

Abstract
Endothelial-monocyte-activating polypeptide II (EMAP II) is a proinflammatory cytokine and a chemoattractant for monocytes and granulocytes. We have previously shown that EMAP II mRNA is strongly expressed at sites of apoptosis in the mouse embryo and that the mature protein is cleaved from its cellular precursor (proEMAP II/p43) by caspase activation to become released from cells. Here we demonstrate in vivo that EMAP II mRNA expression is strongly increased in tumor necrosis factor α (TNF)-treated murine meth A fibrosarcomas and in B16 melanomas, especially in close proximity to areas of tissue necrosis. Furthermore, by means of confocal microscopy, high level expression of proEMAP II/p43 protein correlated predominantly with hypoxic but also with apoptotic cells. In vitro, EMAP II mRNA levels were not increased by hypoxia. However, high amounts of mature EMAP II protein were detected in the supernatants of hypoxic tumor cells. Unlike in apoptotic cells, neither a broad-range caspase inhibitor nor an inhibitor specific for the internal cleavage site was able to inhibit processing of proEMAP II/p43 to the mature EMAP II protein. In conclusion, these data suggest that hypoxia and apoptosis provide two alternative mechanisms of EMAP II generation by tumor cells.
Endothelial-monocyte-activating polypeptide II (EMAP II) was isolated from the conditioned medium of methylcholanthrene A (meth A)-transformed fibrosarcoma cells based on its ability to activate endothelial cells. 1 Injection of EMAP II into the footpad of mice leads to an inflammatory swelling in vivo, which is likely to result from the EMAP II-induced chemotactic migration of mononuclear phagocytes and polymorphonuclear leukocytes observed in vitro, suggesting that EMAP II is a proinflammatory cytokine. 1 In murine tumor models, EMAP II exerts a sensitizing effect on the vasculature of initially tumor necrosis factor α (TNF)-resistant tumors and renders them responsive to the antitumor effects of TNF. 2-5
Cloning of the EMAP II cDNA revealed that the biologically active 23-kd form of EMAP II is the cleavage product of a precursor protein (proEMAP II) which lacks a conventional secretion signal peptide. 2 ProEMAP II has been shown to be identical to the p43 subunit of the mammalian tRNA multisynthetase complex. 6 ProEMAP II/p43 is located in the center of the complex and affects its overall structure and stability. 7 Moreover, proEMAP II/p43 strongly binds to tRNA 6 and may play a role as a co-factor for aminoacylation, 8 an activation step required for the coupling of amino acids to their respective tRNAs. We have demonstrated earlier that induction of apoptosis, but not necrosis, leads to the proteolytic cleavage of proEMAP II/p43 and results in the release of the mature EMAP II protein from cells in vitro. 9 An internal ASTD amino acid sequence at position 144 of the murine protein was identified as the exact cleavage site of proEMAP II/p43 by N-terminal sequencing of the mature protein isolated from cellular supernatants. 1 Recently, we could identify caspase-7 as the proEMAP II/p43 cleaving enzyme and demonstrate that EMAP II cleaved from the tRNA multisynthetase complex displays biological activity. 10,11 In addition to apoptosis, hypoxia was also reported to be a potent inducer of EMAP II release in vitro. 12
During mouse embryogenesis, strong expression of EMAP II mRNA was observed in areas of extensive tissue remodeling and co-localization with apoptotic cells and monocytes was demonstrated. 9 In the adult organism, strong EMAP II mRNA expression is restricted to thymus, testis, and brain. 13 Increase in EMAP II mRNA expression correlates with an accumulation of apoptotic cells in thymus and testis, suggesting a role for EMAP II in the recruitment of phagocytic cells to sites of programmed cell death. However, no apoptotic cells could be detected in the adult brain and it was suggested that EMAP II might contribute to the recruitment of microglial cells and to the immunosurveillance of the brain. 13 In line with this hypothesis, regulation of EMAP II protein expression was found to be linked to inflammatory diseases of the brain. 14 Furthermore, Daemen et al 15 suggested that EMAP II may function as a link between apoptosis and inflammation, as apoptosis was directly correlated with post-translational processing of proEMAP II/p43 to EMAP II and an apoptosis-induced influx of leukocytes in the reperfused kidney.
In the present study, we demonstrate that in vivo EMAP II mRNA expression is increased at sites of necrosis in murine TNF-treated meth A fibrosarcomas and in B16 melanomas. Confocal microscopy studies revealed that increased expression of proEMAP II/p43 in perinecrotic areas of murine B16 melanomas co-localizes with areas of tissue hypoxia and apoptosis. In subsequent in vitro studies we show that hypoxia does not cause an increase in EMAP II mRNA per se but leads to the cleavage of proEMAP II/p43 and the subsequent release of mature EMAP II protein from murine B16 melanoma cells. Unlike in apoptotic cells, under hypoxic conditions this process cannot be abrogated by the addition of caspase inhibitors. We hypothesize that apoptosis and hypoxia employ two distinct mechanisms leading to the release of EMAP II protein.
Materials and Methods
Tissue culture media and supplements were obtained from Gibco (Life Technologies, Eggenstein, Germany), PAA Laboratories (Cölbe, Germany) and Cell Concepts (Umkirch, Germany); reagents, if not otherwise indicated, were obtained from Sigma (Deisenheim, Germany).
Cell Culture and Induction of Apoptosis and Hypoxia In Vitro
BFS-1 cells (methylcholantrene A-transformed murine fibrosarcoma cells) were cultivated in RPMI medium 1640 supplemented with 10% FBS, 4 mmol/L glutamine, 1 mmol/L sodium pyruvate, 100 units/ml penicillin, and 100 μg/ml streptomycin. Murine B16 melanoma cells were cultivated in Dulbecco’s modified Eagle’s medium with glutamine supplemented with 10% FBS, 4 mmol/L glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. Apoptosis was induced by treating confluent cells for 24 hours with a combination of 3 μg/ml cycloheximide and 0.5 nmol/L recombinant huTNF (Cell Concepts, Umkirch, Germany) in serum-free medium. For exposure to a hypoxic environment, subconfluent cells in serum-free medium were incubated in a hypoxic chamber (model 305; Labotect, Göttingen, Germany) containing 1% O2, 5% CO2, and 94% N2 for up to 24 hours. Where indicated, the broad-range caspase inhibitor benzyloxycarbonyl-VAD-fluoromethylketone (Z-VAD-fmk; Bachem, Heidelberg, Germany) or benzyloxycarbonyl-ASTD-fluoromethylketone (Z-ASTD-fmk; Bachem), an inhibitor mimicking the cleavage site of proEMAP II/p43, was added to the medium before incubating the cells in the hypoxic chamber.
