

Abstract
To gain insight into the strategies of the immune system to confer resistance against the development of chronic coxsackievirus B3 (CVB3) myocarditis we compared the course of the disease in C57BL/6 mice, β2-microglobulin knockout (β2m−/−) mice, and perforin-deficient (perforin−/−) mice. We found that perforin−/− mice as well as immunocompetent C57BL/6 mice reveal a resistant phenotype with complete elimination of the virus from the heart in the course of acute myocarditis. In contrast, myocardial CVB3 infection of β2m−/− mice was characterized by a significantly higher virus load associated with a fulminant acute inflammatory response and, as a consequence of virus persistence, by the development of chronic myocarditis. Interferon-γ secretion of stimulated spleen cells was found to be significantly delayed in β2m−/− mice compared to perforin−/− mice and C57BL/6 control mice during acute myocarditis. In addition, generation of virus-specific IgG and neutralizing antibodies were found to be significantly decreased in β2m−/− mice during acute infection. From these results we conclude that protection against the development of chronic myocarditis strongly depends on the expression of β2m, influencing the catabolism of IgG as well as the production of protective cytokines, such as interferon-γ. Moreover, CVB3-induced cardiac injury and prevention of chronic myocarditis was found to be unrelated to perforin-mediated cytotoxicity in our model system.
Enteroviruses, such as coxsackieviruses of group B (CVB), are known to induce a variety of acute and chronic forms of diseases, including myocarditis, meningitis, and pancreatitis. 1 Although most enterovirus infections are bland and self-limiting, acute enterovirus infections of the heart may cause severe congestive heart failure and sudden cardiac death. 2,3 In addition, the detection of persistent enterovirus infection in patients with chronic heart muscle disease, 3-5 as well as the discovery of chronic myocarditis in persistently infected immunocompetent mice, 6,7 indicate that the typically cytolytic CVB are capable of evading immunological surveillance. In previous experimental studies of CVB3 myocarditis it has been demonstrated that permissive mice, such as A/J (H-2a), A.CA/J (H-2f), A.BY/J (H-2b), and SWR/J (H-2q), develop chronic myocarditis that is associated with virus persistence. 6,8 Analysis of persistent myocardial infection by strand-specific in situ hybridization revealed that enterovirus persistence is characterized by restricted viral plus-strand RNA synthesis and gene expression. 6 In contrast to permissive mice, resistant mouse strains, eg, DBA/1J (H-2q) or C57BL/6 (H-2b) eliminate the virus during acute myocarditis, thus preventing the development of chronic heart muscle disease. 6
Although much attention has been focused on the definition of host-specific factors influencing the outcome of myocarditis, 1,9 we still have a rudimentary understanding of how innate and specific immune response mechanisms control myocardial CVB3 infection to prevent chronic myocarditis. In several murine models of CVB3 myocarditis a beneficial role of antibodies has been suggested. CVB3-infected newborn mice were found to be protected from death if CVB3-neutralizing antibodies were given 24 hours before or 2 hours postinfection (p.i.) but not if administered 24 hours p.i. 10,11 More recently, it has been suggested that B cells might also contribute to long-term control of myocarditis because CVB3-infected B cell-deficient mice establish a persistent type of myocardial infection that is associated with severe fibrosis. 12 In agammaglobinemic humans the failure to produce antibodies has been shown to enhance susceptibility to persistent infections with polioviruses, 13 echoviruses, 14 and coxsackieviruses. 15 The protective role of the immune system in limiting viral replication and in clearing virus from the heart was further proven in studies of CVB3-infected SCID mice, illustrating that animals that lack both B and T cells develop severe chronic heart disease. 16 In addition, severe ongoing myocardial damage in association with inflammation was reported in T cell-deficient NMRI mice, 17 suggesting that T lymphocytes execute important antiviral properties. The relevance of CD4+ T cells as essential mediators for virus clearance and for the development of myocarditis has been strengthened by experiments in CD4 knockout mice 18 and MHC class II knockout mice that are also devoid of a CD4+ T cell response. 19
Whereas the protective role of CD4+ T cells in murine CVB3 myocarditis is widely accepted, the role of CD8+ T lymphocytes in the development and progression of chronic myocarditis is controversially discussed. CD8+ cytotoxic T lymphocytes (CTLs) represent the most important defense mechanism against intracellular pathogens. 20 They effectively mediate lysis of virus-infected target cells in a MHC class I-dependent manner. Depending on the target cell, CTLs confer lysis by either a perforin-dependent pathway or by fas-induced apoptosis. 21 Additionally, CD8+ T cells may achieve clearance of acute and also persistent virus infections via production of antiviral cytokines, such as tumor necrosis factor (TNF)-α and interferon (IFN)-γ, possibly via direct interference with viral gene expression. 22,23 Although the role of CTLs in CVB3 myocarditis is not yet defined there is evidence that these cells play an important role in the control of myocardial CVB3 replication. The analysis of acute myocarditis in CD8+ T cell-depleted CD4 knockout mice indicated that viral titers are significantly increased in the absence of functional CTLs. 18 On the other hand, CTLs have been reported to be the major effector cell population of myocardial organ injury in CVB3-infected mice, promoting the relevance of autoimmunity in the pathogenesis of chronic myocarditis. 24
Knockout mice have been proven to provide highly attractive systems for the analysis of specific immune cell functions in cytolytic as well as in noncytolytic virus infections. 20,25 In this study we have used two different knockout mouse strains, perforin (perforin−/−), 26,27 and β2-microglobulin (β2m−/−) knockout mice to dissect the relative contribution of specific immunological components in their role to prevent chronic myocarditis. As a consequence of the failure to express β2m molecules, β2m−/− mice are primarily deficient in CD8+ T cells, 28,29 NK1+ T cells, 30,31 and an IgG-recycling receptor (FcRn), which protects IgG from catabolism. 32-34
Here, we show that chronic myocarditis in β2m−/− mice develops as a consequence of the failure to limit the initial virus load of the heart muscle because of an ineffective antiviral antibody response and a diminished IFN-γ secretion. Moreover, our findings demonstrate that progression from acute to chronic enterovirus myocarditis is independent of perforin-mediated pathways.
Materials and Methods
Virus and Mice
cDNA-generated CVB3 (Nancy strain) 35 was grown and propagated in Vero cells (African Green monkey kidney cells). Stock virus was prepared by three times freezing and thawing and further purified by sucrose gradient centrifugation. Ten 4- to 5-week-old inbred mice of strains C57BL/6, C57BL/6 perforin−/−, and C57BL/6 beta2-microglobulin−/− (β2m−/−) (all H-2b) were infected intraperitoneally with 1 × 105 plaque-forming units of purified CVB3. Animals were sacrificed at different time points p.i., noninfected animals of all strains were used as controls.
Virus Titration
Hearts of infected mice were homogenized in 100 μl of RPMI, tissue debris was removed by centrifugation, and Vero cells were inoculated with serial twofold dilutions of infectious supernatant in four wells per dilution on a 96-cluster plate. After 2 days of incubation at 37°C supernatants were removed and cells were stained with trypan blue. TCID50, defined as that dilution required to infect 50% of inoculated wells, was determined by the method of Reed-Muench.
Tissue Preparation
Samples of aseptically removed tissues (heart, pancreas, spleen, brain) were either fixed for 12 hours in phosphate-buffered (pH 7.2) 4% paraformaldehyde and embedded in paraffin for histology, in situ hybridization, and terminal dUTP nick-end labeling (TUNEL) assays or quick frozen in liquid nitrogen for immunohistochemistry.
