

Abstract
The BL6 subline was derived from the F10 line, which was derived from the B16 mouse melanoma cell line. BL6 cells are more invasive than F10 cells and differ genetically from F10 cells by an alteration of the gene encoding the B56γ regulatory subunit of protein phosphatase 2A (PP2A). This alteration results in the transcription of mRNA encoding a truncated variant of the B56γ1 isoform (Δγ1). When F10 cells were stained with a polyclonal antibody that recognizes three B56γ isoforms, B56γ1, B56γ2, and B56γ3, the immunofluorescent signals co-localized well with the cis-Golgi marker proteins. When BL6 cells were fractionated in a sucrose gradient, B56γ1 and B56γ2, but not B56γ3, were present in the Golgi-enriched fraction. This fraction also contained the catalytic subunit of PP2A. FLAG-tagged Δγ1 preferentially localized to the trans-Golgi area rather than the cis-Golgi. This localization was the same as that of FLAG-tagged B56γ1. NIH3T3 cells stably expressing Δγ1 transported a mutant viral protein from the endoplasmic reticulum to the plasma membrane much faster than wild-type cells. Their directional migration, as assessed by the advance of cells into a cell-free area, was also elevated. As Δγ1 reduces the activity of the B56γ-containing PP2A holoenzymes, these results suggest that the normal holoenzymes suppress vesicle transport and that Δγ1 might increase the invasive ability of BL6 cells by activating Golgi function.
B16 mouse melanoma cells were originally established from a de novo melanoma tumor. 1 These cells underwent 10 rounds of in vivo selection to yield the F10 subline, which in turn went through six rounds of in vitro selection to yield the BL6 subline. 2,3 Every additional round of selection increased the metastatic potential of the cells. Consequently, although both cells metastasize to the lungs after being injected intravenously into mice, BL6 cells can metastasize to the lungs even after being injected subcutaneously. 2,3 When we analyzed the gene expression of the two sublines to identify the differences responsible for the heightened metastatic potential of BL6, 4-9 we found that in BL6 cells, a retrotransposon had been inserted into an intronic region of one allele of the gene encoding the B56γ regulatory subunit of protein phosphatase type 2A (PP2A). 6
PP2A consists of a series of serine/threonine phosphatase holoenzymes that are composed of a common dimeric core of invariable catalytic (C) and structural (A) subunits associated with a variable regulatory (B) subunit. 10 The regulatory subunit is extremely diverse because it is constituted by members from at least three unrelated families, namely, PR55 (or simply B), B56 (B′), and PR72 (B″). 11 Each of these families in turn consists of several subfamilies, each of which contains several proteins resembling each other structurally. B56 seems to be the most complex of the three families as its members are encoded by five distinct mammalian genes that produce at least 13 splicing isoforms. One of these genes is B56γ, which is spliced in three different ways to produce the three isoforms belonging to the B56γ subfamily, namely, B56γ1, B56γ2, and B56γ3. In BL6 cells, the 5′ part of the original B56γ gene is replaced with the retrotransposon sequence, which results in the abundant expression of a chimeric mRNA species that encodes a mutant protein, termed Δγ1, that lacks the N-terminal 65 amino acid residues of the B56γ1 isoform. 6
Okadaic acid (OA) has been used to examine the function of PP2A both in vitro and in vivo. It is a complex fatty acid polyketal 12,13 and inhibits several types of protein phosphatases, including PP1, PP2A, PP4, and PP5. 12-15 When an intact cell is treated with 0.5 μmol/L of OA, the Golgi complex fragments into numerous clusters of Golgi-derived vesicles and tubules that then disperse in the cytoplasm. 16-18 During the Golgi fragmentation, intracellular vesicle transport is arrested. 16,19 Because PP1 and PP2A are the major components of the serine/threonine phosphatase activity in the mammalian cell, 20 and because OA completely inhibits PP2A and PP1 at 1 nmol/L and 5 μmol/L, respectively, 20 it is possible that the Golgi fragmentation caused by the OA treatment is because of the OA-induced inhibition of PP2A. In other words, PP2A may be involved in maintaining the morphology of the Golgi complex and in regulating Golgi complex-mediated vesicle transport. However, this possibility has never been closely examined.
It is believed that the regulatory subunits of PP2A control PP2A functions by directing particular trimeric PP2A holoenzymes into specific subcellular compartments as well as by enhancing the PP2A phosphatase activity on specific substrates. 6,11,21,22 Our previous work showed that both B56γ1 and Δγ1 localize in the perinuclear region when they are expressed exogenously as epitope-tagged proteins. 6 This work also showed that Δγ1 is incapable of promoting the dephosphorylation of specific substrates that is normally mediated by the B56γ subunit-containing PP2A holoenzyme. 6 Thus, although Δγ1 seems capable of acting as a targeting subunit, it lacks the ability to enhance PP2A activity in a substrate-specific manner. Given that PP2A may be crucially involved in Golgi function, we asked whether B56γ1 and Δγ1 actually localize to the Golgi complex, thereby serving to target the PP2A holoenzymes to the Golgi complex. If so, the expression of Δγ1 instead of B56γ1 may interfere with the normal regulation of Golgi function. This possibility may also explain how the expression of Δγ1 could enhance the metastatic potential of BL6 cells. It may be that B56γ1-containing PP2A holoenzymes normally down-regulate Golgi functions such as the transport of various molecules to their destined sites. Efficient vesicular transport is required for establishment of cell polarity and directional cell migration. 23,24 By interfering with the B56γ1-containing PP2A holoenzymes, Δγ1 may accelerate Golgi-mediated vesicle transport, thereby enhancing the migratory properties of BL6 cells.
In the present study, we show that an antibody (Ab) recognizing the endogenous B56γ subunits localizes to the cis-Golgi complex in the interphase. During Golgi fragmentation in mitosis or because of treatment with a microtubule-depolymerizing drug, the B56γ subunit remained associated with the Golgi complex fragments. Of the three isoforms of the B56γ subunit, B56γ1 and B56γ2 were detected in the Golgi-enriched fraction of the cell lysate. When B56γ1 and Δγ1 were transiently expressed as FLAG-tagged proteins, both isoforms localized mainly to the trans-Golgi area rather than the cis-Golgi area. We found that NIH3T3 cells stably expressing Δγ1 transported a viral protein via the Golgi to the plasma membrane faster than wild-type cells and that they were also more proficient in directional migration. These observations suggest that Δγ1 expression might increase the migratory abilities of BL6 cells by disrupting normal Golgi function.
Materials and Methods
Cell Culture
B16 melanoma sublines F10 and BL6 were kindly provided by Dr. I. J. Fidler (University of Texas, Houston, TX). NIH3T3 mouse fibroblastic cells, NRK rat kidney cells, and COS-7 monkey kidney cells were purchased from the American Type Culture Collection (Manassas, VA). All cells were maintained in Dulbecco’s modified Eagle’s medium with 10% fetal calf serum.
Plasmid Construction
The pCX4bsr vector is derived from the pCXbsr vector 25 with a minor modification, and was kindly provided by Dr. T. Akagi (Osaka Bioscience Institute, Osaka, Japan). The AscI-NotI cDNA fragments of Δγ1 or B56γ1 were described previously. 6 The pCX4bsr vector was digested with EcoRI, blunted with T4 DNA polymerase, digested with NotI, and ligated with the AscI-NotI cDNA fragment of Δγ1 or B56γ1 that had been blunted with T4 DNA polymerase at its 5′ end (pCX4bsr-Δγ1 or pCX4bsr-B56γ1). Using the AscI-NotI cDNA fragment of Δγ1 or B56γ1 as a template, polymerase chain reaction was performed using the following primers: sense, 5′-GGCCTGCGTGCTTACATCAGGAAACAGA-3′; anti-sense, 5′-ACGGTACCCCGCAACACTCCCGAGTTACTCTCTTTTTTAG-3′. The anti-sense primer contains a mutation at the third nucleotide of the stop codon of Δγ1 or B56γ1 (TGA to TGC) and a KpnI site at its 3′ end. The 3′ part of the polymerase chain reaction-amplified cDNA fragment was ligated with the 5′ part of the AscI-NotI cDNA fragment at the EcoNI site, and then digested with KpnI. The resulting AscI-KpnI cDNA fragment was inserted into the multiple cloning site of the p3XFLAG-CMV-14 expression vector (Sigma Chemical Co., St Louis, MO) in which a synthetic linker carrying an AscI site (AGCTTGGCGCGCCA) had been inserted into the HindIII site. The plasmid constructs allowed expression of Δγ1 or B56γ1 tagged with FLAG at their C-termini.