In Vivo Tumor Growth
To inoculate tumors, 1.5 × 106 BFS-1 or B16 cells resuspended in phosphate-buffered saline (PBS) were injected subcutaneously into the back of C57B/6 mice. Tumors were allowed to grow to an average diameter of 5 to 10 mm. Mice carrying meth A fibrosarcomas were administered 5 to 10 μg recombinant huTNF intraperitoneally 24 hours before being sacrificed. Control animals received PBS only. Mice inoculated with B16 melanomas were treated with 7-[4′-(2-nitroimidazole-1-yl)-butyl]-theophylline (NITP; kind donation of the Gray Laboratory, Northwood, Middlesex, UK). In hypoxic cells, bioreduction of the nitroimidazole occurs and leads to binding of bioreductive metabolites to intracellular molecules. Because an immunorecognizable side-chain, theophylline, is covalently linked to the nitroimidazole, cells containing bound theophylline can be detected by immunohistochemical methods. 16 A suspension consisting of 70 mg NITP in 0.5 ml 10% dimethylsulfoxide (DMSO) and 4.5 ml peanut oil was prepared and mice were injected 11.25 μl/g body weight intraperitoneally 2 hours before being sacrificed. Excised murine tumors were embedded in Tissue Tek medium (Sakura, Zoeterwoude, Netherlands), snap-frozen, and stored at − 80°C until sectioning. In contrast, a number of tumors used for EMAP II mRNA quantification were directly snap-frozen.
In Situ Hybridization
In situ hybridization on paraformaldehyde-fixed sections (8 μm) was performed as previously described. 17 35S-labeled sense and antisense EMAP II probes were generated from linearized full-length cDNA of mouse EMAP II in pBluescript II SK+ (Stratagene, Amsterdam, Netherlands) using T3 or T7 RNA polymerases (Stratagene). Hybridization was performed overnight at 42°C. Washing steps were carried out in a water bath at 37°C. Slides were coated with a photographic emulsion (Kodak, NTB-2, New York, NY) and exposed for 1 to 2 weeks in the dark. Consecutive sections of one tumor either hybridized with the antisense or the sense probe were exposed for identical periods of time. After development, the slides were counterstained with toluidine blue, mounted in Entellan (Merck, Darmstadt, Germany) and photographed.
Real-Time EMAP II mRNA Quantitation
To extract RNA, 30 to 50 sections of frozen tumor tissue (10 μm each) were collected and transferred into a reaction tube containing 600 μl RNAzol™B (WAK-Chemie, Bad Homburg, Germany). After incubation for 10 minutes at room temperature, 60 μl chloroform/isoamylalcohol (24:1) were added. The samples were vortexed and centrifuged for 15 minutes at 4°C. The aqueous layer was collected, 1 μl glycogen (1 mg/ml) was added, and precipitation was carried out with an equal volume of isopropanol. Samples were incubated for 1 hour at − 20°C and subsequently centrifuged for 15 minutes. The pellets were washed with 75% ethanol, air- dried, and diluted in 10 μl H2O.
Relative mRNA quantitation was performed by real-time PCR using the Sequence Detection System 7700 (PE Applied Biosystems, Foster City, CA) as described in detail previously. 18 For relative quantitation, the target gene was normalized to an internal standard gene, 18S rRNA. Quantitation calculations were performed using the following equation:
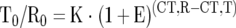 |
T0, initial number of target gene mRNA copies; R0, initial number of standard gene mRNA copies; E, efficiency of amplification; CT,T, threshold cycle of target gene; CT,R, threshold cycle of standard gene; K, constant.
For cDNA synthesis and real-time PCR, reagents were applied as recently described. 19 Two μl cDNA were used for each sample. Primers were added to a final concentration of 900 nmol/L each and hybridization probes to a final concentration of 200 nmol/L in a final volume of 50 μl. Cycling conditions were 95°C for 6 minutes, followed by 45 cycles of 95°C for 20 seconds, 59°C for 30 seconds, and 73°C for 30 seconds. The primer and probe sequences are as follows (in all cases, the first oligonucleotide is the forward PCR primer, the second one is the reverse PCR primer and the third one is the TaqMan probe): 1) EMAP II (amplicon size: 117 bp), 5′-TGTGGGAGAAGCAGCCCC-3′, 5′-CGCATCTTTGCAGGCTTCA-3′,5′-TCATGTTCCTCTAGAACAGATGCAAAATCGT-3′ 2) 18S rRNA (amplicon size: 88 bp), 5′-AAAACCAACCCGGTGAGCT-3′, 5′-CGATCGGCCCGAGGTTATCT-3′, 5′-CGCCGGCGGCTTTGGTGACT-3′. Accession numbers of the sequences taken from GenBank are NM007926 for EMAP II and K01364 for 18S rRNA.
In Situ Detection of Apoptosis
On frozen sections, DNA strand breaks were detected by the terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) technique, as described by Gavrieli et al. 20 We used a TUNEL kit from Roche Molecular Biochemicals (Hoffmann LaRoche, Basel, Switzerland). The fluorescein-labeled DNA fragments were detected by confocal microscopy.
Immunohistochemistry
Cryosections (5 μm) of B16 melanomas were double-stained for p43/proEMAP II expression, hypoxia, and apoptosis in all three possible combinations. Sections were fixed in 4% paraformaldehyde for 30 minutes and washed. To perform double-staining involving the detection of apoptotic cells the TUNEL assay was used according to the manufacturer’s protocol. Sections were consecutively incubated in Tris-buffered saline (TBS)/1% bovine serum albumin (BSA)/5% donkey serum for 30 minutes to block unspecific binding. To detect proEMAP II/p43 protein, sections were incubated with the EMAP II-specific rabbit polyclonal antiserum SA 2847 diluted 1:80 in TBS/1% BSA. For the detection of hypoxic cells, a theophylline-specific goat antiserum (Fitzgerald Industries, Concord, MA) was used in a 1:500 dilution. Sections were incubated with primary antibodies overnight at 4°C. After washing with TBS, indocarbocyanin (Cy 3)-conjugated donkey anti-rabbit (Chemicon, Hofheim, Germany) and Cy 3-conjugated donkey anti-goat IgG antibodies (Jackson Immuno Research, West Grove, PA) (each diluted 1:100 in TBS/1% BSA), were applied to the sections for 45 minutes at room temperature. After three final washing steps the sections were mounted in Mowiol (Calbiochem, Bad Soden, Germany). Analysis of the sections was performed by confocal microscopy. On sections that were double-stained for proEMAP II/p43 and theophylline, the EMAP II-specific antiserum SA 2847 was applied as source for the first primary antibodies overnight and Cy 3-conjugated donkey anti-rabbit IgG was used to detect EMAP II-specific antibodies as described above. After another blocking step, theophylline-specific goat antiserum was applied for 2 hours at room temperature. Detection of bound primary antibodies occurred by incubating the sections with fluorescein isothiocyanate (FITC)-conjugated donkey anti-goat IgG (Jackson Immuno Research) diluted 1:200 in TBS/1% BSA.
Confocal Microscopy
Sections of the tumors were examined using laser scanning confocal microscopy (LEICA TCS SP, Leitz, Germany). Series of confocal sections (0.5 μm interval) from tumor were used for consecutive three-dimensional reconstruction at a Silicon Graphics workstation “Obtaine” (SGI Silicon Graphics, Munich, Germany) using the 3-D multichannel image processing software “Imaris” (Bitplane, Zürich, Switzerland).