Histopathology
Histological analysis was performed on deparaffinized 5-μm-thick tissue sections that were stained with hematoxylin and eosin (H&E) to assess inflammation and myocyte injury or with Sirius Red to visualize the degree of fibrosis. The extent of myocardial lesions comprising cell necrosis, inflammation, and scarring was quantified on H&E-stained transverse sections of both heart chambers. Myocardial damage was quantified by selection of 30 visual fields per heart tissue section by systematic random sampling at a magnification of ×160. Area fractions of myocardial injury (area of damage per total area of myocardium in μm2/mm2) were calculated by point counting according to standard morphometric procedures. 6
In Situ Hybridization
CVB3-positive strand genomic RNA in tissues was detected using single-stranded 35S-labeled RNA probes that were synthesized from the dual-promoter plasmid pCVB3-R1. 6 Control RNA probes were obtained from the vector pSPT18. Pretreatment, hybridization, and washing procedures of dewaxed 5-μm paraffin tissue sections were performed as described previously. 6 Slide preparations were subjected to autoradiography, exposed for 3 weeks at 4°C, and counterstained with H&E.
For quantitative evaluation of hybridized heart tissue sections, in situ autoradiographs (n = 7 mice per mouse strain) were processed by an interactive image analyzing system, applying the Optimas software (Stemmer, Pucheim, Germany). Slide preparations were analyzed using a black and white video camera mounted on a microscope at a primary magnification of ×10 and ×40, respectively. Video signals were digitized resulting in images of 512 × 512 pixels with a gray-value range of 0 to 255 for each pixel. By applying a chain-code algorithm the autoradiographic signals were segmented from the background. Thereafter, areas of infected cells were automatically analyzed within 50 visual fields (each 100,000 μm2) per tissue section that were selected by systematic random sampling. Area fractions of infected tissues are expressed as arithmetic means ± SEM of seven animals per time point.
Immunhistochemistry
For characterization of heart muscle-infiltrating immune cells, cryostat heart tissue sections were fixed for 10 minutes in−20°C cold acetone and incubated for 1 hour at room temperature with rat anti-mouse antibodies against murine Mac-1 (clone M1/70, Roche, Grenzach-Wyhlen, Germany), Ia (clone M5/114, Roche), CD4 (clone H129.19, Pharmingen, Hamburg, Germany), CD8a (clone 53-6.7, Pharmingen), CD45R/B220 (clone RA3-6B2, Pharmingen), followed by three washing steps in Tris-buffered saline and incubation for 1 hour at room temperature with a secondary antibody (biotinylated anti-rat immunoglobulin; DAKO, Hamburg, Germany). Visualization of positive cells was completed by treatment with a streptAB-complex/AP (DAKO) and new fuchsin (DAKO). Slides were counterstained with hematoxylin. Quantification of immunohistochemically positive cells was achieved by counting at a magnification of ×400 in 150 randomly selected quadrants (mean area, 62,500 μm2) per tissue section. Results are expressed as arithmetic means of seven animals per strain ± SEM. 6
TUNEL Assay
Five-μm-thick paraffin-embedded heart muscle sections taken from all three inbred mouse strains at 4, 8, 12, and 28 days p.i. were deparaffinized and rehydrated. For the detection of apoptotic cardiac cells the ApopTag in situ apoptosis detection kit (Oncor, Heidelberg, Germany) was applied according to the manufacturer’s protocol with new fuchsin as chromogen. Slides were counterstained with hematoxylin.
Detection of Virus-Specific Antibodies by Enzyme-Linked Immunosorbent Assay (ELISA)
Detection of virus-specific serum IgM and IgG immunoglobulins was performed by an indirect ELISA. Microtiter plates (Polysorb; Nunc, Wiesbaden, Germany) were coated with purified CVB3 antigen (200 ng/well in 100 μl of 0.1 mol/L carbonate, pH 9.6) 36 and incubated overnight at room temperature. Plates were blocked with 5% bovine serum albumin (Sigma, Taufkirchen, Germany) in phosphate-buffered saline for 1 hour and subsequently incubated with twofold serial dilutions of sera obtained from infected mice at different time points p.i. After an incubation at room temperature for 2 hours the plates were washed and incubated with either horseradish peroxidase-conjugated goat anti-mouse IgG or peroxidase-conjugated goat anti-mouse IgM secondary antibody (Dianova, Hamburg, Germany) for an additional hour. Antibody binding was detected after a substrate reaction for 15 minutes with 3,5,3′,5′-tetramethylbenzidine. The reaction was stopped with 1 mol/L of H2SO4 and the absorbance at 450 nm was measured with a plate reader (Sanofi, Berlin, Germany). CVB3-specific antibody titers were defined as the highest dilution of serum showing an OD greater than the mean OD of sera obtained from naive mice plus threefold the SEM.
In Vitro Neutralization Assay
To define in vitro neutralization titers of sera taken from CVB3-infected mice, infectious virus corresponding to 100 TCID50/well was incubated with twofold serial dilutions of sera on a microtiter plate and incubated for 2 hours at 37°C. Vero cells (104 per well) were added and plates were incubated for additional 48 hours. Cells were stained with trypan blue and neutralization titers were defined as the highest serum dilutions leading to complete protection against CVB3-induced cytolysis.
Flow Cytometry
For flow cytometric analysis of cytokine production mouse splenocytes (2 × 106/ml) were stimulated for 4 hours with 5 ng/ml of phorbol 12-myristate-13-acetate and 500 ng/ml of ionomycin in the presence of Brefeldin A (GolgiPlug; Pharmingen). Cells were harvested and Fc receptors were blocked with Fc Block (Pharmingen). To identify the relevant cell populations cells were incubated with fluorochrome-conjugated anti-CD4-PerCP and anti-CD3-APC antibodies, respectively. For intracellular cytokine staining cells were fixed and permeabilized (Cytofix/Cytoperm Plus, Pharmingen), washed, and incubated with anti-IFN-γ fluorescein isothiocyanate and anti-interleukin (IL)-4 phycoerythrin or with the appropriate isotype controls (Pharmingen). The percentage of cytokine-expressing CD4+ T cells was determined in a four-color analysis using a FACSCalibur (Becton Dickinson, Heidelberg, Germany) flow cytometer.
Determination of Cytokines
Single-cell suspensions of spleen cells from CVB3-infected mice and from uninfected controls were prepared at indicated time points after infection and restimulated with 2 × 107 plaque-forming unit equivalents of heat-inactivated (20 minutes at 65°C) CVB3. After 4 days of restimulation, cell culture supernatants were screened for cytokine production, ie, IFN-γ and IL-4 by commercially available ELISA kits (OptEIA kits for mouse IFN-γ, IL-4; Becton-Dickinson). Tests were performed according to the manufacturer’s instructions.
Results
Outcome of CVB3 Infection
To determine whether lack of β2m and perforin expression influences the outcome of CVB3 myocarditis, β2m−/− and perforin−/− mice as well as C57BL/6 control mice were infected intraperitoneally with 105 plaque-forming units of purified CVB3 and sacrificed at days 4, 6, 8, 10, 12, and 28 p.i. Clinical signs of illness, such as weight loss, were observed in all three mouse strains during acute infection with up to 25% loss of weight in β2m−/− mice. Mortality was less than 5% in all strains and only observed during acute myocarditis. Whereas perforin−/− mice and C57BL/6 control mice completely recovered after acute infection, β2m−/− mice consistently revealed clinical symptoms, eg, rough hair coat and hunched posture at any time of infection as observed up to 28 days p.i. (data not shown).