Plasmid Transfection
NIH3T3 cells were transfected with pCX4bsr-Δγ1 or empty pCX4bsr vector by using the Fugene 6 transfection regent (Roche Diagnostics Co. Ltd., Indianapolis, IN) according to the manufacturer’s instructions. After transfection, cells were selected by resistance to blasticidin (3 μg/ml; Invitrogen, Carlsbad, CA) for 3 weeks to obtain single colonies. For transient expression of exogenous FLAG-tagged proteins, similar procedures were performed using p3XFLAG-CMV-14-Δγ1 or p3XFLAG-CMV-14-B56γ1 vector. After culturing for 24 hours, cells were processed for immunofluorescence.
Antibodies
Two Abs specific for the B56γ subunits were used. Their production has been described previously. 6 Briefly, Δγ1 and the B56γ isoforms B56γ1 and B56γ2 were produced as recombinant proteins as described previously, 6 and were used to immunize rabbits. Affinity-purified IgG fractions from the rabbit sera were examined for their specificity by Western blot analysis as described below. Anti-pan-B56γ Ab was derived from the rabbit immunized with recombinant B56γ1, whereas anti-B56γ1/2 Ab was from the rabbit immunized with a mixture of recombinant B56γ1, B56γ2, and Δγ1. Other primary Abs used are specific for giantin (polyclonal Ab 26 ), GM130 (clone 35; Transduction Laboratories, Lexington, KY), Rab8 (clone 4, Transduction Laboratories), p115 (clone 46, Transduction Laboratories), Golgi 58K (58K-8, Sigma), the PP2A C subunit (clone 46, Transduction Laboratories), syntaxin-6 (clone 30, Transduction Laboratories), calnexin (clone 37, Transduction Laboratories), lamin B (M-20; Santa Cruz Biotechnology, Santa Cruz, CA), α-tubulin (DM 1A, Sigma), and FLAG (polyclonal Ab, Sigma). Secondary Abs used are the peroxidase-labeled anti-rabbit or anti-mouse IgG Abs (MBL Co. Ltd., Nagoya, Japan), Cy2- and Cy5-labeled anti-rabbit IgG Abs (Jackson ImmunoResearch, West Grove, PA), and Cy2-, Cy3-, and Cy5-labeled anti-mouse IgG Abs (Jackson ImmunoResearch).
Immunofluorescence
Cells were grown on coverslips in Dulbecco’s modified Eagle’s medium containing 10% fetal calf serum. In some cases, cells were incubated with nocodazole (5 μg/ml) for 2 hours at 37°C before fixation. After washing with phosphate-buffered saline (PBS), cells were fixed and mildly permeabilized with methanol at −20°C for 10 minutes. No further permeabilization of cells was performed with disruptive agents such as Triton X-100. Cells were blocked with 2% bovine serum albumin (fraction V, Sigma) in PBS and then incubated in the blocking solution with anti-B56γ1/2 Ab at 1:400 dilution (the approximate protein concentration is 0.6 μg/ml), another Ab as indicated at 1:500 dilution, or both. Control experiments included two procedures. One was that after blocking, cells were incubated with preimmune rabbit serum at 1:20 dilution instead of the anti-B56γ1/2 Ab. The second involved diluting the anti-B56γ1/2 Ab at 1:400 in the blocking solution and adding this to a tube containing the recombinant B56γ2 protein at a concentration of 0.1 to 6.0 μg/ml. After leaving the tube for 1 hour at room temperature, the mixture was poured onto the coverslips bearing the cells that had just been blocked with 2% bovine serum albumin. The reaction with the primary Abs or preimmune serum was continued for 2 hours at 4°C. After washing with PBS, the cells were stained with a mixture of Cy5-labeled anti-rabbit IgG Ab and Cy2-labeled anti-mouse IgG Ab. To visualize DNA, after the cells had been incubated with the primary Abs, they were incubated with 0.5 mg/ml of DNase-free RNase for 1 hour in the blocking solution. The cells were then stained with a mixture of Cy5-labeled anti-rabbit IgG Ab, Cy3-labeled anti-mouse IgG Ab, and the nucleic acid stain SYTO13 (500 nmol/L; Molecular Probes, Eugene, OR). Cells were visualized using a confocal laser-scanning microscope (LSM510; Carl Zeiss, OberKochen, Germany).
Cell Cycle Synchronization
NIH3T3 cells were plated onto coverslips and the following day, cells growing in log phase were incubated in the presence of 2.5 mmol/L of thymidine for 20 hours to arrest the cells at the G1/S phase. Cells were then incubated for 10 hours in fresh medium without thymidine to promote the growth of unarrested cells. Subsequently, 5 μg/ml of aphidicolin was added to the medium and the incubation was continued for 15 hours. Cells were washed several times to remove the drug and incubated in fresh medium for 6 to 9 hours to allow progression into mitosis. Cells on coverslips were stained as described above.
Preparation of Golgi-Enriched Fraction
BL6 cells were homogenized in 0.25 mol/L of sucrose and protease inhibitor mixture with a nitrogen bombardment apparatus (Parr Instrument Co. Ltd., Moline, IL.), followed by centrifugation at 1000 × g for 10 minutes. The supernatant was separated by the method of Balch and colleagues 27 to obtain the Golgi fraction. Briefly, the postnuclear supernatant was adjusted to 1.4 mol/L of sucrose by the addition of ice-cold 2.3-mol/L sucrose containing 10 mmol/L Tris-HCl and 1 mmol/L ethylenediaminetetraacetic acid. It was then loaded into a SW28 tube and overlaid with 1.2 mol/L and 0.8 mol/L of sucrose containing 10 mmol/L of Tris-HCl. The gradients were centrifuged at 90,000 × g for 2.5 hours in the SW28 rotor. The turbid band at the 0.8/12-mol/L sucrose interface was harvested by syringe puncture.
Subcellular Fractionation of NIH3T3 Cells
NIH3T3 cells were grown in one 150-mm dish to 90% confluency. The cells were washed four times with a prechilled solution containing 10 mmol/L Tris (pH 7.5) and 0.25 mol/L sucrose and then scraped from the plates. The cells in an ∼1-ml solution were supplemented with 10 μl of protease inhibitor cocktail (Sigma) and passed through a 25G3/4 needle 15 times on ice. The cell lysate was centrifuged at 1500 × g for 10 minutes at 4°C. The resulting postnuclear supernatant was mixed with 2 mol/L of sucrose to a final concentration of 1.6 mol/L of sucrose. The mixture (∼2 ml) was loaded at the bottom of a centrifuge tube and overlaid with 1.3, 1.0, and 0.8 sucrose in 10 mmol/L of Tris (pH 7.5) (2.5 ml of each sucrose density), and 0.5 mol/L of sucrose (1.5 ml). Equilibrium centrifugation was performed with a RPS40T rotor (Hitachi, Tokyo, Japan) at 135,000 × g for 14 hours at 4°C. Fractions (∼1 ml per fraction) were collected from the top.
Western Blot Analysis
Cells were lysed in buffer containing 10 mmol/L Tris-HCl (pH 8.0), 1 mmol/L ethylenediaminetetraacetic acid, 0.5% Nonidet P-40, and 1 mmol/L phenylmethylsulfonyl fluoride. Equal quantities of cell lysates or fractionated cell extracts were denatured in sodium dodecyl sulfate gel loading buffer, separated on 10% sodium dodecyl sulfate-acrylamide gels, transferred to Immobilon (Millipore, Bedford, MA), and reacted with the primary Ab indicated. After washing, the blots were incubated with peroxidase-labeled secondary Ab and then reacted with Renaissance reagents (NEN, Boston, MA) before exposure.