Western Blotting
Proteins from the interstitial fluid of murine tumors were extracted as previously described. 13 Briefly, the material was diced with a scalpel and mixed with a mild chaotropic extraction buffer. Cell lysates from in vitro experiments were obtained by mixing the cells with a buffer containing 10 mmol/L Tris/HCl (pH 7.5) + 1% SDS. After centrifugation, the supernatant was collected, the protein content was determined and the samples were subjected to SDS/PAGE and immunoblotting (see below). Cell supernatants from in vitro experiments were collected and proteins were concentrated and precipitated by addition of trichloroacetic acid. SDS/PAGE and immunoblotting were performed as described. 21 In brief, samples were mixed with Laemmli buffer, boiled at 95°C for 10 minutes, and loaded onto 15% SDS/PAGE gels. Proteins were separated by electrophoresis and blotted onto nitrocellulose (Schleicher & Schüll, Dassel, Germany) using a semi-dry blotting apparatus. Unspecific binding was reduced by blocking the membrane in TBS/0.1% Tween 20/5% nonfat dry milk. The primary antibody (rabbit anti-EMAP II antiserum SA 2847, diluted 1:1000 in TBS/0.1% Tween 20/5% BSA) was applied overnight at 4°C. After washing, the membranes were incubated in a peroxidase-coupled goat anti-rabbit IgG (Jackson Immuno Research; diluted 1:3500 in blocking buffer) for 1 hour at room temperature and developed using an enhanced chemiluminescence kit (Amersham Pharmacia Biotech, Freiburg, Germany). As a control for equal protein loading, the blot of B16 cell lysates was reprobed with mouse anti-GAPDH antibody (Biogenesis, Poole, UK). After incubation in Pierce Restore Western blot stripping buffer (Pierce, Sankt Augustin, Germany) for 30 minutes at 37°C, the membrane was extensively washed, blocking was repeated, and the GAPDH-specific antibody (diluted 1:1000 in TBS/0.1% Tween 20/5% BSA) was applied overnight at 4°C. The primary antibody was detected using peroxidase-coupled goat anti-mouse IgG (Jackson Immuno Research; diluted 1:10000 in blocking buffer).
DNA Fragmentation Analysis
After induction of apoptosis or exposure to hypoxia, 1 × 106 cells were harvested, washed in PBS, and processed as described. 9 Briefly, cells were lysed and DNA was extracted from the supernatants in two sequential extraction steps using phenol and a mixture of phenol/chloroform/isoamylalcohol, respectively. After precipitation, the DNA was redissolved, subjected to RNase digestion, and analyzed on an agarose gel.
Results
EMAP II mRNA Is Strongly Expressed in TNF-Treated Meth A Fibrosarcomas and in B16 Melanomas
Previously, we have shown high level EMAP II mRNA expression at sites of tissue remodeling in the murine embryo, where it correlates with areas of apoptosis. 9 To investigate EMAP II mRNA expression under pathological conditions, we analyzed murine meth A fibrosarcomas, in which systemic TNF application induces hemorrhagic necroses. 22 In comparison to normal skin, in which no hybridization signal for EMAP II mRNA could be detected (Figure 1A ▶ , asterisks), control tumors (injected with PBS), displayed EMAP II mRNA expression, albeit at a low level (Figure 1A ▶ , arrows). Hematoxylin and eosin (H&E) staining revealed no tissue necrosis in these tumors (Figure 1G) ▶ . However, in response to a single intraperitoneal injection of 5 μg recombinant huTNF meth A fibrosarcomas developed extensive necroses (Figure 1H ▶ , arrows). In situ hybridization revealed prominent signals for EMAP II mRNA in TNF-treated meth A fibrosarcomas in a rim of perinecrotic cells surrounding the necrotic center of the tumor (Figure 1B) ▶ , which are stronger than those in control tumors (Figure 1A) ▶ . Murine B16 melanomas were the second tumor model system chosen to investigate EMAP II mRNA expression. Beginning with a diameter of about 5 mm, B16 melanomas spontaneously formed necroses (Figure 1I ▶ , arrows) that became more pronounced with increasing size of the tumor. We detected strongest hybridization signals for EMAP II mRNA in cell clusters located in close proximity to necrotic areas (Figure 1C ▶ , arrows).
Figure 1.
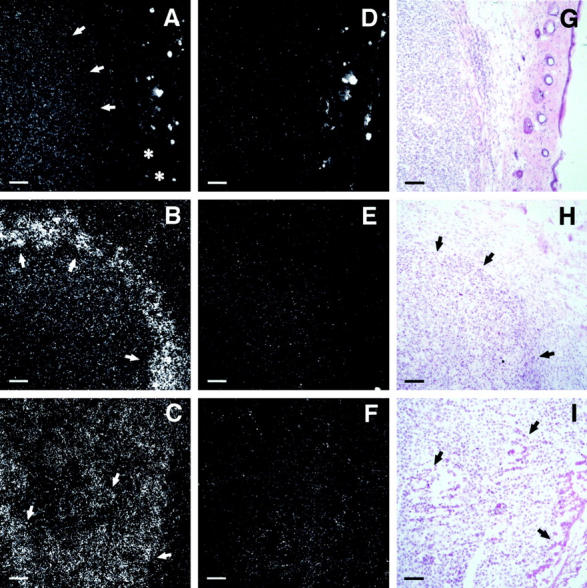
In situ hybridization analysis of EMAP II mRNA expression in PBS- versus TNF-treated meth A fibrosarcomas and in an untreated B16 melanoma. Shown are 8 μm cryosections of the tumors which were hybridized with an 35S-UTP antisense riboprobe for the detection of EMAP II mRNA (A–C). A sense probe was used as a control (D–F). Adjacent sections were stained with hematoxylin and eosin (G–I). A: Moderate EMAP II mRNA expression can be seen in the PBS-treated control meth A fibrosarcoma (arrows) in contrast to the skin tissue (asterisks) surrounding the tumor. Bright hybridization signals within the skin result from unspecific binding of the riboprobe to hair follicles and is likewise seen in Figure 1D ▶ . B: Increased expression of EMAP II mRNA can be detected in a rim of cells (arrows) surrounding the necrotic center of the TNF-treated meth A fibrosarcoma compared to control tumors (Figure 1A) ▶ . C: EMAP II mRNA expression is shown in perinecrotic areas (arrows) in the B16 melanoma. Almost no signals can be detected on tumor sections incubated with the sense riboprobe (D–F). Size markers correspond to 80 μm.
As a rapid and sensitive technique for precise EMAP II mRNA quantitation, real-time PCR of sectioned tumor material was performed. To exclude amplification of genomic DNA, suitable intron-spanning primers were selected for EMAP II. For 18S rRNA, used as the internal control gene, cDNA synthesis without reverse transcriptase added revealed no significant signals (data not shown). The primer/probe-sets were selected to work under identical PCR cycling conditions with an amplification efficiency that was approximately equal and amounted to 1.0 (100%).
