging of Alzheimer-related changes. Acta Neuropathol (Berl) 1991, 82:239-259 [DOI] [PubMed] [Google Scholar]
- 25.Shii K, Roth RA: Inhibition of insulin degradation by hepatoma cells after microinjection of monoclonal antibodies to a specific cytosolic protease. Proc Natl Acad Sci USA 1986, 83:4147-4151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McMillan PJ, Leverenz JB, Dorsa DM: Specific down-regulation of presenilin 2 gene expression is prominent during early stages of sporadic late-onset Alzheimer’s disease. Brain Res Mol Brain Res 2000, 78:138-145 [DOI] [PubMed] [Google Scholar]
- 27.Bernstein HG, Ansorge S, Riederer P, Reiser M, Frolich L, Bogerts B: Insulin-degrading enzyme in the Alzheimer’s disease brain: prominent localization in neurons and senile plaques. Neurosci Lett 1999, 263:161-164 [DOI] [PubMed] [Google Scholar]
- 28.Standl E, Kolb HJ: Insulin degrading enzyme activity and insulin binding of erythrocytes in normal subjects and type 2 (non-insulin-dependent) diabetic patients. Diabetologia 1984, 27:17-22 [DOI] [PubMed] [Google Scholar]
- 29.Abraham R, Myers A, Wavrant-DeVrieze F, Hamshere ML, Thomas HV, Marshall H, Compton D, Spurlock G, Turic D, Hoogendoorn B, Kwon JM, Petersen RC, Tangalos E, Norton J, Morris JC, Bullock R, Liolitsa D, Lovestone S, Hardy J, Goate A, O’Donovan M, Williams J, Owen MJ, Jones L: Substantial linkage disequilibrium across the insulin-degrading enzyme locus but no association with late-onset Alzheimer’s disease. Hum Genet 2001, 109:646-652 [DOI] [PubMed] [Google Scholar]
- 30.Leibson CL, Rocca WA, Hanson VA, Cha R, Kokmen E, O’Brien PC, Palumbo PJ: Risk of dementia among persons with diabetes mellitus: a population-based cohort study. Am J Epidemiol 1997, 145:301-308 [DOI] [PubMed] [Google Scholar]
- 31.Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM: Diabetes mellitus and the risk of dementia: the Rotterdam Study. Neurology 1999, 53:1937-1942 [DOI] [PubMed] [Google Scholar]
- 32.Peila R, Rodriguez BL, Launer LJ: Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: the Honolulu-Asia Aging Study. Diabetes 2002, 51:1256-1262 [DOI] [PubMed] [Google Scholar]
- 33.Iwata N, Tsubuki S, Takaki Y, Watanabe K, Sekiguchi M, Hosoki E, Kawashima-Morishima M, Lee HJ, Hama E, Sekine-Aizawa Y, Saido TC: Identification of the major Aβ-42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat Med 2000, 6:143-150 [DOI] [PubMed] [Google Scholar]
- 34.Shirotani K, Tsubuki S, Iwata N, Takaki Y, Harigaya W, Maruyama K, Kiryu-Seo S, Kiyama H, Iwata H, Tomita T, Iwatsubo T, Saido TC: Neprilysin degrades both amyloid β peptides 1–40 and 1–42 most rapidly and efficiently among thiorphan- and phosphoramidon-sensitive endopeptidases. J Biol Chem 2001, 276:21895-21901 [DOI] [PubMed] [Google Scholar]
- 35.Iwata N, Tsubuki S, Takaki Y, Shirotani K, Lu B, Gerard NP, Gerard C, Hama E, Lee HJ, Saido TC: Metabolic regulation of brain Aβ by neprilysin. Science 2001, 292:1550-1552 [DOI] [PubMed] [Google Scholar]
- 36.Yasojima K, Akiyama H, McGeer EG, McGeer PL: Reduced neprilysin in high plaque areas of Alzheimer brain: a possible relationship to deficient degradation of β-amyloid peptide. Neurosci Lett 2001, 297:97-100 [DOI] [PubMed] [Google Scholar]