Vesicular Stomatitis Virus G Protein-Green Fluorescent Protein (VSVG-GFP) Transport Assay
The expression plasmid pCDM8.1 vector (Invitrogen) encoding VSVG from the temperature-sensitive (ts) 045 mutant strain fused with the enhanced GFP (Clontech, Palo Alto, CA) at its C-terminus (VSVG-GFP) 28 was kindly provided by Dr. J. Lippincott-Schwartz (National Institutes of Health, Bethesda, MD). Cells were transiently transfected with pCDM8.1-VSVG-GFP using the Fugene 6 transfection reagent (Roche Diagnostics). After transfection, the cells were cultured at 40°C for 16 hours. Thirty minutes before shifting the temperature from 40°C to 32°C, 100 μg/ml of cycloheximide was added to each dish according to the methods of Toomre and colleagues. 29 The cells were subsequently fixed at the indicated times after the temperature shift and then processed for fluorescence microscopy and quantification as follows.
Quantification of Fluorescence
Methanol-fixed cells were incubated with the anti-p115 Ab and stained with Cy5-labeled anti-mouse IgG Ab to define the Golgi area. Images were created on the computer monitor of a confocal laser-scanning microscope (LSM510) through a filter set suitable for detecting fluorescein and Cy5 signals. The fluorescence intensity of VSVG-GFP in the Golgi area was measured with an image processing system of LSM510. Using the histogram tools in this system, the Cy5-labeled Golgi area and the whole cell area were measured in a unit of μm2 by delineating the margins of both areas with a digital cursor. The mean GFP fluorescence intensities in both areas were also measured in arbitrary units and the mean background fluorescence intensity was subtracted from both values. The corrected mean intensities multiplied by the delineated areas yielded the total GFP fluorescence intensities in the Golgi area and in the whole cell area. Proportions of the former to the latter were then calculated. The proportion at time 0 was expressed as 1. Proportions at other time points were converted into values relative to the proportion at time 0. We examined 25 cells at each time point and calculated the mean and SD. Experiments were repeated three times with similar results.
Wound Closure Assay
The wound closure assay was performed according to the methods of Goodman and colleagues 30 Cells were cultured in 12-well plates in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum. The initial plating was adjusted after 24 hours to yield subconfluent monolayers at the same cell density. The monolayers were wounded by scratching the bottom of the culture plates with a plastic scraper so that a cross with 1-mm-long lines was made. Phase contrast images of the wounded monolayers were photographed with a camera of a Diaphot microscope (Nikon, Tokyo, Japan). After washing, the monolayers were incubated for 10 hours in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum and then photographed again. By using the center point of the wound cross as a guide, two photos taken before and after the 10-hour incubation were compared. A Coolscan III film scanner (Nikon) was used to create photo images on a monitor of a Macintosh computer (Power Mac G4) equipped with an image processing software (Adobe Photoshop Ver 5.0; Adobe System Inc., Mountain View, CA) and a digital cursor. On the monitor, the width of the initial wounds was ∼5 cm. A cursor was used to manually delineate the front line of cells that had advanced into the cell-free space from randomly chosen 1-mm segments of the initial wound border. The area sandwiched between the delineation and the initial wound border was expressed in pixel units. The mean area and SD were calculated by observing 20 sites on the wounds for each cell type. Experiments were repeated three times with similar results.
Results
The B56γ Subunit-Specific Ab Localizes to the cis-Golgi Complex
Lysates of cultured NRK rat kidney cells, NIH3T3 mouse fibroblasts, and the F10 and BL6 melanoma lines were separated on a gel and blotted with two Abs against the B56γ subunit that have varying affinities for the three isoforms. The anti-pan-B56γ Ab recognizes all three isoforms in all four lysates but has the highest affinity for B56γ3 (Figure 1 ▶ , left). On the Western blot, this Ab recognized B56γ2 as a doublet of bands. The anti-B56γ1/2 Ab recognized B56γ2 strongly, B56γ1 moderately, and B56γ3 weakly in the lysates of F10 and BL6 cells (Figure 1 ▶ , right) but did not recognize B56γ1 in the NRK and NIH3T3 lysates. This suggests that B56γ1 may be modified in a cell type-specific manner. Expression of Δγ1 in BL6 cells was undetectable by either Ab. This was consistent with our previous observations showing that Δγ1 expression is detectable only when BL6 cells grow in vivo. 6
Figure 1.

Detection of the three B56γ isoforms by two B56γ-specific types of Abs. NRK, NIH3T3, F10, and BL6 cell lysates were analyzed by Western blotting using the anti-pan-B56γ (left) and anti-B56γ1/2 (right) Abs. After stripping, the blot was probed with the anti-α-tubulin Ab to indicate the total amount of proteins loaded per lane.
Previous reports suggest that B56γ3 localizes to the nucleus, B56γ1 to the cytoplasm, and B56γ2 to both compartments. 6,31,32 We used the anti-B56γ1/2 Ab to characterize the cytoplasmic localization of the B56γ isoforms in F10 cells in more detail. These cells were only mildly permeabilized so that the Ab would not penetrate into the nucleus. The Ab stained a stack-like structure in the perinuclear region of F10 cells, suggesting that the B56γ subunits recognized by this Ab are localized to the Golgi (Figure 2) ▶ . Given that the anti-B56γ1/2 Ab most prominently stains the B56γ1 and B56γ2 subunits in Western blotting (Figure 1) ▶ , and that the B56γ3 subunit has been reported to have a purely nuclear distribution, 6,31,32 these observations suggest that the B56γ1 and B56γ2 subunits are localized to the Golgi complex. Supporting the notion that the B56γ2 subunit is localized at the Golgi is that preincubation of the anti-B56γ1/2 Ab with the recombinant B56γ2 protein at concentrations exceeding 1.0 μg/ml almost completely abolished the stack-like staining (data not shown). That the anti-B56γ1/2 Ab specifically recognizes the B56γ subunits in the immunofluorescence assays was confirmed by the fact that incubation with preimmune rabbit serum in place of the anti-B56γ1/2 Ab resulted in no specific staining (data not shown). To confirm that the anti-B56γ1/2 Ab labels the Golgi complex, we double stained F10 cells with the anti-B56γ1/2 Ab and an Ab against either GM130 33 or p115, 33 which are both cis-Golgi markers (Figure 2) ▶ . The anti-B56γ1/2 Ab-specific signals co-localized well with the signals for both GM130 and p115 (Figure 2) ▶ . When BL6, NRK, NIH3T3, and COS-7 cells were examined, the same staining patterns were observed (data not shown).
Figure 2.

The endogenous B56γ1 and γ2 isoforms localize to the cis-Golgi area. F10 cells were mildly permeabilized so that the Abs would not penetrate into the nucleus and detect nucleus-localized B56γ subunits. The cells were then double labeled with the anti-B56γ1/2 Ab and either the anti-GM130 (A) or the anti-p115 Ab (B). The anti-B56γ1/2 Ab was stained with Cy5 (red, left) and the other two Abs with Cy2 (green, middle). Cy5 and Cy2 images were then merged (right).
The B56γ Subunit-Specific Ab Stains Fragment and Disperse in the Cytoplasm during Golgi Fragmentation
To confirm that the anti-B56γ1/2 Ab specifically recognizes the Golgi complex, this Ab was used to stain cells with a fragmented Golgi complex. The Golgi complex undergoes extensive fragmentation as the cell progresses into mitosis. 34 Thus, NIH3T3 cells at different stages of mitosis were triple labeled with the anti-B56γ1/2 Ab, the anti-GM130 Ab, and the nucleic acid stain SYTO13 (Figure 3A) ▶ . The mitotic stages were identified by the patterns of the SYTO13-labeled DNA, and Golgi complex fragmentation was monitored by the GM130 signals. The Golgi complex started to break down into smaller fragments in prophase, and during metaphase and anaphase it was dispersed throughout the cytoplasm. In late telophase, it started to reassemble. As shown in Figure 3A ▶ , labeling with the anti-B56γ1/2 Ab coincided well with the GM130 signals in prophase, metaphase, and late telophase. In anaphase and telophase, the anti-B56γ1/2 Ab signals became as diffuse in the cytoplasm as the GM130 signals.
Figure 3.