Histopathology of Hearts from CVB3-Infected β2m−/−, Perforin−/−, and C57BL/6 Mice
Serial stained tissue sections from the hearts including the right and left ventricle of all three mouse strains were morphologically investigated after staining with H&E and Sirius Red at indicated time points after infection. At day 4 p.i. the myocardium of β2m−/−, perforin−/−, and C57BL/6 mice was characterized by small disseminated foci of inflammation in association with single infected myocytes in both ventricles. In the course of infection the virus-induced mononuclear cell infiltrates as well as myocyte necroses increased in all animals. The maximum of inflammatory cardiac lesions was observed in all three mouse strains at day 12 p.i., however with considerable differences regarding the extent of virus-induced myocardial injury (Figure 1 ▶ ; A, B, and C; H&E stains). As shown in Figure 1A ▶ , in β2m−/− mice numerous mononuclear cells as well as damaged myocytes were observed. In contrast, perforin−/− (Figure 1B) ▶ and C57BL/6 mice (Figure 1C) ▶ revealed only small scattered foci of inflammatory lesions. The morphometrical quantitation of heart muscle damage comprising myocytolysis, inflammation, and fibrosis at day 12 p.i. revealed an area percentage of damage in β2m−/− mice of 19.6 ± 4.2%, whereas perforin−/− mice and C57BL/6 control animals exhibited myocardial injury in only 3.6 ± 1.2% and 1.2 ± 0.9%, respectively. As demonstrated by stainings of heart muscle tissue sections with Sirius Red, the cardiac inflammation was primarily replaced by fibrosis in all three mouse strains at 28 days p.i. (Figure 1 ▶ ; D, E, and F). Whereas the hearts of β2m−/− mice disclosed large disseminated fibrotic areas (Figure 1D) ▶ in association with inflammatory cells (compare Figure 2D ▶ ), the myocardia of perforin−/− mice (Figure 1E) ▶ and C57BL/6 control mice (Figure 1F) ▶ were characterized by comparably small areas of scarring in the absence of notable inflammation at 28 days p.i.
Figure 1.
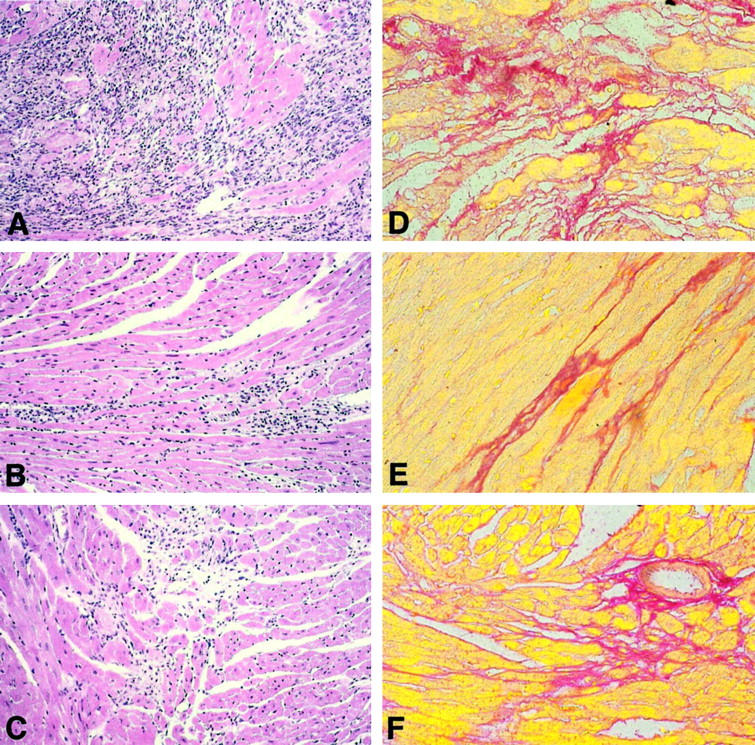
H&E stains of hearts from CVB3-infected β2m−/− mice (A), perforin−/− mice (B), and C57BL/6 mice (C) 12 days p.i. illustrate marked differences in the extent of inflammatory lesions with the most severe myocardial damage in β2m−/− mice (A). Correspondent to the findings of inflammation, Sirius Red stains from hearts 28 days p.i. reveal the largest areas of fibrosis in β2m−/− mice (D). Scarring was found to be slightly but not significantly diminished in perforin−/− mice (E) compared to C57BL/6 mice (F). Original magnifications, ×85.
Figure 2.
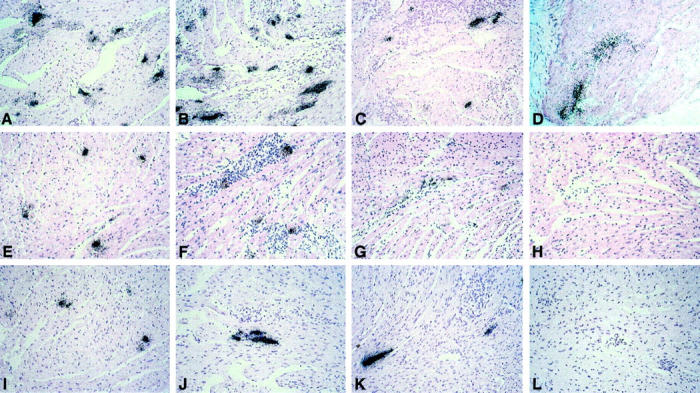
In situ hybridization for the detection of CVB3 infection in the myocardium of β2m−/− mice (A, 4 days p.i.; B, 8 days p.i. C, 12 days p.i.; D, 28 days p.i.), perforin−/− mice (E, 4 days p.i., F, 8 days p.i.; G, 12 days p.i.; H, 28 days p.i.), and C57BL/6 mice (I, 4 days p.i.; J, 8 days p.i.; K, 12 days p.i.; L, 28 days p.i.). The most CVB3 RNA-positive cells are observed in β2m−/− mice 8 days p.i. (B). In contrast to perforin−/− mice (H) and C57BL/6 mice (L), β2m−/− mice (D) still reveal CVB3 RNA in the myocardium at day 28 p.i., demonstrating viral persistence. Results are representative for a total of seven mice per strain. Counterstain, H&E. Original magnifications, ×70.
Analysis of Myocardial CVB3 Infection in β2m−/−, Perforin−/−, and C57BL/6 Mice
To compare the extent of myocardial virus infection in the knockout mouse strains [Figure 2 ▶ ; A to D (β2m−/− mice) and Figure 2 ▶ ; E to H (perforin−/− mice)], with that in C57BL/6 control mice (Figure 2 ▶ ; I to L) in the course of the disease, RNA/RNA in situ hybridizations were performed from paraffin-embedded hearts. In all three mouse strains the acute phase of myocarditis was found to be characterized by increasing numbers of infected myocytes [Figure 2 ▶ ; A, E, and I (4 days p.i.); B, F, and J (8 days p.i.)]. During acute infection (8 days p.i.) the highest amounts of in situ hybridization-positive myocardial foci were observed in β2m−/− mice (Figure 2B) ▶ . In contrast, perforin−/− mice (Figure 2F) ▶ revealed the typical infection patterns of resistant C57BL/6 control mice (Figure 2J) ▶ with only small areas of infection, indicating that perforin is not a relevant factor for limiting virus spread. Consistent with in situ hybridization results, concomitant TCID50 assays revealed a significant elevation of virus titers in hearts of β2m−/− mice (TCID50 1 × 104/g cardiac tissue) compared to those of perforin−/− mice (TCID50 1.1 × 103/g cardiac tissue) and C57BL/6 mice (TCID50 1.7 × 103/g cardiac tissue) at day 8 p.i. Twelve days p.i., in all three mouse strains a considerable decline of myocardial infection was noted [Figure 2 ▶ ; C, G, and K (12 days p.i.)]. However, the immune response was not found to be capable of eliminating CVB3 completely from the hearts of β2m−/− mice, thus inducing a persistent type infection in the myocardium (Figure 2D ▶ , 28 days p.i.). Moreover, virus persistence was not only observed in heart muscle but also in lymphatic tissue of spleens and lymph nodes and in single cells of the pancreata and brains from β2m−/− mice. By contrast, in none of the organs from perforin−/− mice or C57BL/6 control mice was a persistent type of infection noted in in situ hybridization experiments (data not shown).