Figure 2 ▶ shows that skin tissue surrounding tumors was negative for EMAP II mRNA. In contrast, a low mean expression of EMAP II mRNA was detectable in PBS-treated BFS-1 fibrosarcomas, confirming the data obtained by in situ hybridization. The relative amount of EMAP II mRNA could be significantly enhanced by treatment of the BFS-1 fibrosarcomas with TNF compared to the PBS-treated control tumors (EMAP II, P = 0.036; Mann-Whitney two-sample rank test). As no differences in expression levels of EMAP II mRNA and the extent of necroses within tumors after treatment with either 5 μg (n = 9) or 10 μg (n = 3) TNF were observed, TNF-treated tumors are depicted as one single group. The mean relative expression of EMAP II mRNA in B16 melanomas was comparable to the EMAP II mRNA expression level in TNF-treated fibrosarcomas. Taken together, our results show that EMAP II mRNA is strongly expressed in two different experimental tumors, the TNF-treated BFS-1 fibrosarcoma and the B16 melanoma, mainly in areas adjacent to necroses.
Figure 2.

Relative EMAP II mRNA expression in tumors quantitated by real-time PCR. For relative quantitation, the EMAP II gene was normalized to the internal control gene 18S rRNA as described in Materials and Methods. Skin tissue (n = 3; mean = 1) surrounding the subcutaneously implanted tumors was negative for EMAP II mRNA. In contrast, a low mean EMAP II mRNA expression was detected in control BFS-1 fibrosarcomas after PBS-treatment (n = 7; mean = 16.6). Treating mice for 24 hours with TNF (n = 12; mean = 34.3) led to a significant increase in intratumoral EMAP II mRNA levels compared to the control tumors (EMAP II, P = 0.036; Mann-Whitney two-sample rank test). The relative EMAP II mRNA expression in B16 melanomas (n = 4; mean = 38.8) was similar to the amount of EMAP II mRNA detected in the BFS-1 + TNF group. K, constant (see Materials and Methods).
Strong ProEMAP/p43 II Expression Correlates with Apoptosis and Hypoxia in B16 Melanomas
Having shown earlier that EMAP II mRNA expression co-localizes with areas of apoptosis in mouse embryos, 9 sections of B16 melanomas were double-stained with the TUNEL technique to detect apoptotic cells (Figure 3A) ▶ and with an EMAP II-specific antiserum (Figure 3B) ▶ . Strong proEMAP II/p43 protein expression was detectable as a bright red staining of the cells in perinecrotic areas of B16 melanomas. In contrast, the surrounding tumor cells stained less bright, indicating a lower expression level. An overlay of both stainings showed that cells positive for both TUNEL staining and strong proEMAP II/p43 expression accumulate in perinecrotic areas (Figure 3C) ▶ , although a detailed analysis revealed that TUNEL-positive cells were less abundant and located closer to the necrotic core than cells showing proEMAP II/p43 staining. Of note, the resulting overlay between EMAP II-specific staining and TUNEL staining cannot lead to yellow fluorescence because proEMAP II/p43 is mainly located in the cytosol as part of the tRNA multisynthetase complex whereas TUNEL staining detects fragmented DNA within the nucleus.
Figure 3.
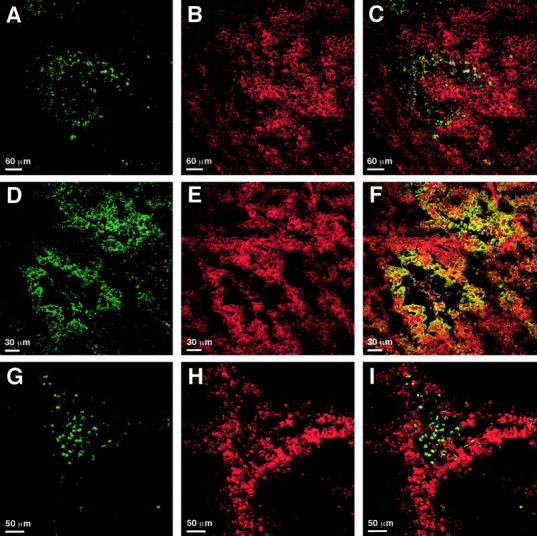
Detection of apoptosis, hypoxia, and proEMAP II/p43 expression in B16 melanomas by immunofluorescent staining and confocal laser-scanning microscopy. 5 μm sections of B16 melanomas were stained with the TUNEL technique (A and G), EMAP II- (B and E) or theophylline-specific (D and H) antisera. C, F, and I represent the merged images of the respective panels in the left and middle column. C: ProEMAP II/p43 expressing (Cy 3, red) and TUNEL-positive (FITC, green) cells partially overlap in perinecrotic areas of the tumor. High proEMAP II/p43 expression is predominantly detectable in areas that are devoid of apoptotic cells. F: Strong expression of proEMAP II/p43 (Cy 3, red) and accumulation of the hypoxia marker NITP (FITC, green) occurs in cells in perinecrotic areas of the tumor. Double-positive cells appear yellow. Most of the hypoxic cells show proEMAP II/p43 expression. I: TUNEL-positive cells (FITC, green) in perinecrotic areas are surrounded by a rim of hypoxic (Cy 3, red) cells. Of note, only double-staining of proEMAP II/p43- positive and hypoxic cells gives rise to a full (yellow) overlay due to the common cytosolic expression of these antigens in the cytosol, whereas this is not the case for overlays with TUNEL, which stains the nuclear compartment. For technical reasons, hypoxic cells are visualized in green color in panels D and F, whereas they appear red in panels H and I.
With respect to its localization close to necrotic areas of tumors, the pattern of EMAP II mRNA expression levels resembles the distribution of VEGF mRNA in human glioblastomas. 23 Since the expression of VEGF is regulated by hypoxia, 24 we investigated whether strong proEMAP II/p43 expression could be linked to hypoxia in the B16 melanomas. Sections of B16 melanomas were double-stained with an antibody directed against theophylline, an immunorecognizable group coupled to NITP, which is a marker for hypoxic cells (Figure 3D) ▶ , and with the polyclonal antiserum directed against EMAP II (Figure 3E) ▶ . Confocal microscopy analysis demonstrated tissue hypoxia in perinecrotic areas of the B16 melanomas. A large proportion of the hypoxic cells also displayed high level proEMAP II/p43 expression, which is indicated by yellow staining (Figure 3F) ▶ .
To determine the relation between hypoxia and apoptosis within the B16 melanomas, double-staining for apoptotic (Figure 3G) ▶ and hypoxic cells (Figure 3H) ▶ was performed. Subsequent analysis by confocal microscopy showed that the apoptotic cells were located close to the necrotic core, surrounded by a broad band of hypoxic cells (Figure 3I) ▶ . There was only limited overlap of both stainings, indicating that hypoxic and apoptotic cells are distinct populations.