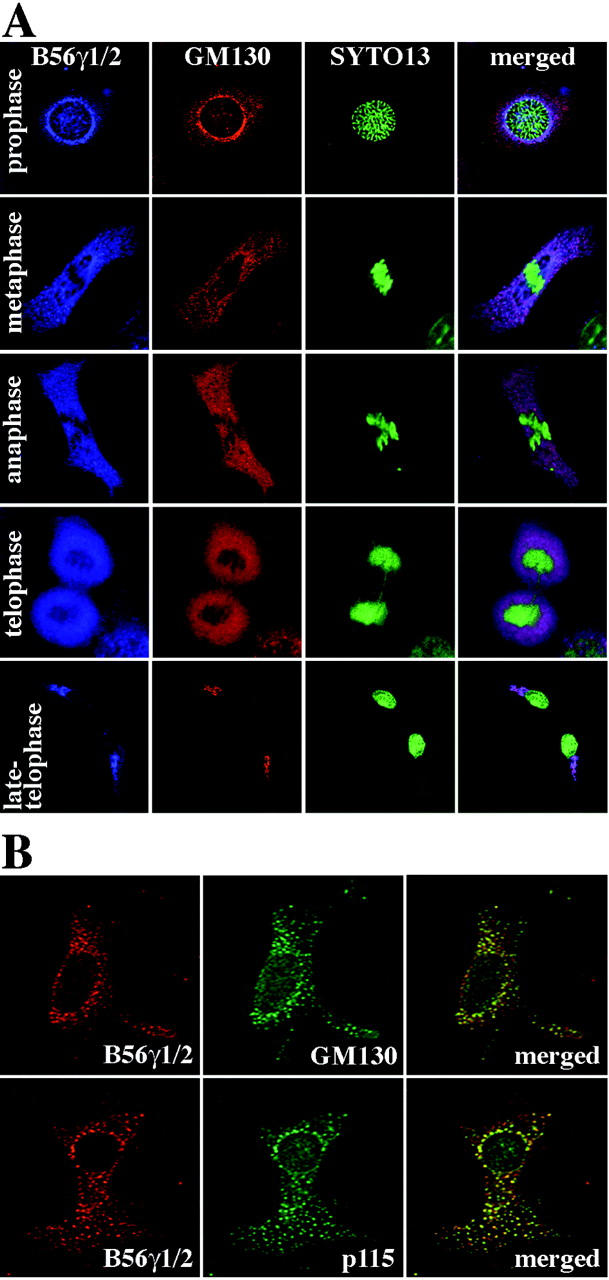
Association of endogenous B56γ subunits with cis-Golgi marker proteins during Golgi fragmentation. A: NIH3T3 cells were synchronized so that the cell population was enriched with mitotic cells. The cells were then fixed with methanol and triple-labeled with anti-B56γ1/2 Ab (Cy5, blue), anti-GM130 Ab (Cy3, red), and SYTO13 (green). The three images were then merged (right). Cells in prophase, metaphase, anaphase, telophase, and late telophase are shown. B: NIH3T3 cells were treated with nocodazole for 2 hours and double labeled with anti-B56γ1/2 Ab and either anti-GM130 Ab (top row) or anti-p115 Ab (bottom row). The anti-B56γ1/2 Ab was stained with Cy5 (red, left) and the other two Abs with Cy2 (green, middle). Cy5 and Cy2 images were then merged (right).
Nocodazole is a drug that depolymerizes microtubules and converts the Golgi stack into ministacks dispersed throughout the cytoplasm. 16 NIH3T3 cells were treated with this drug, and were double labeled with the anti-B56γ1/2 Ab and an Ab against either GM130 or p115 (Figure 3B) ▶ . Again, the anti-B56γ1/2 Ab-specific signals co-localized well with the signals for both GM130 and p115 as punctate structures dispersed throughout the cytoplasm. These immunofluorescence results suggest that the anti-B56γ1/2 Ab specifically recognizes the Golgi component, and that one or more of the B56γ isoforms localize to the cis-Golgi complex in interphase.
The B56γ1 and B56γ2 Isoforms Are Present in a Golgi-Enriched Cell Lysate Fraction
To confirm the Golgi localization of the B56γ subunit, BL6 whole cell lysates and a Golgi-enriched fraction purified from the lysates were separated on a gel and blotted with Abs specific for the subcellular localization markers syntaxin-6 35 (Golgi membrane), Rab8 36 [trans-Golgi network (TGN)], calnexin 37 [endoplasmic reticulum (ER)], lamin B (nucleus), and α-tubulin (cytoplasm). The Golgi-enriched fraction reacted strongly with the syntaxin-6 Ab, faintly with the calnexin Ab, and not at all with the lamin B or α-tubulin Abs, indicating successful fractionation (Figure 4A) ▶ . Reblotting with the anti-pan-B56γ Ab showed that B56γ1 and B56γ2, but not B56γ3, were contained in the Golgi-enriched fraction (Figure 4A) ▶ . Comparison of the band intensities of the total cell lysate and the Golgi-enriched fraction indicated that B56γ1 localized to the Golgi more profoundly than the γ2 isoform. In addition, use of an Ab against the PP2A C subunit revealed a considerable amount of this subunit was present in the Golgi-enriched fraction, suggesting that functional B56γ-containing PP2A heterotrimers might be present in the Golgi complex (Figure 4A) ▶ .
Figure 4.

Subcellular localization of the B56γ and C subunits of PP2A. A: Localization of the B56γ1 and γ2 isoforms and the PP2A C subunit to Golgi membranes. Cell lysate and Golgi-enriched fraction (Golgi) of BL6 cells were separated on a 10% sodium dodecyl sulfate-acrylamide gel and blotted with the Abs indicated. B: The components of nucleus-depleted homogenates of NIH3T3 cells were separated in a discontinuous gradient by equilibrium centrifugation. Fractions (1 to 10) were collected from the top. An aliquot of the loaded cell lysate and the fractions were separated on a 10% sodium dodecyl sulfate-acrylamide gel and blotted with the Abs indicated. Fraction numbers 8, 9, and 10 correspond to the position of sample loading.
The components of nucleus-depleted homogenates of NIH3T3 cells were fractionated in a discontinuous gradient by equilibrium centrifugation (Figure 4B) ▶ . The integral Golgi-resident protein giantin 26 was predominantly recovered in the lower density sucrose (fractions 4 to 6), whereas most of the peripherally-associated Golgi protein p115 remained at the bottom of the gradient (fractions 8 to 10) that represented the high-density sucrose load and contained soluble cytosolic proteins. The majority of the B56γ and C subunits of PP2A was recovered in the high-density sucrose load (fractions 8 to 10). However, a small but detectable amount of both subunits also floated to the lower density sucrose fractions 5 to 7, with a peak observed in the fraction 6 (Figure 4B) ▶ . These results suggest that although most of the B56γ-containing PP2A heterotrimers are soluble in the cytoplasm, some are peripherally associated with the Golgi membrane more strongly than p115.
Subcellular Localization of Δγ1
As Δγ1 interferes with the B56γ-containing PP2A holoenzymes, 6 examining the effect of expressing this mutant isoform on cellular function could reveal the function of the normal B56γ subunit in the Golgi complex. We generated an NIH3T3 subclone that stably expresses exogenous Δγ1 (NIH3T3Δγ1). Western blotting of NIH3T3Δγ1 cell lysates with the anti-pan-B56γ Ab showed that Δγ1 was expressed at similar levels as the endogenous B56γ1 isoform and that the expression levels of the three normal B56γ isoforms were not altered in the NIH3T3Δγ1 cells (Figure 5A ▶ , left). In contrast, Western blotting with the anti-B56γ1/2 Ab failed to detect either B56γ1 or Δγ1 in NIH3T3Δγ1 cell lysates (Figure 5A ▶ , right). This Ab also did not show signals specific to NIH3T3Δγ1 cells when it was used in immunocytochemistry (Figure 5B) ▶ . This suggests that although Δγ1 expression does not alter the localization of the endogenous B56γ isoforms in the Golgi complex, it is difficult to determine the localization of Δγ1 in NIH3T3Δγ1 cells.
Figure 5.