To compare myocardial infection quantitatively in the three mouse strains we used an interactive image analysis system to measure the extent of in situ-positive myocardium (μm2) per mm2 of cardiac cross-tissue sections (Figure 3) ▶ . As soon as 4 days p.i. higher area fractions of infection were observed in β2m−/− mice than in the other two mouse strains. At the maximum of cardiac infection (day 8 p.i.) the area fractions of infection were found to be seven times higher in β2m−/− mice than in perforin−/− and C57BL/6 mice. At day 12 p.i. still three times more virus-positive cardiac tissue was present in β2m−/− mice than in perforin−/− and control mice, indicating impaired resolution of the virus from the hearts of β2m−/− mice.
Figure 3.

Kinetics of area fractions of infection (μm2/mm2) representing in situ hybridization-positive tissue within a transverse cardiac tissue section of β2m−/−, perforin−/−, and C57BL/6 mice in the course of myocarditis. Area fractions of infected tissues are expressed as arithmetic means of seven animals ± SEM per time point.
Characterization of Cardiac Immune Cells in CVB3 Myocarditis of β2m−/−, Perforin−/−, and C57BL/6 Mice
To identify immune cell populations of myocardial infiltrates in the course of CVB3 myocarditis immunohistochemical stainings were performed from frozen heart muscle-tissue sections using antibodies to Mac-1 (macrophages, natural killer cells), Ia (activated macrophages, B cells, dendritic cells), CD4 (T helper cells), CD8 (cytotoxic T cells), and CD45R/B220 (B cells and lytically-active subsets of lymphokine-activated killer cells). As soon as 4 days p.i. small inflammatory foci consisted primarily of Mac-1 and Ia-positive cells in all three mouse strains. In the course of infection an additional cardiac infiltration of CD4+ (β2m−/−) or CD4+ and CD8+ T lymphocytes (perforin−/− and C57BL/6 mice), respectively, was noted. The maximum of myocardial mononuclear cell infiltrates was reached around day 12 p.i. in all mice (Figure 4) ▶ . As illustrated in Figure 4 ▶ , the hearts of β2m−/− mice revealed significantly more Mac-1+, Ia+, and CD4+ cells than the two resistant mouse strains. At day 28 p.i. chronic myocardial inflammation consisting of macrophages and CD4+T lymphocytes were observed only in β2m−/− mice.
Figure 4.

Visualization of immunohistochemically stained Mac-1+ (A, E, and I), Ia+ (B, F, and J), CD4+ (C, G, and K), and CD8+ (D, H, and L) cells in hearts of β2m−/− (A–D), perforin−/− (E–H), and C57BL/6 (I–L) mice 12 days p.i. Results are representative for a total of seven mice per strain. Counterstain, hematoxylin. Original magnifications, ×50.
Quantitative Analysis of Immune Cell Subsets
At the maximum of cardiac inflammation (day 12 p.i.), the immunohistochemical stainings from the hearts of the knockout and control mice were quantitated by an interactive image analysis system. The number of mononuclear cells per mm2 heart tissue section was found to be four times higher in β2m−/− mice (795 ± 115 cells/mm2) than in perforin−/− (191 ± 63 cells/mm2) and C57BL/6 control mice (180 ± 48 cells/mm2). Table 1 ▶ illustrates the relative contribution of immune cell subsets in the murine hearts. In all three mouse strains the majority of infiltrating cells was found to be Mac-1+ with 43.2 ± 2.6% in C57BL/6, 55.9 ± 2.3% in perforin−/−, and 58.1 ± 1.8% in β2m−/− mice. Ia+ immune cells were present in β2m−/− and perforin−/− mice in 24.8 ± 2.3 and in 29.7 ± 3.1%, respectively, and in the control mice in 17.0 ± 3.4%. Relatively more CD4+ T lymphocytes were detected in C57BL/6 (42.7 ± 2.5%) and β2m−/− (38.3 ± 1.7%) mice compared to those in perforin−/− mice with 29.2 ± 3.1%. Regarding CD8+ T lymphocytes comparable relative amounts were observed in perforin−/− (11.3 ± 1.4%) and C57BL/6 (10.2 ± 0.4%) mice whereas, as expected, in hearts of β2m−/− mice only a small number of CD8+ T cells (1.4 ± 0.3%) was present. CD45R/B220+ B cells represented only a minor cell population (2.2 to 3.9%) in the hearts of all CVB3-infected mice.
Table 1.
Characterization of Cardiac Cell Infiltrates in CVB3-Infected Mice 12 Days p.i.
| Mouse strain | Cells/mm2 | % Positive cells | ||||
|---|---|---|---|---|---|---|
| Mac-1 | Ia | CD4 | CD45R/B220 | CD8a | ||
| β2m−/− | 795 ± 115 | 58.1 ± 1.8 | 24.8 ± 2.3 | 38.3 ± 1.7 | 2.2 ± 0.3 | 1.4 ± 0.3 |
| Perforin−/− | 191 ± 63 | 55.9 ± 2.3 | 29.7 ± 3.1 | 29.2 ± 3.1 | 3.6 ± 0.8 | 11.3 ± 1.4 |
| C57BL/6 | 180 ± 48 | 43.2 ± 2.6 | 17.0 ± 3.4 | 42.7 ± 2.5 | 3.9 ± 0.5 | 10.2 ± 0.4 |
Contribution of immune cell subsets (Mac-1, Ia, CD4, CD45R/B220, CD8a) in the inflammatory response of hearts from β2m−/−, perforin−/−, and C57BL/6 mice 12 days after CVB3 infection (n = 7 per mouse strain).
Visualization of Apoptosis in CVB3-Infected Hearts
To determine whether apoptotic events induced by CD8+ T cells may contribute to the pathogenesis of acute and chronic myocarditis in the three different mouse strains, the hearts were analyzed by TUNEL assays in the course of CVB3 infection. As soon as 4 days p.i. cardiac apoptotic endothelial cells were randomly detected in single animals, independently of the mouse strain (Figure 5 ▶ , inset). In the course of myocarditis the vast majority of TUNEL-positive cells were identified in all three mouse strains within inflammatory lesions, representing apoptotic immune cells as demonstrated in Figure 5 ▶ in the heart of a β2m−/− mouse 8 days p.i. After day 12 p.i. only β2m−/− mice revealed single apoptotic immune cells in areas of chronic inflammation. Single myocytes undergoing apoptosis (Figure 5 ▶ , arrow) were noted in β2m−/− and perforin−/− mice as well as in control animals at a very low frequency (one apoptotic myocyte per mid-cardiac tissue section).