ProEMAP II/p43 Protein Is Processed in B16 Melanomas In Vivo and EMAP II Is Released under Hypoxic Conditions in Vitro
Next, we analyzed the processing of proEMAP II/p43 protein in B16 melanomas in vivo. The polyclonal antiserum directed against EMAP II detects both the proEMAP II/p43 and the mature 23-kd form of EMAP II. 9 Since the immunohistological analysis of B16 tumor sections alone does not provide sufficient evidence that mature EMAP II protein is present within the tumor, protein extracts from B16 tumors were subjected to Western blot analysis to allow the distinction between proEMAP II/p43 and mature EMAP II protein based on their different molecular weights. As shown in Figure 4A ▶ , four bands were detected by the antiserum. The most prominent band corresponding to a protein with a molecular weight of 43-kd was attributed to proEMAP II/p43. The bands of intermediate size, 34-kd and 30- to 32-kd are likely to represent cleavage products of proEMAP II/p43, whereas the band of 23-kd has the size of the mature EMAP II protein. 1 This demonstrates that proEMAP II/p43 is processed to mature EMAP II in B16 melanomas in vivo.
Figure 4.

A: Expression of EMAP II protein in the B16 melanoma in vivo. Protein extracts were analyzed by Western blotting using the EMAP II-specific polyclonal antiserum SA 2847. ProEMAP II/p43 and EMAP II proteins are indicated by arrows. B: Release of EMAP II protein from hypoxic B16 melanoma cells in vitro. B16 melanoma cells were cultured at an oxygen concentration of 1% in a hypoxic chamber for the times indicated. Culture supernatants were subjected to Western blot analysis using the EMAP II-specific antiserum. Only the band corresponding to the EMAP II protein is depicted. Release of EMAP II protein under hypoxia was observed in five independent experiments. C: Hypoxic cells release EMAP II protein in the absence of DNA fragmentation. Cells corresponding to the supernatants subjected to Western blotting in B were analyzed for DNA fragmentation indicative of apoptosis (left panel). As a positive control, fragmentation of B16 melanoma cells, treated with a combination of cycloheximide and TNF (right panel) to induce apoptosis, is shown.
Having shown that strongly proEMAP II/p43 expressing cells are located to areas of tissue hypoxia in the B16 melanoma, we investigated whether hypoxia is a direct mechanism leading to increased expression of EMAP II by means of quantitative real-time PCR technology in vitro. Confluent B16 melanoma cells were either cultivated for 1 hour, 4 hours, 8 hours, or 16 hours under hypoxic (1% O2) or normoxic conditions. We did not observe any differences in EMAP II mRNA expression between hypoxic and normoxic cells at any of the time points analyzed (data not shown).
Because we have demonstrated cleavage of proEMAP II/p43 in B16 tumors, we wanted to know whether hypoxia could be a mechanism to cause cleavage of proEMAP II/p43 and release of mature EMAP II protein. By Western blot analysis, mature EMAP II protein was detected in the supernatants of cells exposed to hypoxia for 20 hours and 24 hours, whereas no EMAP II protein was released from hypoxic B16 cells at the earlier time point of 16 hours (Figure 4B) ▶ . Control cells kept under normoxic conditions did not release EMAP II protein (Figure 5) ▶ . Since apoptotic cells can cleave proEMAP II/p43 leading to the release of EMAP II protein from cells, 9 we investigated whether hypoxic B16 melanoma cells processing proEMAP II/p43 protein undergo apoptosis in parallel. Lysates from corresponding supernatants were analyzed for apoptosis by DNA fragmentation analysis. 25 As shown in Figure 4C ▶ , cells exposed to hypoxia for 16 to 24 hours did not reveal the typical pattern of DNA fragmentation. In contrast, 24 hours of treatment with a combination of cycloheximide and TNF led to a prominent DNA-laddering in these cells. These data demonstrate that proEMAP II/p43 cleavage in hypoxic cells occurs in the absence of apoptosis.
Figure 5.
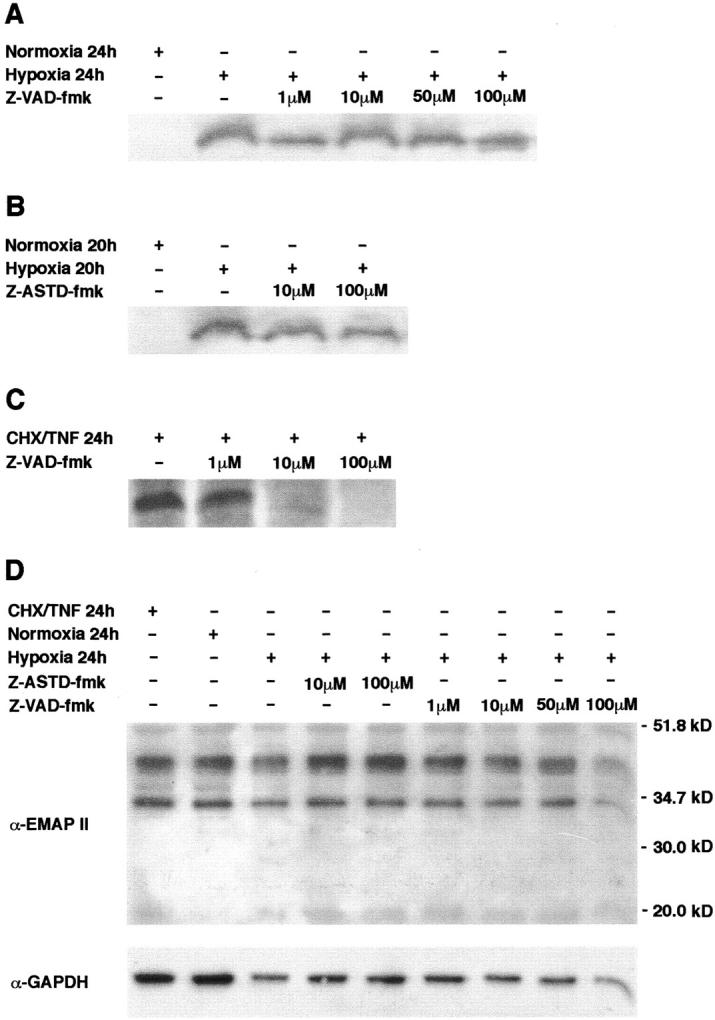
The peptide-based caspase inhibitors Z-VAD-fmk (A) and Z-ASTD-fmk (B) do not inhibit formation of mature EMAP II in hypoxic B16 melanoma cells. Cells were incubated at 1% oxygen in a hypoxic chamber for the times indicated. Increasing concentrations of the inhibitors were applied. Control cells were grown in a normoxic environment. Supernatants of the cultured cells were subjected to Western blot analysis using the EMAP II-specific antiserum. Only the band corresponding to the mature 23-kd EMAP II protein is shown. C: Z-VAD-fmk inhibits cleavage of proEMAP II/p43 in apoptotic B16 melanoma cells. Apoptosis was induced in B16 melanoma cells and where indicated, Z-VAD-fmk was added at increasing concentrations. The culture medium was analyzed for EMAP II protein by Western blotting. D: Western blot analysis of total lysates of B16 melanoma cells. After stimulation as indicated, cells were lysed and 20 μg total protein were analyzed by Western blotting using the EMAP II-specific antiserum (upper panel). The membrane was reprobed with a GAPDH-specific antibody to ensure equal protein loading (lower panel).