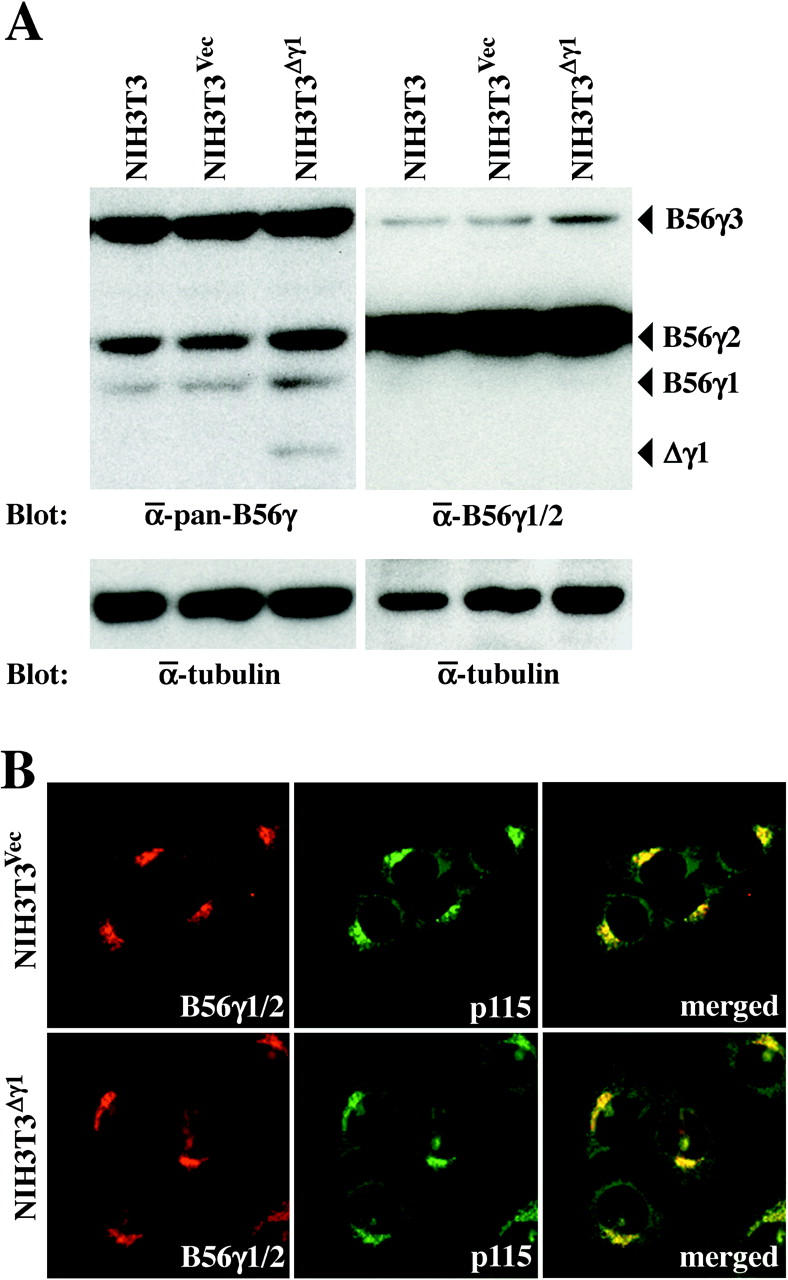
Expression of Δγ1 in NIH3T3Δγ1 cells. A: Western blot analysis of NIH3T3, NIH3T3Vec, and NIH3T3Δγ1 cells with the anti-pan-B56γ (left) and anti-B56γ1/2 (right) Abs. After stripping, the blot was probed with the anti-α-tubulin Ab to indicate the total amount of proteins loaded per lane. B: Immunofluorescence of NIH3T3Vec and NIH3T3Δγ1 cells. Cells were double labeled with the anti-B56γ1/2 Ab (Cy5, red) and anti-p115 Ab (Cy2, green). Cy5 and Cy2 images were then merged (right).
To better characterize the localization of Δγ1, we transiently transfected NIH3T3 cells with the cDNA encoding Δγ1 fused with FLAG at its C-terminus (Δγ1-FLAG) and detected the fusion protein with the anti-FLAG Ab. Δγ1-FLAG did not co-localize with the cis-Golgi marker p115 but was distributed in the vicinity of the p115 signals (Figure 6A) ▶ . This pattern suggests that Δγ1-FLAG localizes to organelles close to the cis-Golgi complex. This was further assessed by double labeling the cells transiently transfected with Δγ1-FLAG cDNA with the Abs against FLAG and calnexin, an ER marker. There was no detectable co-localization between the two signals (Figure 6B) ▶ . However, when the cells were double labeled with Abs against FLAG and TGN38, 38 a marker of the TGN area, the FLAG-specific signals co-localized well with those of TGN38, although they also extended beyond the anti-TGN38 Ab-labeled region (Figure 6C) ▶ . When we examined the subcellular localization of B56γ1 fused with FLAG at its C-terminus (B56γ1-FLAG), we found that B56γ1-FLAG was also detected in a limited perinuclear region, and double staining revealed that the localization of B56γ1-FLAG was similar to that of Δγ1-FLAG (data not shown). Thus, both Δγ1-FLAG and B56γ1-FLAG localize mainly to the TGN area. This differs from the cis-Golgi localization of the endogenously expressed normal B56γ isoforms that was detected by immunocytochemistry using the anti-B56γ1/2 Ab (Figure 2) ▶ .
Figure 6.

Transiently expressed Δγ1-FLAG protein preferentially localizes to the TGN area rather than to the cis-Golgi area. NIH3T3 (A and B) or NRK (C) cells were transfected with p3XFLAG-CMV-14-Δγ1 and incubated for 24 hours. Cells were then double labeled with anti-FLAG Ab and an Ab against p115 (A), calnexin (B), or TGN38 (C). The anti-FLAG Ab was stained with Cy2 (green, left) and the other Abs with Cy5 (red, middle). Cy2 and Cy5 images were then merged (right).
Δγ1 Expression Accelerates VSVG-GFP Transport
NIH3T3Δγ1 cells and NIH3T3 cells stably transformed with the empty vector (NIH3T3Vec) were assessed for the efficiency with which their vesicle transport systems transport the GFP-linked viral protein VSVG (VSVG-GFP) to the plasma membrane. In this system, VSVG-GFP is transported from the ER only at the permissive temperature. Thus, the cells were transiently transfected with a plasmid expressing VSVG-GFP and kept at the restrictive temperature (40°C). The fusion protein accumulated in the ER (Figure 7A ▶ , 0 minutes) to equivalent levels in the two cell types. Cycloheximide was added to the culture to stop protein synthesis before shifting the temperature to 32°C. This allowed us to chase a defined pulse of VSVG-GFP that had already accumulated in the ER. To determine when VSVG-GFP entered and exited the Golgi, the cells were stained with the p115 Ab at various intervals after the temperature shift. After 30 minutes, both cell types showed that most of the VSVG-GFP had moved into the Golgi area and there was very high co-localization between VSVG-GFP and p115 (Figure 7A ▶ , 30 minutes). After this period, VSVG-GFP and p115 gradually stopped co-localizing while the cell periphery simultaneously began to display GFP fluorescence, indicating insertion of VSVG-GFP into the plasma membrane. This pattern was true for both cell types except that while GFP fluorescence at the cell periphery was detectable in NIH3T3Δγ1 cells 1 hour after the temperature reduction, in NIH3T3Vec cells this was detected 2 hours later (Figure 7A ▶ , compare 60 minutes with 120 minutes). Even after 3 hours, a considerable amount of VSVG-GFP was still retained in the Golgi area of NIH3T3Vec cells but there was little or no co-localization between VSVG-GFP and p115 in NIH3T3Δγ1 cells as early as 2 hours after the temperature shift (Figure 7A ▶ , compare 120 minutes and 180 minutes).
Figure 7.

Transport of VSVG-GFP from the Golgi area to the plasma membrane is accelerated in NIH3T3Δγ1 cells. A: Immunofluorescence micrographs of NIH3T3Vec and NIH3T3Δγ1 cells expressing VSVG-GFP. Cells were transfected with pCDM8.1-VSVG-GFP and incubated for 16 hours at 40°C, during which time the VSVG-GFP accumulated in the ER (0 minutes). Cells were then treated with 100 μg/ml of cycloheximide for 30 minutes to stop protein synthesis and then shifted to 32°C for 30, 60, 120, and 180 minutes to allow the nascent VSVG-GFP to be transported by the secretory pathway. The cells were then fixed with methanol and stained with anti-p115 Ab (Cy5, red) to define the Golgi area. Fluorescein and Cy5 images were merged. The yellow area indicates the retention of VSVG-GFP in the Golgi. B: Quantification of VSVG-GFP fluorescence retained in the Golgi area. The immunofluorescence micrographs in A were examined for GFP fluorescence intensities in the Golgi areas and in the whole cell areas as described in Materials and Methods. The proportion of GFP fluorescence intensity in Golgi area to that in whole cell area at time 0 is expressed as 1. The relative proportions of GFP fluorescence intensity in the Golgi area at the time points indicated are plotted, with bars indicating the SD. *, P < 0.05 by t-test when the values of the NIH3T3 and NIH3T3Vec cells were compared.