Figure 5.

In situ detection of apoptotic cells by TUNEL assay in the hearts of CVB3-infected mice. In the course of infection single endothelial cells (inset) and myocytes (arrow) are found to be TUNEL-positive. The majority of apoptotic cells, however, represent mononuclear inflammatory cells as exemplary demonstrated in a β2m−/− mouse heart. Counterstain, hematoxylin. Original magnification, ×400.
Different Cytokine Production in Knockout and Control Mice
Previous investigations have demonstrated that CD8+ T cells may play a role in the regulation of CD4+ Th1/Th2 activity. 37 Therefore, we determined in our model systems the accumulation of Th1/Th2 cytokines IFN-γ and IL-4 in CD4+ T cells after polyclonal stimulation by fluorescence-activated cell sorting analysis. At any time of infection, the number of IL-4-producing T cells was found to be below background levels in β2m−/− as well as in perforin−/− and C57BL/6 mice (data not shown). In contrast, in all three mouse strains a typical Th1 response developed with measurable IFN-γ levels as observed up to day 28 days p.i. (Figure 6) ▶ . At all time points p.i. investigated significantly lower numbers of IFN-γ producing T cells were observed in β2m−/− mice after mitogenic stimulation of spleen cells compared to those of perforin−/− and C57BL/6 mice. Interestingly, the number of splenic IFN-γ-expressing CD4+ T cells obtained from β2m−/− mice were significantly reduced at the maximum of virus replication (day 6 and 8 p.i.).
Figure 6.

Frequency of IFN-γ-producing CD4+ T cells as determined by fluorescence-activated cell sorting analysis. Spleen cells from β2m−/−, perforin−/−, and C57BL/6 mice, respectively, were isolated from CVB3-infected animals at different time points p.i. Cells were stimulated for 4 hours with phorbol 12-myristate-13-acetate and ionomycin in the presence of Brefeldin A, and INF-γ production of CD4+ T cells was subsequently analyzed by flow cytometry. Values represent mean frequency of INF-γ-secreting cells ± SEM, P < 0001, between β2m−/− and perforin−/− mice at day 6 p.i., and P < 0.01 between β2m−/− and C57BL/6 mice at day 6 p.i. as determined by one-way analysis of variance and Tukey-Kramer multiple comparisons test.
To evaluate whether reduced IFN-γ production observed after polyclonal stimulation in β2m−/− mice also affects virus-specific T cells, spleen cells of infected animals were isolated 4, 8, 10, and 28 days p.i. and stimulated with inactivated CVB3 antigen. Supernatants were analyzed for IFN-γ production by standard ELISA techniques. As shown in Table 2 ▶ , virus-specific T cells obtained 8 days p.i. from perforin−/− and also from C57BL/6 mice were found to produce detectable amounts of IFN-γ. However, INF-γ production by CVB3-specific T cells from β2m−/− mice reached only background levels at this time point. Despite the delay of IFN-γ production in β2m−/− mice during acute infection, an ongoing antiviral cytokine response was observed in these animals, most likely because of the persistence of viral antigen with detectable levels of IFN-γ at day 28 p.i.
Table 2.
INF-γ Production of CVB3-Specific T Cells
| Mouse strain | Days after infection | ||||
|---|---|---|---|---|---|
| 0 | 4 | 8 | 10 | 28 | |
| β2m−/− | 123 ± 17 | 39 ± 2 | 67 ± 18 | 2018 ± 241 | 1150 ± 152 |
| Perforin−/− | 114 ± 40 | 62 ± 4 | 1889 ± 195 | 2966 ± 308 | 248 ± 79 |
| C57BL/6 | 48 ± 12 | 21 ± 3 | 775 ± 321 | 2254 ± 235 | 798 ± 113 |
Spleen cells from CVB3-infected β2m−/−, perforin−/−, and C57BL/6 mice were prepared at indicated time points p.i. and restimulated with 2 × 107 PFU-equivalents of inacti-vated CVB3. Culture supernatants were harvested after 4 days and IFN-γ secretion was determined by ELISA. Values represent pg/ml ± SEM.
CVB3-Specific Antibody Responses in β2m−/−, Perforin−/−, and C57BL/6 Control Mice
It is known that a defective CD8+ T cell response may influence the appearance or maintenance of antiviral serum antibodies either because of a low concentration of free antigen because of inefficient lysis of infected cells or because of an abnormal cytokine expression pattern. Because an efficient antibody response is essential for the control of infections with cytolytic viruses, the antibody responses after CVB3 infection in β2m−/−, perforin−/−, and C57BL/6 mice were analyzed by ELISA and neutralization assays.
In all three mouse strains virus-specific IgM responses were effectively induced with comparable titers. Maximum serum IgM levels were detected at day 6 p.i. and declined to background values until day 12 p.i. (data not shown). In contrast, serum IgG responses were found to be significantly different in β2m−/− mice compared to those of perforin−/− and C57BL/6 mice (Figure 7A) ▶ . Whereas the course of induction of virus-specific IgG antibodies was comparable during the early phase of infection (up to day 6 p.i.) in all animals, β2m−/− mice showed a dramatic decline in serum IgG between day 6 and 8 p.i. Also at later time points (day 12 and 28 p.i.) IgG concentrations were significantly lower in β2m−/− mice than in the two resistant mouse strains.
Figure 7.

Defective antibody responses in permissive β2m−/− mice. Sera from CVB3-infected β2m−/−, perforin−/−, and C57BL/6 mice were collected at various time points p.i. and titers of virus-specific IgG antibodies (A) as well as in vitro neutralization activity (B) were determined by CVB3-specific ELISAs and neutralization assays, respectively. Values are mean titers (log2) ± SEM, P < 0001, in IgG titers between β2m−/− and perforin−/− mice at days 8 and 28 p.i., and P < 0.01 between β2m−/− and C57BL/6 mice at day 8 p.i., and P < 0001 between β2m−/− and C57BL/6 at day 28 p.i. as determined by one-way analysis of variance and Tukey-Kramer multiple comparisons test.
To determine whether the reduction of CVB3-specific IgG antibodies in β2m−/− mice interferes with effective virus control, in vitro neutralization assays were performed. As shown in Figure 7B ▶ , virus-specific neutralizing antibodies (nAbs) can be detected as early as 6 days p.i. in all three mouse strains. Whereas nAb concentrations gradually increased with time in resistant C57BL/6 and perforin−/− mice, there was a highly significant decrease in nAb titers in β2m−/− mice after 8 days p.i., which correlates with the reduction of CVB3-specific IgG during the acute infection (Figure 7A) ▶ . At later stages of infection (day 28 p.i.) the nAbs reached their highest amounts in all three mouse strains, but the levels were still significantly reduced in β2m−/− mice compared to the resistant mouse strains.
Discussion
The aim of this study was to elucidate strategies of the immune system relevant to prevent the transition of acute myocarditis into the chronic form of the disease. For this purpose we compared the course of CVB3 infection in β2m−/− mice with that in perforin−/− mice and immunocompetent C57BL/6 control mice. Importantly, we found that in contrast to C57BL/6 and perforin−/− mice, β2m−/− mice are not capable of eliminating the virus from the heart, thus resulting in chronic myocarditis. As β2m−/− mice are primarily devoid of CD8+ T cells and previous investigations suggested that CD8+ T lymphocytes are required to clear CVB4 infection from the pancreas 38 we attempted to discriminate the role of CD8+ T cells in the murine models of CVB3 myocarditis.