Hypoxia-Induced Processing of ProEMAP II/p43 Is Not Abrogated by the Caspase Inhibitors Z-VAD-fmk and Z-ASTD-fmk
One member of the family of cysteine proteases, caspase-7, has been identified as the proEMAP II/p43 cleaving enzyme, 10,11 linking apoptosis to EMAP II release. To address the question whether caspases are also involved in the formation of EMAP II protein during hypoxia, B16 melanoma cells were subjected to hypoxia for 24 hours in the presence of increasing concentrations of the broad-range caspase inhibitor Z-VAD-fmk. Western blot analysis of the supernatants demonstrated that even at the maximal concentration of 100 μmol/L Z-VAD-fmk did not interfere with the processing of proEMAP II/p43 in hypoxic B16 cells (Figure 5A) ▶ . As proEMAP II/p43 is cleaved via an internal ASTD motif, 10 a tetrapeptide representing the ASTD cleavage site coupled to afluoromethylketone (Z-ASTD-fmk) was added to B16 melanoma cells at increasing concentrations before incubation under hypoxia for 20 hours. Addition of Z-ASTD-fmk did not inhibit the formation of EMAP II in hypoxic cells as shown by Western blot analysis of the supernatants (Figure 5B) ▶ . In contrast, Z-VAD-fmk abrogated the cleavage of proEMAP II/p43 in apoptotic cells in a dose-dependent manner (Figure 5C) ▶ . These data suggest that caspases are not involved in the formation of EMAP II protein under hypoxic conditions in vitro. To address the question whether changes in cytosolic proEMAP II/p43 or EMAP II levels correlate with the release of EMAP II into the supernatant of hypoxic cells, Western blot analysis of lysates of B16 melanoma cells under normoxic, apoptotic, or hypoxic conditions, in the presence or absence of fluoromethylketone-inhibitors, were performed (Figure 5D ▶ , upper panel). In none of the lysates could mature EMAP II protein be detected, whereas proEMAP II/p43 was abundantly present. As a control for equal protein loading, the membrane was reprobed with a GAPDH-specific antibody (Figure 5D ▶ , lower panel). As changes in the proEMAP II/p43 levels merely reflect small differences in the protein loading, we can conclude that none of the conditions has any impact on translation of the proEMAP II/p43 protein.
Discussion
We have analyzed the expression of EMAP II mRNA in two different murine tumor models, the meth A fibrosarcoma and the B16 melanoma model, by in situ hybridization and real-time PCR. Whereas real-time PCR was used to quantify overall EMAP II mRNA levels, the semi-quantitative technique of in situ hybridization allowed us to detect enhanced transcription of the EMAP II gene and further to specifically attribute these transcripts to certain cell populations within the tumors and control tissue. Since EMAP II mRNA is detectable in every cell due to its function as the gene for a housekeeping protein we have chosen conditions which allow only the detection of increased EMAP II mRNA expression levels versus basal levels. This explains the absence of EMAP II mRNA-specific hybridization signals in the skin surrounding the tumor in Figure 1A ▶ . Although meth A fibrosarcomas in general displayed a higher expression of EMAP II mRNA than skin tissue (Figure 1A ▶ and Figure 2 ▶ ) the expression of EMAP II mRNA could be significantly enhanced by treatment of the tumors with TNF (Figure 1B ▶ and Figure 2 ▶ ). EMAP II transcripts were most abundant in a prominent rim of cells surrounding the necrotic center of the TNF-treated tumor compared to control tumors (Figure 1B) ▶ . To clarify whether high level EMAP II mRNA expression in perinecrotic areas of tumors is a general phenomenon or whether it is a specific response to TNF, B16 melanomas, which spontaneously develop necroses, were chosen as a second model system to analyze EMAP II mRNA expression. Indeed, B16 melanomas also showed increased levels of EMAP II mRNA in perinecrotic areas of the tumors compared to control tissue (Figure 1C ▶ and Figure 2 ▶ ). In conclusion, EMAP II mRNA could be shown to be up-regulated in the tumors analyzed and specific up-regulation was found at sites of necrosis.
Previously, we have shown that cells expressing high levels of EMAP II mRNA co-localize with areas of apoptosis in embryos as well as in thymus and testis of the adult mouse. 9,13 To investigate whether likewise a correlation of proEMAP II/p43 expression and apoptosis can be observed in tumors, double-staining with an EMAP II-specific antiserum and the TUNEL technique were performed on sections of B16 melanomas (Figure 3, A–C) ▶ . In confirmation of our in situ hybridization data, strong proEMAP II/p43 expression was observed in perinecrotic areas of the tumor. The weak staining of the surrounding tumor tissue can be explained by the fact that proEMAP II/p43 is part of the tRNA-multisynthetase complex and therefore present in every cell. 6 TUNEL-positive cells were mainly found in regions of developing necroses or in perinecrotic areas of established necroses. This raised the issue whether the staining was apoptosis-specific, as it is known that the TUNEL assay might not only label apoptotic cells but also random strand breaks in late- stage necrotic cells. However, positive TUNEL staining was largely absent from cells displaying swelling of the cytoplasm, a feature of necrotic cell death. Moreover, the intense staining of our tissue sections indicates large amounts of DNA strand breaks, which are only found in apoptotic cells (as reviewed by Stadelmann et al 26 ). We are therefore convinced that the TUNEL staining of our sections truly detected apoptosis and not necrosis. Confocal microscopy demonstrated that apoptotic cells co-localized with strongly proEMAP II/p43 expressing cells in close proximity to necrotic areas as also demonstrated on mRNA levels. Therefore, these data confirm our observation that cells expressing high levels of EMAP II mRNA are found in areas of tissue apoptosis. However, this correlation is more striking in the developing mouse embryo 9 than in the perinecrotic areas of tumors.
With this study, we are the first to analyze the correlation between apoptosis, hypoxia, and proEMAP II/p43 expression. The analysis of tissue hypoxia in B16 melanomas revealed no significant overlap between hypoxic and apoptotic areas (Figure 3, G–I) ▶ . Whereas almost all TUNEL-positive cells were found in regions of developing necroses or in perinecrotic areas of established necroses, maximal binding of NITP, indicating severe hypoxia, was detected at the boundary of necrotic/apoptotic regions. This spatial separation of apoptosis and hypoxia in experimental tumors in vivo confirms a previous report. 27 In contrast, a very strong and consistent overlap can be observed between markers of hypoxia and prominent EMAP II staining in perinecrotic areas of B16 melanomas. This is the first demonstration of a correlation between hypoxia and enhanced proEMAP II/p43 expression in a pathological setting in vivo (Figure 3, D–F) ▶ . As we did not observe increases in proEMAP II/p43 mRNA expression in tumor cells by hypoxia in vitro, hypoxia may not be the direct cause of the elevation of proEMAP II/p43 mRNA levels, although lack of an activity in vitro can have many reasons including genetic instability of the tumor cells, additional factors or conditions, which are only present in the in vivo situation. The proEMAP II/p43 promoter contains sequences which appear to resemble hypoxia responsive elements (J. Clifford Murray, personal communication), but expression of the hypoxia inducible transcription factors could be different in vivo and in vitro.