To examine the kinetics of VSVG-GFP movement into and out of the Golgi more quantitatively, we measured the intensity of GFP fluorescence that co-localized with p115 at the indicated times after the temperature shift and normalized it against the intensity before the temperature shift (time 0). NIH3T3, NIH3T3Vec, and NIH3T3Δγ1 cells all showed a 3.5-fold increase in the GFP fluorescence intensity in the Golgi area 30 minutes after the temperature shift (Figure 7B) ▶ . Thereafter, the GFP fluorescence intensity in the Golgi area decreased as VSVG-GFP exited from the Golgi. In intact NIH3T3 cells and NIH3T3Vec cells, this decrease occurred relatively slowly because it took 2 hours for the GFP fluorescence intensity to halve and 3 hours to be reduced by two-thirds (Figure 7B) ▶ . In contrast, in NIH3T3Δγ1 cells, VSVG-GFP exited from the Golgi more rapidly as the GFP fluorescence intensity in the Golgi area had already returned to time 0 levels 2 hours after the temperature shift (Figure 7B) ▶ . Thus, Δγ1 appears to accelerate the vesicle transport of VSVG-GFP from the Golgi to the plasma membrane.
NIH3T3Δγ1 Cells Show Increased Directional Migration
We examined whether Δγ1 can affect directional cell migration by an in vitro wound closure assay. 23,30 Wounds 1 mm long were made in subconfluent monolayers of NIH3T3Vec and NIH3T3Δγ1 cells and the cells were then allowed to migrate into the cell-free area. Cell migration was quantified by measuring the area occupied by cells that had advanced into the cell-free area from the initial border of the wound. Ten hours after the wounding, NIH3T3Δγ1 cells migrated 30% faster than NIH3T3Vec cells (Figure 8) ▶ .
Figure 8.
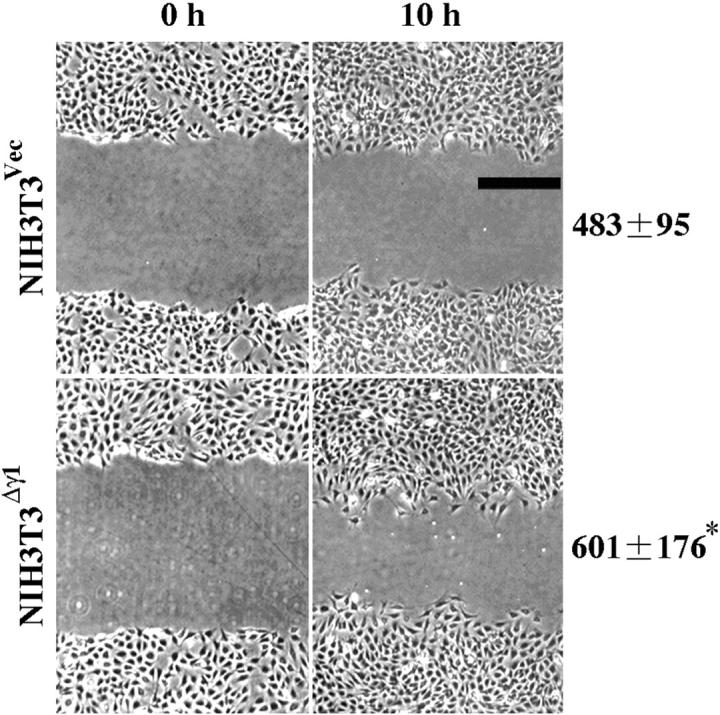
NIH3T3Δγ1 cells show increased directional migration ability. Directional cell migration was measured by an in vitro wound closure assay. Subconfluent monolayers of NIH3T3Vec and NIH3T3Δγ1 cells were wounded with a plastic scraper at time 0 and the cells were allowed to migrate into the cell-free area for 10 hours. Phase-contrast images of the same wound at time 0 and 10 hours are shown. The level of cell migration was quantified as described in Materials and Methods and expressed in pixel units as mean ± SD (see right side of the image). *, P < 0.05 by t-test when compared with the value of NIH3T3Vec cells. Scale bar, 100 μm.
Discussion
In the present study, we examined the subcellular localization of the B56γ subunit in the cytoplasm to probe its function. Our immunocytochemical and biochemical observations indicated that the B56γ1 and B56γ2 isoforms localize to the Golgi complex. Previous studies with cells overexpressing epitope-tagged B56γ isoforms have suggested that B56γ1 may be localized in the cytoplasm, B56γ3 in the nucleus, and B56γ2 in both compartments. 6,31,32 One report by van Lookeren Campagne and colleagues 39 has more precisely assessed the localization of the endogenous isoforms by staining neurons with an antiserum recognizing all three isoforms of the B56γ subunit. Specific signals in the perinuclear region as well as in the nucleus were observed. Our observations strongly suggest that the perinuclear signals are probably the B56γ1 and B56γ2 isoforms present in the Golgi complex. Thus, the endogenous B56γ1 and B56γ2 isoforms in the cytoplasm appear to localize mainly to the Golgi complex.
The B56γ subunits recognized by the anti-B56γ1/2 Ab remained associated with the Golgi complex even when the Golgi was fragmented in the early and late mitotic stages and by nocodazole treatment, indicating that the subunit binds to bona fide Golgi sites. Furthermore, when the nucleus-depleted homogenate of BL6 cells was separated in a discontinuous gradient, a proportion of the B56γ1 and B56γ2 isoforms was recovered in the fraction enriched with Golgi membranes and TGN (Figure 4A) ▶ . Notably, however, the majority of both isoforms was retained in the soluble cytosolic fraction (Figure 4B) ▶ . In this regard, the B56γ1 and B56γ2 isoforms seem to resemble p115. It has been shown that p115 is recovered predominantly in the cytosol after cell disruption, although it is absent from the cytosolic pool in intact cells. 40,41 This discrepancy is considered to be because of the release of p115 from Golgi membranes during cell disruption. 40 Our immunocytochemical assays revealed that the localization of the B56γ isoforms was restricted to the Golgi area with little diffuse cytoplasmic staining and thus we favor the hypothesis that the B56γ isoforms are associated with Golgi membranes in vivo but that, as with p115, this association is not maintained during cell disruption.
It is likely that B56γ1 and B56γ2 bind to Golgi membranes by protein-protein interactions because neither isoform contains a motif capable of mediating direct lipid interactions. Lowe and colleagues 42 have demonstrated that the PP2A heterotrimer containing the Bα regulatory subunit is responsible for dephosphorylation of the Golgi protein GM130 in the mitotic telophase. They suggested that this PP2A holoenzyme might dissociate from and reassociate with Golgi membranes in a cell-cycle stage-dependent manner. These observations together with our own suggest that particular molecules that function as receptors for B56γ and Bα might be present on the Golgi membrane, although their identity is currently unknown.
When NIH3T3Δγ1 cells stably expressing exogenous Δγ1 were immunostained with the anti-B56γ1/2 Ab, their staining pattern was indistinguishable not only from that of the parental NIH3T3 cells, but also from that of F10 cells. Because the anti-B56γ1/2 Ab recognizes B56γ1 in the lysate of F10 cells but fails to recognize it in the lysate of NIH3T3 cells, the labeling of the cis-Golgi complex with this Ab may be attributable to B56γ2. In the immunofluorescence of the endogenous B56γ isoforms shown here, B56γ1 may not be well visualized because of its lower protein levels or the lower levels of its immunoreactivity with the anti-B56γ1/2 Ab. When Δγ1-FLAG and B56γ1-FLAG were expressed transiently in NIH3T3 cells, both proteins were detected mainly in the TGN area rather than in the cis-Golgi area. This suggests that B56γ1 and Δγ1 may in fact be localized differently than B56γ2. Although the precise localization of these proteins remains to be clarified, it is notable that Δγ1 and B56γ1 appear to localize to the same compartment of the Golgi complex and differ from B56γ2 in their localization sites within the Golgi complex.