CD8+ cytotoxic T cells have been described to mediate in vivo anti-viral effects either by direct lysis of cells via perforin or, to a minor degree, by fas-dependent mechanisms 21,39 and/or by the release of anti-viral cytokines. 22,23,39 For noncytolytic viruses, eg, lymphocytic choriomeningitis virus it has been shown that clearance of virus infection is strongly dependent on perforin-mediated pathways. 39 In contrast, for cytolytic viruses, such as vaccinia virus, vesicular stomatitis virus, and semliki forest virus neither perforin nor fas-related mechanisms were found to be required for the resolution of infection. 39 In the present study we demonstrate that prevention of chronic myocarditis is not mediated by elimination of CVB3-infected cells via perforin-mediated cytotoxicity. Perforin was previously described as a molecule that is not relevant for CVB3 clearance but for exacerbation of the severity of myocarditis. 40 In our quantitative investigations we could demonstrate that the area fractions of infection and of myocardial lesions and well as the inflammatory response in perforin−/− mice surpass those of C57BL/6 control mice. In contrast to findings of Gebhard and colleagues, 40 in our own, and in the experimental systems of others 19 immunocompetent C57BL/6 mice were found to be not susceptible for the development of chronic CVB3 myocarditis or severe fibrosis. Our finding that perforin does not influence the severity of myocarditis is substantiated by our former observation that DBA/1J mice that also possess the perforin gene reveal a resistant phenotype that is characterized by a mild acute myocarditis and the absence of chronic myocarditis or late fibrosis. 6 However, discrepancies with regard to the various model systems might reflect divergent virus-specific virulence factors of the different CVB3 strains (CVB3 Woodruff strain 40 versus the cDNA-generated cardiotropic CVB3 Nancy strain 6,17 ) used in these studies.
A potential anti-viral mechanism alternative to CD8+ T cell-dependent perforin-mediated lysis, eg, mediated by fas-dependent cytotoxicity, was also discussed for CVB3 myocarditis in perforin−/− mice 40 because fas is abundantly expressed in heart tissue. 41 Fas-mediated signaling belongs to the most potent stimuli that mediate cardiomyocyte apoptosis. 42 Therefore, we evaluated apoptotic events in the course of CVB3 myocarditis by TUNEL assays. In C57BL/6 control mice and β2m−/− mice as well as in perforin−/− mice we found that the majority of apoptotic cells represent inflammatory cells during acute myocarditis. These observations are supported by the findings of Henke and colleagues 43 that the murine proapoptotic protein Siva is the mediator of apoptotic processes within inflammatory areas of CVB3-infected murine heart and pancreas tissues and that apoptosis in myocytes is usually not detected. 18 Whereas in all three investigated mouse strains apoptosis of inflammatory cells was consistently observed during acute myocarditis only single cardiac myocytes revealed signs of apoptosis. However, in comparison to Gebhard and colleagues 40 who found apoptotic myocytes only in C57BL/6 control mice but not in perforin−/− mice, we observed TUNEL-positive myocytes in perforin−/− mice and β2m−/− mice as well as in C57BL/6 control animals at a very low frequency (one apoptotic myocyte per mid-cardiac tissue section). In a recent study, Peng and colleagues 44 demonstrated the presence of 15 apoptotic myocytes per mid-ventricle heart tissue section in acutely CVB3-infected permissive mice. In contrast, quantitative analyses from Mall and colleagues 45 in permissive CVB3-infected mice revealed that already before the cellular immune response invades the heart more than 250 myocytes per cross tissue section undergo cytolysis within 24 hours, confirming that rather cytolysis than apoptosis determines myocyte injury during acute CVB3 infection. 46 In addition, experimental data of Huber and colleagues 47 from CVB3-infected fas-deficient lpr/lpr mice and fas ligand-deficient gld/gld mice support the concept that the fas system is not required to prevent chronic myocarditis.
Despite the failure of CD8+ cytotoxic T cells to eliminate CVB3-infected cells via perforin or fas-mediated pathways, there is evidence from our observations in CD8+ T cell-deficient β2m−/− mice that CD8+ T lymphocytes do contribute to the control of virus infection in the acute phase of CVB3 myocarditis. Recent observations in CVB-infected CD8 knockout and CD4 knockout mice as well as in CD8+ T cell-depleted mice point to a time-dependent role of CD8+ T cells with beneficial effects during acute CVB infections and detrimental effects during chronic myocarditis. 38,48
As known from other cytolytic virus infections, CD8+ T lymphocytes cells can elaborate a variety of effector mechanisms against the infection by induction of a cytokine-based local antiviral state in the direct neighborhood of the infected cells and by promoting the generation of neutralizing antibodies. 39,49 Regulatory CD8+ T cells have been shown to clear virus infection also in a noncytolytic manner via production of antiviral cytokines, such as IFN-γ and TNF-α. 50 Both cytokines are also synthesized by infiltrating cells in hearts of immunocompetent mice with CVB3 myocarditis. 51 IFN-γ, a cytokine that can be produced by CD8+ and CD4+ T cells and also by natural killer cells belongs to one of the most potent regulators of inflammation and immune function. 52 Because previous reports provided evidence for a role of CD8+ T cells in the regulation of CD4+ T-cell cytokine response, 37 we analyzed CD4+ T cells for the secretion of IFN-γ and IL-4 in our model systems. In contrast to IL-4, which is not detected in CVB3-infected C57BL/6 mice, 53 perforin−/− mice, or β2m−/− mice, IFN-γ secretion could be measured in all three mouse strains during acute myocarditis. However, the very low levels of IFN-γ in β2m−/− mice during the maximum of myocardial virus replication at day 6 and 8 p.i. indicate that in absence of CD8+ T cells IFN-γ is not efficiently secreted by CD4+ T helper cells to reduce viral load during acute myocarditis. This idea is supported by the recent findings that the development of IFN-γ-producing Th1 cells is dependent on an efficient initial IFN-γ production by CD8+ T cells. 54,55 Moreover, the relevance for these results in virus infections was confirmed by Peterson and colleagues 56 who demonstrated that this helper effect of CD8+ T cells in the development of CD4 Th1 cell responses appears to be an important mechanism in generating protective immunity in infection with Friend murine retrovirus. Altered kinetics of IFN-γ production with a delay of 2 days compared to control mice was also observed in lymphocytic choriomeningitis virus-infected β2m−/− mice. 57
The importance of IFN-γ in the induction of an optimal anti-viral response was also confirmed in our investigations of immunocompetent mice. We found that in susceptible ABY/SnJ mice the IFN-γ production is considerably delayed, reaching maximum levels of IFN-γ expression only 16 days p.i., whereas resistant C57BL/6 mice revealed the highest IFN-γ levels already 12 days p.i. In addition, in ABY/SnJ mice that develop chronic myocarditis a significant delay of virus-specific antibody production is observed during acute myocarditis (G Szalay and colleagues, submitted).