We could demonstrate in vitro that hypoxia leads to the cleavage of proEMAP II/p43 and the subsequent release of the mature EMAP II protein (Figure 4B) ▶ for which the proinflammatory biological activities are described. 1 Two possibilities arise from the observation that strong proEMAP II/p43 expression is found in hypoxic tissue areas. First, due to additional factors or conditions present in vivo, proEMAP II/p43 expression is increased in hypoxic cells and the hypoxic cells could subsequently die by apoptosis, a process known to lead to the release of mature EMAP II. Alternatively, hypoxia and apoptosis could be two independent mechanisms, each leading to the maturation of proEMAP II/p43 protein. In fact, our experiments demonstrate that in vitro hypoxic B16 melanoma cells release mature EMAP II without being apoptotic (Figure 4C) ▶ as shown by the absence of DNA fragmentation. 25 Likewise, no PARP-cleavage was observed (data not shown) and neither one of the two caspase-inhibitors, Z-VAD-fmk and Z-ASTD-fmk, potent inhibitors of proEMAP II/p43 cleavage in apoptotic cells, had any effect on the formation of mature EMAP II protein under hypoxia (Figure 5, A and B) ▶ . In principle, hypoxia and apoptosis could also act on different phases of the same cascade to release EMAP II. However, mature EMAP II protein was not detected in apoptotic, normoxic, or hypoxic cells (Figure 5D) ▶ . This argues against the possibility that, in hypoxia, preformed accumulated EMAP II simply is released, which would explain why caspase-inhibitors are not effective in the release of mature EMAP II protein from hypoxic cells. In conclusion, our findings suggest that by an as yet unknown mechanism, proEMAP II/p43 expression is elevated in perinecrotic tumor areas. The precursor protein is subsequently cleaved to the mature EMAP II protein by enzymes activated either during apoptosis or under hypoxic conditions. Caspase activation is not involved in the cleavage of proEMAP II/p43 in hypoxia, which supports the hypothesis that apoptosis and hypoxia are independent mechanisms for the formation of mature EMAP II protein in tumor cells. This can be best explained by an alternative cleavage enzyme responsible for the processing of proEMAP II/p43 in hypoxia. Hypoxia-regulated proteases and possible candidates for the cleavage of proEMAP II/p43 are calpain, 28 cathepsin, 29 or metalloproteinases. 30 Investigations are under way for the identification of this protease.
Our identification of hypoxia as an alternative caspase-independent mechanism of proEMAP II/p43 processing in tumor cells may be of prognostic or therapeutic value based on the antitumorigenic potential reported previously for mature EMAP II. For example, growth of tumors, which had been retrovirally transduced with EMAP II cDNA, is retarded 31 and, furthermore, inhibition of primary and metastatic tumor growth after intraperitoneal application of recombinant EMAP II protein was observed. 32 These and other therapeutic applications suggest that EMAP II is a negative modulator of tumor growth. Growth inhibition was proposed to be due to an antiangiogenic activity of EMAP II as demonstrated by the inhibition of endothelial cell proliferation and angiogenesis in vitro. Antiangiogenesis mediated by EMAP II may result from its ability to induce apoptosis in endothelial cells, a process that was shown to be magnified by concomitant hypoxia. 32 It can be hypothesized that hypoxia is involved in antitumor activities by both causing release of EMAP II as well as by sensitizing endothelial cells to respond to EMAP II to undergo apoptosis.
In conclusion, we hypothesize that, depending on the cellular context, EMAP II may have different roles within an organism: first, involvement in the removal of cellular debris during tissue remodeling in the embryo and in adult thymus and testis by chemotactic attraction of monocytes without provoking inflammation 9,13 and second, participation in the host defense mechanism against tumors by provoking an inflammatory response that ultimately results in the destruction and regression of the tumor. Here we could demonstrate that mature EMAP II is generated via two distinct mechanisms in the tumor cell, namely apoptosis and hypoxia. EMAP II generation by hypoxia may be one mechanism to evoke an inflammatory host response not only in tumors but also in ischemic diseases. It remains to be elucidated which protease(s) is (are) responsible for the observed formation of mature EMAP II protein under hypoxia.
Acknowledgments
We thank D. Männel for kindly providing us with the murine BFS-1 fibrosarcoma and B16 melanoma cells and for helpful discussions. We also thank R.M. Bohle for giving us the opportunity to use the SDS7700 and M.M. Stein for skillful technical assistance concerning real-time mRNA quantitation.
Footnotes
Address reprint requests to M. Clauss, Max-Planck-Institute for Physiological and Clinical Research, Parkstr. 1, 61231 Bad Nauheim, Germany. E-mail: M.Clauss@Kerckhoff.mpg.de.
Supported by the Deutsche Forschungsgesellschaft (DFG) (SFB 547, C5 to M.C. and Z1 to L.F.) and the European Community (EC) (BMH4-CT96–3277).
Ulrike E. Knies’ current address is the Imperial Cancer Research Fund, Oxford, UK.
Heike A. Behrensdorf’s current address is Nanogen Recognomics GmbH (Industrial Park Hoechst), Frankfurt, Germany.