In our previous work, we showed that both B56γ1 and Δγ1 localize, in addition to the perinuclear region, to cell adhesion sites, and both isoforms co-localize with paxillin cytoskeletal proteins there. 6 The different localization pattern of B56γ1 and Δγ1 between our previous and present experiments may simply be attributable to the different types of tagged epitopes and different tagging sites used. B56γ1 and Δγ1 were tagged with the hemagglutinin (HA) epitope at their N-terminus in the previous study, whereas they were tagged with the FLAG epitope at their C-terminus in the present study. The HA tagging at the N-terminus may emphasize the localization of B56γ1 and Δγ1 at cell adhesion sites while the FLAG tagging at the C-terminus may emphasize their Golgi localization. It is intriguing that B56γ1 and Δγ1 may localize to both the Golgi complex and cell adhesion sites, because a considerable amount of paxillin molecules is pooled in the Golgi complex and paxillin moves out of this pool to cell adhesion sites probably via vesicle transport. 43,44 The association of paxillin with B56γ1 or Δγ1 may already occur when paxillin is pooled in the Golgi complex. This association may be maintained during the delivery of paxillin molecules from the Golgi pool to cell adhesion sites.
The transport of VSVG-GFP from the ER to the plasma membrane was accelerated in NIH3T3Δγ1 cells. This suggested that the B56γ-containing PP2A holoenzymes play a role in the function of the Golgi complex. As we showed previously, Δγ1 interferes with the dephosphorylation of specific substrates by the B56γ-containing PP2A holoenzymes in cells. 6 This is supported by a recent study showing that expression of N-terminally truncated B56γ subunits in Drosophila results in phenotypes similar to those produced by loss of function of B56γ. 45 N-terminally truncated isoforms of the B56γ subunit, including Δγ1, appear to function as dominant-negative mutants that specifically interfere with B56γ subunit-containing PP2A holoenzymes. Thus, in NIH3T3Δγ1 cells, Δγ1 may reduce the activity of the B56γ-containing PP2A holoenzymes present in the Golgi area and thereby accelerate vesicle transport. This hypothesis presumes that normal B56γ-containing PP2A holoenzymes suppress vesicle transport. This notion may seem to be contradicted by the fact that OA, which inhibits PP2A, arrests vesicle transport by inducing Golgi fragmentation. However, this discrepancy can probably be explained by the possibility that OA inhibits all PP2A holoenzymes while Δγ1 interferes only with those containing the B56γ subunit. Thus, Δγ1 may accelerate the efficacy of intracellular vesicle transport by reducing the suppressive role of the B56γ-containing PP2A holoenzymes. We showed previously that paxillin efficiently localizes to cell adhesion sites at an early stage of NIH3T3Δγ1 cell adhesion. We attributed this phenomenon then to an enhanced phosphorylation of paxillin. The observations in the current study suggest that accelerated vesicle transport in NIH3T3Δγ1 cells may also contribute to this phenomenon.
NIH3T3Δγ1 cells also exhibited an increased directional migration ability. This is consistent with the fact that efficient vesicle transport is necessary for directional cell migration. 23,24 As the intracellular delivery of adhesion molecules, such as NCAM, 46 paxillin, 44 and integrins, 47,48 to the cell surface is mediated directly by vesicle transport, more efficient vesicle transport may facilitate a more rapid recruitment of adhesion molecules to cell contact sites. Δγ1 is expressed by BL6 cells, a highly metastatic subline, but not by F10 cells, the parental subline. Although further studies are necessary to clarify the relationship between vesicle transport and metastasis, the observations reported in this study suggest that Δγ1 might contribute to the increased invasive ability of BL6 cells by up-regulating Golgi function.
Acknowledgments
We thank I. J. Fidler for the B16 melanoma sublines, J. Lippincott-Schwartz for the VSVG-GFP plasmid construct, P. Hughes for critically reading the manuscript, T. Akagi for pCX4bsr, and M. Kohara for technical assistance.
Footnotes
Address reprint requests to Akihiko Ito, M.D., Department of Pathology, Osaka University Medical School, 2-2 Yamada-oka, Suita, Osaka 565-0871, Japan. E-mail: aito@patho.med.osaka-u.ac.jp.
Supported by grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan; the Osaka Cancer Society; the Sagawa foundation for promotion of cancer research; the Naito Foundation; and the Japanese Association for Metastasis Research.
A. I. and Y. K. contributed equally to this work.
References
- 1.Fidler IJ: Biological behavior of malignant melanoma cells correlated to their survival in vivo. Cancer Res 1975, 35:218-224 [PubMed] [Google Scholar]
- 2.Hart IR: The selection and characterization of an invasive variant of the B16 melanoma. Am J Pathol 1979, 97:587-600 [PMC free article] [PubMed] [Google Scholar]
- 3.Poste G, Doll J, Hart IR, Fidler IJ: In vitro selection of murine B16 melanoma variants with enhanced tissue-invasive properties. Cancer Res 1980, 40:1636-1644 [PubMed] [Google Scholar]
- 4.Nakaji T, Kataoka TR, Watabe K, Nishiyama K, Nojima H, Shimada Y, Sato F, Matsushima H, Endo Y, Kuroda Y, Kitamura Y, Ito A, Maeda S: A new member of the GTPase superfamily that is upregulated in highly metastatic cells. Cancer Lett 1999, 147:139-147 [DOI] [PubMed] [Google Scholar]
- 5.Kataoka TR, Ito A, Asada H, Watabe K, Nishiyama K, Nakamoto K, Itami S, Yoshikawa K, Ito M, Nojima H, Kitamura Y: Annexin VII as a novel marker for invasive phenotype of malignant melanoma. Jpn J Cancer Res 2000, 91:75-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ito A, Kataoka TR, Watanabe M, Nishiyama K, Mazaki Y, Sabe H, Kitamura Y, Nojima H: A truncated isoform of the PP2A B56 subunit promotes cell motility through paxillin phosphorylation. EMBO J 2000, 19:562-571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ito A, Katoh F, Kataoka TR, Okada M, Tsubota N, Asada H, Yoshikawa K, Maeda S, Kitamura Y, Yamasaki H, Nojima H: A role for heterologous gap junctions between melanoma and endothelial cells in metastasis. J Clin Invest 2000, 105:1189-1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watabe K, Ito A, Asada H, Endo Y, Kobayashi T, Nakamoto K, Itami S, Takao S, Shinomura Y, Aikou T, Yoshikawa K, Matsuzawa Y, Kitamura Y, Nojima H: Structure, expression and chromosome mapping of MLZE, a novel gene which is preferentially expressed in metastatic melanoma cells. Jpn J Cancer Res 2001, 92:140-151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakamoto K, Ito A, Watabe K, Koma Y, Asada H, Yoshikawa K, Shinomura Y, Matsuzawa Y, Nojima H, Kitamura Y: Increased expression of a nucleolar Nop5/Sik family member in metastatic melanoma cells: evidence for its role in nucleolar sizing and function. Am J Pathol 2001, 159:1363-1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Usui H, Imazu M, Maeta K, Tsukamoto H, Azuma K, Takeda M: Three distinct forms of type 2A protein phosphatase in human erythrocyte cytosol. J Biol Chem 1988, 263:3752-3761 [PubMed] [Google Scholar]
- 11.Virshup DM: Protein phosphatase 2A: a panoply of enzymes. Curr Opin Cell Biol 2000, 12:180-185 [DOI] [PubMed] [Google Scholar]
- 12.Takai A, Bialojan C, Troschka M, Ruegg JC: Smooth muscle myosin phosphatase inhibition and force enhancement by black sponge toxin. FEBS Lett 1987, 217:81-84 [DOI] [PubMed] [Google Scholar]
- 13.Bialojan C, Ruegg JC, Takai A: Effects of okadaic acid on isometric tension and myosin phosphorylation of chemically skinned guinea-pig taenia coli. J Physiol 1988, 398:81-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang X, Cheng A, Honkanen RE: Genomic organization of the human PP4 gene encoding a serine/threonine protein phosphatase (PP4) suggests a common ancestry with PP2A. Genomics 1997, 44:336-343 [DOI] [PubMed] [Google Scholar]