The relevance of IFN-γ in CVB3 myocarditis was further supported by the observation that expression of IFN-γ from recombinant CVB3 variants protect mice from lethal disease and virus-induced tissue damage. 58 From a study of Opavsky and colleagues 48 it has been concluded that local IFN-γ expression may contribute to a restricted cell-to-cell spread of CVB3 in the heart, thus limiting the extent of myocardial organ injury. Furthermore, IFN-γ has also been shown to protect pancreatic cells from invading CVB4 by activation of resident macrophages. 59
Besides the finding of reduced IFN-γ expression we also observed a severe deficiency in production of IgG and neutralizing antibodies in CVB3-infected β2m−/− mice. Despite the fact that class I molecules and CD8+ T cells do not play a direct role in promoting antibody responses it has been demonstrated that β2m−/− mice infected with vaccinia virus, 60 rotavirus, 61 and influenza virus 62 develop lower virus-specific antibody titers than do control mice. In correspondence to our findings of normal serum IgM titers but reduced levels of IgG, also vaccinia virus-infected β2m−/− mice were found to be less efficient in antigen-specific IgG production than their β2m+/− littermates. 60 To date, no mechanisms have been defined to be responsible for the decreased production of virus-specific antibody titers in these knockout mice. Recently it has been shown that β2m−/− mice that are deficient in the MHC class I-related Fc receptor (FcRn), reveal an increased degradation of IgG 34 and have therefore lower levels of serum IgG than wild-type mice. 32,63 Thus, it is likely that the failure of the FcRn-mediated protection of IgG from catabolism 63 might explain our findings of reduced IgG titers and neutralizing antibodies in CVB3-infected β2m−/− mice.
As demonstrated by in situ hybridization experiments the extent of myocardial infection in β2m−/− is seven times higher than in perforin−/− mice or C5/BL/6 mice. These observations support the concept that a reduced early antibody response in β2m−/− mice leads to a high cardiac viral load that cannot be eliminated by the invading natural killer cells, macrophages, and CD4+ T cells, thus inducing a persistent type of infection. Studies with MHC class II knockout mice also support an important role for antibodies in the control of CVB3 infection. It has been suggested that because of their failure to generate IgG and neutralizing antibodies, MHC class II knockout mice develop chronic CVB3 myocarditis in association with persistent heart muscle infection. 19 The extraordinary role of antibodies in virus clearance was further substantiated by the finding of high-viral organ titers and chronic infections of heart, liver, pancreas, and spleen in CVB3-infected B cell-deficient mice. 12 Moreover, previous studies provided evidence that treatment of CVB3-infected mice with polyclonal immunoglobulin protects mice against cardiac damage and inflammation and improves ventricular remodeling. 11,64
In summary, we have demonstrated the relevance of β2m-dependent immune-mediated mechanisms in the outcome of CVB3 myocarditis. From our data we conclude that the early limitation of the viral load by neutralizing IgG antibodies and the restriction of intramyocardial virus spread by IFN-γ, as demonstrated for C57BL/6 mice as well as for perforin−/− mice, reduce the extent of acute virus replication and inflammation, thus providing protective mechanisms against the development of chronic myocarditis. In contrast, the failure to eliminate infected myocytes during acute stages of infection as observed in β2m−/− mice and also in permissive immunocompetent mice 6 results in a persistent type of infection in association with ongoing chronic myocardial inflammation. Chronic inflammation because of persistent viral infection is detrimental for myocardial function because of expression of cardiotoxic cytokines, such as TNF-α and IL-1, 51,65 leading to cardiac fibrosis, the dominant hallmark of chronic myocarditis and dilated cardiomyopathy.
Acknowledgments
We thank Sandra Bundschuh and Carmen Ruoff for excellent technical assistance.
Footnotes
Address reprint requests to Dr. Karin Klingel, Department of Molecular Pathology, Institute for Pathology, University Hospital Tuebingen, Liebermeisterstr.8, 72076, Tuebingen, Germany. E-mail: karin.klingel@med.uni-tuebingen.de.
Supported by grants from the Federal Ministry of Education, Science Research, and Technology (grant 01KS9602); the Interdisciplinary Center of Clinical Research, Tuebingen; and the Deutsche Forschungsgemeinschaft (grant SCHN 628/1).
K. K. and J. J. S. contributed equally to this study.
References
- 1.Melnick JL: Enteroviruses: poliovirus, coxsackieviruses, echoviruses, and newer enteroviruses. Fields BN Knipe DM Howley P eds. Fields Virology. 1996:pp 655-712 Lippincott-Raven, New York
- 2.Martino TA, Liu P, Sole MJ: Viral infection and the pathogenesis of dilated cardiomyopathy. Circ Res 1994, 74:182-188 [DOI] [PubMed] [Google Scholar]
- 3.Baboonian C, Treasure T: Meta-analysis of the association of enteroviruses with human heart disease. Heart 1997, 78:539-543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kandolf R, Klingel K, Zell R, Selinka HC, Raab U, Schneider-Brachert W, Bültmann B: Molecular pathogenesis of enterovirus-induced myocarditis: virus persistence and chronic inflammation. Intervirology 1993, 35:140-151 [DOI] [PubMed] [Google Scholar]
- 5.Kandolf R, Sauter M, Aepinus C, Schnorr J-J, Selinka HC, Klingel K: Mechanisms and consequences of enterovirus persistence in cardiac myocytes and cells of the immune system. Virus Res 1999, 62:149-158 [DOI] [PubMed] [Google Scholar]
- 6.Klingel K, Hohenadl C, Canu A, Albrecht M, Seemann M, Mall G, Kandolf R: Ongoing enterovirus-induced myocarditis is associated with persistent heart muscle infection: quantitative analysis of virus replication, tissue damage, and inflammation. Proc Natl Acad Sci USA 1992, 89:314-318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andreoletti L, Hober D, Becquart P, Belaich S, Copin MC, Lambert V, Wattre P: Experimental CVB3-induced chronic myocarditis in two murine strains: evidence of interrelationships between virus replication and myocardial damage in persistent cardiac infection. J Med Virol 1997, 52:206-214 [DOI] [PubMed] [Google Scholar]
- 8.Koide H, Kitaura Y, Deguchi H, Ukimura A, Kawamura K, Hirai K: Genomic detection of enteroviruses in the myocardium-studies on animal hearts with coxsackievirus B3 myocarditis and endomyocardial biopsies from patients with myocarditis and dilated cardiomyopathy. Jpn Circ J 1992, 56:1081-1093 [DOI] [PubMed] [Google Scholar]
- 9.Loria RM: Host conditions affecting the course of coxsackievirus infections. Bendinelli M Friedman H eds. Coxsackieviruses: A General Update. 1988:pp 135-157 Plenum Press, New York
- 10.Rager-Zisman B, Allison AC: The role of antibody and host cells in the resistance of mice against infection by coxsackie B3 virus. J Gen Virol 1973, 19:329-338 [DOI] [PubMed] [Google Scholar]
- 11.Takada H, Kishimoto C, Hiraoka Y: Therapy with immunoglobulin suppresses myocarditis in a murine coxsackievirus B3 model. Antiviral and anti-inflammatory effects. Circulation 1995, 92:1604-1611 [DOI] [PubMed] [Google Scholar]