References
- 1.Kao J, Ryan J, Brett G, Chen J, Shen H, Fan YG, Godman G, Familletti PC, Wang F, Pan YC, Stern D, Clauss M: Endothelial monocyte-activating polypeptide II: a novel tumor-derived polypeptide that activates host-response mechanisms. J Biol Chem 1992, 267:20239-20247 [PubMed] [Google Scholar]
- 2.Kao J, Houck K, Fan Y, Haehnel I, Libutti SK, Kayton ML, Grikscheit T, Chabot J, Nowygrod R, Greenberg S, Kuang WJ, Leung DW, Haywards JR, Kisiel W, Heath M, Brett J, Stern DM: Characterization of a novel tumor-derived cytokine: endothelial-monocyte activating polypeptide II. J Biol Chem 1994, 269:25106-25119 [PubMed] [Google Scholar]
- 3.Marvin MR, Libutti SK, Kayton M, Kao J, Hayward J, Grikscheit T, Fan Y, Brett J, Weinberg A, Nowygrod R, LoGerfo P, Feind C, Hansen KS, Schwartz M, Stern D, Chabot J: A novel tumor-derived mediator that sensitizes cytokine-resistant tumors to tumor necrosis factor. J Surg Res 1996, 63:248-255 [DOI] [PubMed] [Google Scholar]
- 4.Gnant MF, Berger AC, Huang J, Puhlmann M, Wu PC, Merino MJ, Bartlett DL, Alexander HR, Jr, Libutti SK: Sensitization of tumor necrosis factor α-resistant human melanoma by tumor-specific in vivo transfer of the gene encoding endothelial monocyte-activating polypeptide II using recombinant vaccinia virus. Cancer Res 1999, 59:4668-4674 [PubMed] [Google Scholar]
- 5.Wu PC, Alexander HR, Huang J, Hwu P, Gnant M, Berger AC, Turner E, Wilson O, Libutti SK: In vivo sensitivity of human melanoma to tumor necrosis factor (TNF)-α is determined by tumor production of the novel cytokine endothelial-monocyte activating polypeptide II (EMAPII). Cancer Res 1999, 59:205-212 [PubMed] [Google Scholar]
- 6.Quevillon S, Agou F, Robinson JC, Mirande M: The p43 component of the mammalian multi-synthetase complex is likely to be the precursor of the endothelial monocyte-activating polypeptide II cytokine. J Biol Chem 1997, 272:32573-32579 [DOI] [PubMed] [Google Scholar]
- 7.Norcum MT, Warrington JA: The cytokine portion of p43 occupies a central position within the eukaryotic multisynthetase complex. J Biol Chem 2000, 275:17921-17924 [DOI] [PubMed] [Google Scholar]
- 8.Kaminska M, Deniziak M, Kerjan P, Barciszewski J, Mirande M: A recurrent general RNA binding domain appended to plant methionyl-tRNA synthetase acts as a cis-acting cofactor for aminoacylation. EMBO J 2000, 19:6908-6917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Knies UE, Behrensdorf HA, Mitchell CA, Deutsch U, Risau W, Drexler HC, Clauss M: Regulation of endothelial monocyte-activating polypeptide II release by apoptosis. Proc Natl Acad Sci USA 1998, 95:12322-12327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Behrensdorf HA, van de Craen M, Knies UE, Vandenabeele P, Clauss M: The endothelial monocyte-activating polypeptide II (EMAP II) is a substrate for caspase-7. FEBS Lett 2000, 466:143-147 [DOI] [PubMed] [Google Scholar]
- 11.Shalak V, Kaminska M, Mitnacht-Kraus R, Vandenabeele P, Clauss M, Mirande M: The EMAPII cytokine is released from the mammalian multisynthetase complex after cleavage of its p43/proEMAPII component. J Biol Chem 2001, 276:23769-23776 [DOI] [PubMed] [Google Scholar]
- 12.Barnett G, Jakobsen AM, Tas M, Rice K, Carmichael J, Murray JC: Prostate adenocarcinoma cells release the novel proinflammatory polypeptide EMAP-II in response to stress. Cancer Res 2000, 60:2850-2857 [PubMed] [Google Scholar]
- 13.Knies UE, Kroger S, Clauss M: Expression of EMAP II in the developing and adult mouse. Apoptosis 2000, 5:141-151 [DOI] [PubMed] [Google Scholar]
- 14.Schluesener HJ, Seid K, Zhao Y, Meyermann R: Localization of endothelial-monocyte-activating polypeptide II (EMAP II), a novel proinflammatory cytokine, to lesions of experimental autoimmune encephalomyelitis, neuritis, and uveitis: expression by monocytes and activated microglial cells. Glia 1997, 20:365-372 [DOI] [PubMed] [Google Scholar]
- 15.Daemen MA, van’t Veer C, Denecker G, Heemskerk VH, Wolfs TG, Clauss M, Vandenabeele P, Buurman WA: Inhibition of apoptosis induced by ischemia-reperfusion prevents inflammation. J Clin Invest 1999, 104:541-549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hodgkiss RJ, Jones G, Long A, Parrick J, Smith KA, Stratford MR, Wilson GD: Flow cytometric evaluation of hypoxic cells in solid experimental tumours using fluorescence immunodetection. Br J Cancer 1991, 63:119-125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Breier G, Albrecht U, Sterrer S, Risau W: Expression of vascular endothelial growth factor during embryonic angiogenesis and endothelial cell differentiation. Development 1992, 114:521-532 [DOI] [PubMed] [Google Scholar]
- 18.Fink L, Seeger W, Ermert L, Hanze J, Stahl U, Grimminger F, Kummer W, Bohle RM: Real-time quantitative RT-PCR after laser-assisted cell picking. Nat Med 1998, 4:1329-1333 [DOI] [PubMed] [Google Scholar]
- 19.Fink L, Kinfe T, Seeger W, Ermert L, Kummer W, Bohle RM: Immunostaining for cell picking and real-time mRNA quantitation. Am J Pathol 2000, 157:1459-1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gavrieli Y, Sherman Y, Ben-Sasson SA: Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol 1992, 119:493-501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Towbin H, Staehelin T, Gordon J: Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA 1979, 76:4350-4354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B: An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci USA 1975, 72:3666-3670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Plate KH, Breier G, Weich HA, Risau W: Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature 1992, 359:845-848 [DOI] [PubMed] [Google Scholar]
- 24.Damert A, Machein M, Breier G, Fujita MQ, Hanahan D, Risau W, Plate KH: Up-regulation of vascular endothelial growth factor expression in a rat glioma is conferred by two distinct hypoxia-driven mechanisms. Cancer Res 1997, 57:3860-3864 [PubMed] [Google Scholar]
- 25.Cohen JJ, Duke RC: Glucocorticoid activation of a calcium-dependent endonuclease in thymocyte nuclei leads to cell death. J Immunol 1984, 132:38-42 [PubMed] [Google Scholar]
- 26.Stadelmann C, Lassmann H: Detection of apoptosis in tissue sections. Cell Tissue Res 2000, 301:19-31 [DOI] [PubMed] [Google Scholar]
- 27.Koch CJ, Chasan JE, Jenkins WT, Chan CY, Laughlin KM, Evans SM: Co-localization of hypoxia and apoptosis in irradiated and untreated HCT116 human colon carcinoma xenografts. Adv Exp Med Biol 1998, 454:611-618 [DOI] [PubMed] [Google Scholar]
- 28.Vanderklish PW, Bahr BA: The pathogenic activation of calpain: a marker and mediator of cellular toxicity and disease states. Int J Exp Pathol 2000, 81:323-339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cuvier C, Jang A, Hill RP: Exposure to hypoxia, glucose starvation, and acidosis: effect on invasive capacity of murine tumor cells and correlation with cathepsin (L + B) secretion. Clin Exp Metastasis 1997, 15:19-25 [DOI] [PubMed] [Google Scholar]
- 30.Canning MT, Postovit LM, Clarke SH, Graham CH: Oxygen-mediated regulation of gelatinase and tissue inhibitor of metalloproteinases-1 expression by invasive cells. Exp Cell Res 2001, 267:88-94 [DOI] [PubMed] [Google Scholar]
- 31.Berger AC, Alexander HR, Tang G, Wu PS, Hewitt SM, Turner E, Kruger E, Figg WD, Grove A, Kohn E, Stern D, Libutti SK: Endothelial monocyte activating polypeptide II induces endothelial cell apoptosis and may inhibit tumor angiogenesis. Microvasc Res 2000, 60:70-80 [DOI] [PubMed] [Google Scholar]
- 32.Schwarz MA, Kandel J, Brett J, Li J, Hayward J, Schwarz RE, Chappey O, Wautier JL, Chabot J, Lo Gerfo P, Stern D: Endothelial-monocyte activating polypeptide II, a novel antitumor cytokine that suppresses primary and metastatic tumor growth and induces apoptosis in growing endothelial cells. J Exp Med 1999, 190:341-354 [DOI] [PMC free article] [PubMed] [Google Scholar]