- 15.Dobson S, Kar B, Kumar R, Adams B, Barik S: A novel tetratricopeptide repeat (TPR) containing PP5 serine/threonine protein phosphatase in the malaria parasite, plasmodium falciparum. BMC Microbiol 2001, 1:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dinter A, Berger EG: Golgi-disturbing agents. Histochem Cell Biol 1998, 109:571-590 [DOI] [PubMed] [Google Scholar]
- 17.Thyberg J, Moskalewski S: Disorganization of the Golgi complex and the cytoplasmic microtubule system in CHO cells exposed to okadaic acid. J Cell Sci 1992, 103:1167-1175 [DOI] [PubMed] [Google Scholar]
- 18.Lucocq J, Berger E, Hug C: The pathway of Golgi cluster formation in okadaic acid-treated cells. J Struct Biol 1995, 115:318-330 [DOI] [PubMed] [Google Scholar]
- 19.Chou CF, Omary MB: Mitotic arrest with anti-microtubule agents or okadaic acid is associated with increased glycoprotein terminal GlcNAc’s. J Cell Sci 1994, 107:1833-1843 [DOI] [PubMed] [Google Scholar]
- 20.Cohen P: Classification of protein-serine/threonine phosphatases: identification and quantification in cell extracts. Methods Enzymol 1991, 201:389-398 [DOI] [PubMed] [Google Scholar]
- 21.Kamibayashi C, Estes R, Slaughter C, Mumby MC: Subunit interactions control protein phosphatase 2A: effects of limited proteolysis, N-ethylmaleimide, and heparin on the interaction of the B subunit. J Biol Chem 1991, 266:13251-13260 [PubMed] [Google Scholar]
- 22.Kamibayashi C, Estes R, Lickteig RL, Yang S-I, Craft C, Mumby MC: Comparison of heterotrimeric protein phosphatase 2A containing different B subunits. J Biol Chem 1994, 269:20139-20148 [PubMed] [Google Scholar]
- 23.Bershadsky AD, Futerman AH: Disruption of the Golgi apparatus by brefeldin A blocks cell polarization and inhibits directed cell migration. Proc Natl Acad Sci USA 1994, 91:5686-5689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kulkarni SV, Gish G, van der Geer P, Henkemeyer M, Pawson T: Role of p120 Ras-GAP in directed cell movement. J Cell Biol 2000, 149:457-470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akagi T, Shishido T, Murata K, Hanafusa H: v-Crk activates the phosphoinositide 3-kinase/AKT pathway in transformation. Proc Natl Acad Sci USA 2000, 97:7290-7295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Misumi Y, Sohda M, Tashiro A, Sato H, Ikehara Y: An essential cytoplasmic domain for the Golgi localization of coiled-coil proteins with a COOH-terminal membrane anchor. J Biol Chem 2001, 276:6867-6873 [DOI] [PubMed] [Google Scholar]
- 27.Balch WE, Dunphy WG, Braell WA, Rothman JE: Reconstitution of the transport of protein between successive compartments of the Golgi measured by the coupled incorporation of N-acetylglucosamine. Cell 1984, 39:405-416 [DOI] [PubMed] [Google Scholar]
- 28.Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJ, Lippincott-Schwartz J: ER-to-Golgi transport visualized in living cells. Nature 1997, 389:81-85 [DOI] [PubMed] [Google Scholar]
- 29.Toomre D, Keller P, White J, Olivo JC, Simons K: Dual-color visualization of trans-Golgi network to plasma membrane traffic along microtubules in living cells. J Cell Sci 1999, 112:21-33 [DOI] [PubMed] [Google Scholar]
- 30.Goodman SL, Vollmers HP, Birchmeier W: Control of cell locomotion: perturbation with an antibody directed against specific glycoproteins. Cell 1985, 41:1029-1038 [DOI] [PubMed] [Google Scholar]
- 31.Tehrani MA, Mumby MC, Kamibayashi C: Identification of a novel protein phosphatase 2A regulatory subunit highly expressed in muscle. J Biol Chem 1996, 271:5164-5170 [DOI] [PubMed] [Google Scholar]
- 32.McCright B, Rivers AM, Audlin S, Virshup DM: The B56 family of protein phosphatase 2A (PP2A) regulatory subunits encodes differentiation-induced phosphoproteins that target PP2A to both nucleus and cytoplasm. J Biol Chem 1996, 271:22081-22089 [DOI] [PubMed] [Google Scholar]
- 33.Nakamura N, Lowe M, Levine TP, Rabouille C, Warren G: The vesicle docking protein p115 binds GM130, a cis-Golgi matrix protein, in a mitotically regulated manner. Cell 1997, 89:445-455 [DOI] [PubMed] [Google Scholar]
- 34.Lucocq JM, Berger EG, Warren G: Mitotic Golgi fragments in HeLa cells and their role in the reassembly pathway. J Cell Biol 1989, 109:463-474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bock JB, Lin RC, Scheller RH: A new syntaxin family member implicated in targeting of intracellular transport vesicles. J Biol Chem 1996, 271:17961-17965 [DOI] [PubMed] [Google Scholar]
- 36.Zerial M, McBride H: Rab proteins as membrane organizers. Nat Rev Mol Cell Biol 2001, 2:107-117 [DOI] [PubMed] [Google Scholar]
- 37.Wada I, Rindress D, Cameron PH, Ou WJ, Doherty JJ, II, Louvard D, Bell AW, Dignard D, Thomas DY, Bergeron JJ: SSR alpha and associated calnexin are major calcium binding proteins of the endoplasmic reticulum membrane. J Biol Chem 1991, 266:19599-19610 [PubMed] [Google Scholar]
- 38.Horn M, Banting G: Okadaic acid treatment leads to a fragmentation of the trans-Golgi network and an increase in expression of TGN38 at the cell surface. Biochem J 1994, 301:69-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Lookeren Campagne M, Okamoto K, Prives C, Gill R: Developmental expression and co-localization of cyclin G1 and the B′ subunits of protein phosphatase 2a in neurons. Brain Res Mol Brain Res 1999, 64:1-10 [DOI] [PubMed] [Google Scholar]
- 40.Gao YS, Alvarez C, Nelson DS, Sztul E: Molecular cloning, characterization, and dynamics of rat formiminotransferase cyclodeaminase, a Golgi-associated 58-kDa protein. J Biol Chem 1998, 273:33825-33834 [DOI] [PubMed] [Google Scholar]
- 41.Sohda M, Misumi Y, Yano A, Takami N, Ikehara Y: Phosphorylation of the vesicle docking protein p115 regulates its association with the Golgi membrane. J Biol Chem 1998, 273:5385-5398 [DOI] [PubMed] [Google Scholar]
- 42.Lowe M, Gonatas NK, Warren G: The mitotic phosphorylation cycle of the cis-Golgi matrix protein GM130. J Cell Biol 2000, 149:341-356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mazaki Y, Hashimoto S, Okawa K, Tsubouchi A, Nakamura K, Yagi R, Yano H, Kondo A, Iwamatsu A, Mizoguchi A, Sabe H: An ADP-ribosylation factor GTPase-activating protein Git2-short/KIAA0148 is involved in subcellular localization of paxillin and actin cytoskeletal organization. Mol Biol Cell 2001, 12:645-662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Norman JC, Jones D, Barry ST, Holt MR, Cockcroft S, Critchley DR: ARF1 mediates paxillin recruitment to focal adhesions and potentiates Rho-stimulated stress fiber formation in intact and permeabilized Swiss 3T3 fibroblasts. J Cell Biol 1998, 143:1981-1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hannus M, Feiguin F, Heisenberg CP, Eaton S: Planar cell polarization requires Widerborst, a B′ regulatory subunit of protein phosphatase 2A. Development 2002, 129:3493-3503 [DOI] [PubMed] [Google Scholar]
- 46.Musch A, Cohen D, Kreitzer G, Rodriguez-Boulan E: cdc42 regulates the exit of apical and basolateral proteins from the trans-Golgi network. EMBO J 2001, 20:2171-2179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ng T, Shima D, Squire A, Bastiaens PI, Gschmeissner S, Humphries MJ, Parker PJ: PKCα regulates β1 integrin-dependent cell motility through association and control of integrin traffic. EMBO J 1999, 18:3909-3923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Johanna I, Whelan RDH, Watson R, Parker PJ: PKCe controls the traffic of b1 integrins in motile cells. EMBO J 2002, 21:3608-3619 [DOI] [PMC free article] [PubMed] [Google Scholar]