- 12.Mena I, Perry CM, Harkins S, Rodriguez F, Gebhard J, Whitton JL: The role of B lymphocytes in coxsackievirus B3 infection. Am J Pathol 1999, 155:1205-1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kew OM, Sutter RW, Nottay BK, McDonough MJ, Prevots DR, Quick L, Pallansch MA: Prolonged replication of a type 1 vaccine-derived poliovirus in an immunodeficient patient. J Clin Microbiol 1998, 36:2893-2899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Misbah SA, Spickett GP, Ryba PC, Hockaday JM, Kroll JS, Sherwood C, Kurtz JB, Moxon ER, Chapel HM: Chronic enteroviral meningoencephalitis in agammaglobulinemia: case report and literature review. J Clin Immunol 1992, 12:266-270 [DOI] [PubMed] [Google Scholar]
- 15.Geller TJ, Condie D: A case of protracted coxsackie virus meningoencephalitis in a marginally immunodeficient child treated successfully with intravenous immunoglobulin. J Neurol Sci 1995, 129:131-133 [DOI] [PubMed] [Google Scholar]
- 16.Chow LH, Beisel KW, McManus BM: Enteroviral infection of mice with severe combined immunodeficiency. Evidence for direct viral pathogenesis of myocardial injury. Lab Invest 1992, 66:24-31 [PubMed] [Google Scholar]
- 17.Kandolf R, Ameis D, Kirschner P, Canu A, Hofschneider PH: In situ detection of enteroviral genomes in myocardial cells by nucleic acid hybridization: an approach to the diagnosis of viral heart disease. Proc Natl Acad Sci USA 1987, 84:6272-6276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Henke A, Huber S, Stelzner A, Whitton JL: The role of CD8+ T lymphocytes in coxsackievirus B3-induced myocarditis. J Virol 1995, 69:6720-6728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leipner C, Borchers M, Merkle I, Stelzner A: Coxsackievirus B3-induced myocarditis in MHC class II-deficient mice. J Hum Virol 1999, 2:102-114 [PubMed] [Google Scholar]
- 20.Kägi D, Ledermann B, Burki K, Zinkernagel RM, Hengartner H: Molecular mechanisms of lymphocyte-mediated cytotoxicity and their role in immunological protection and pathogenesis in vivo. Annu Rev Immunol 1996, 14:207-232 [DOI] [PubMed] [Google Scholar]
- 21.Lowin B, Hahne M, Mattmann C, Tschopp J: Cytolytic T-cell cytotoxicity is mediated through perforin and Fas lytic pathways. Nature 1994, 370:650-652 [DOI] [PubMed] [Google Scholar]
- 22.Ramsay AJ, Ruby J, Ramshaw IA: A case for cytokines as effector molecules in the resolution of virus infection. Immunol Today 1993, 14:155-157 [DOI] [PubMed] [Google Scholar]
- 23.Ruby J, Ramshaw I: The antiviral activity of immune CD8+ T cells is dependent on interferon-gamma. Lymphokine Cytokine Res 1991, 10:353-358 [PubMed] [Google Scholar]
- 24.Huber SA: Autoimmunity in myocarditis: relevance of animal models. Clin Immunol Immunopathol 1997, 83:93-102 [DOI] [PubMed] [Google Scholar]
- 25.Sanna PP, Burton DR: Role of antibodies in controlling viral disease: lessons from experiments of nature and gene knockouts. J Virol 2000, 74:9813-9817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kägi D, Ledermann B, Bürki K, Seiler P, Odermatt B, Olsen KJ, Podack ER, Zinkernagel RM, Hengartner H: Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature 1994, 369:31-37 [DOI] [PubMed] [Google Scholar]
- 27.Walsh CM, Matloubian M, Liu CC, Ueda, Kurahara CG, Christensen JL, Huang MT, Young JD, Ahmed R, Clark WR: Immune function in mice lacking the perforin gene. Proc Natl Acad Sci USA 1994, 91:10854-10858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koller BH, Marrack P, Kappler JW, Smithies O: Normal development of mice deficient in beta 2M, MHC class I proteins, and CD8+ T cells. Science 1990, 248:1227-1230 [DOI] [PubMed] [Google Scholar]
- 29.Zijlstra M, Bix M, Simister NE, Loring JM, Raulet DH, Jaenisch R: Beta 2-microglobulin deficient mice lack CD4–8+ cytolytic T cells. Nature 1990, 344:742-746 [DOI] [PubMed] [Google Scholar]
- 30.Bendelac A, Killeen N, Littman DR, Schwartz RH: A subset of CD4+ thymocytes selected by MHC class I molecules. Science 1994, 263:1774-1778 [DOI] [PubMed] [Google Scholar]
- 31.Coles MC, Raulet DH: Class I dependence of the development of CD4+ CD8− NK1.1+ thymocytes. J Exp Med 1994, 180:395-399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghetie V, Hubbard JG, Kim JK, Tsen MF, Lee Y, Ward ES: Abnormally short serum half-lives of IgG in beta 2-microglobulin-deficient mice. Eur J Immunol 1996, 26:690-696 [DOI] [PubMed] [Google Scholar]
- 33.Junghans RP, Anderson CL: The protection receptor for IgG catabolism is the beta2-microglobulin-containing neonatal intestinal transport receptor. Proc Natl Acad Sci USA 1996, 93:5512-5516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Israel EJ, Wilsker DF, Hayes KC, Schoenfeld D, Simister NE: Increased clearance of IgG in mice that lack beta 2-microglobulin: possible protective role of FcRn. Immunology 1996, 89:573-578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kandolf R, Hofschneider PH: Molecular cloning of the genome of a cardiotropic Coxsackie B3 virus: full-length reverse-transcribed recombinant cDNA generates infectious virus in mammalian cells. Proc Natl Acad Sci USA 1985, 82:4818-4822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pasch A, Küpper JH, Wolde A, Kandolf R, Selinka HC: Comparative analysis of virus-host cell interactions of haemagglutinating and non-haemagglutinating strains of coxsackievirus B3. J Gen Virol 1999, 80:3153-3158 [DOI] [PubMed] [Google Scholar]
- 37.Hahn S, Gehri R, Erb P: Mechanism and biological significance of CD4-mediated cytotoxicity. Immunol Rev 1995, 146:57-79 [DOI] [PubMed] [Google Scholar]
- 38.Ramsingh AI, Lee WT, Collins DN, Armstrong LE: T cells contribute to disease severity during coxsackievirus B4 infection. J Virol 1999, 73:3080-3086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kägi D, Seiler P, Pavlovic J, Ledermann B, Bürki K, Zinkernagel RM, Hengartner H: The roles of perforin- and Fas-dependent cytotoxicity in protection against cytopathic and noncytopathic viruses. Eur J Immunol 1995, 25:3256-3262 [DOI] [PubMed] [Google Scholar]
- 40.Gebhard JR, Perry CM, Harkins, Lane T, Mena I, Asensio VC, Campbell IL, Whitton JL: Coxsackievirus B3-induced myocarditis: perforin exacerbates disease, but plays no detectable role in virus clearance. Am J Pathol 1998, 153:417-428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Watanabe-Fukunaga R, Brannan CI, Itoh N, Yonehara S, Copeland NG, Jenkins NA, Nagata S: The cDNA structure, expression, and chromosomal assignment of the mouse Fas antigen. J Immunol 1992, 148:1274-1279 [PubMed] [Google Scholar]
- 42.Feuerstein GZ, Young PR: Apoptosis in cardiac diseases: stress- and mitogen-activated signaling pathways. Cardiovasc Res 2000, 45:560-569 [DOI] [PubMed] [Google Scholar]
- 43.Henke A, Launhardt H, Klement K, Stelzner A, Zell R, Munder T: Apoptosis in coxsackievirus B3-caused diseases: interaction between the capsid protein VP2 and the proapoptotic protein siva. J Virol 2000, 74:4284-4290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peng T, Sadusky T, Li Y, Coulton GR, Zhang H, Archard LC: Altered expression of Bag-1 in Coxsackievirus B3 infected mouse heart. Cardiovasc Res 2001, 50:46-55 [DOI] [PubMed] [Google Scholar]
- 45.Mall G, Klingel K, Albrecht M, Seemann M, Rieger P, Kandolf R: Natural history of Coxsackievirus B3-induced myocarditis in ACA/Sn mice: viral persistence demonstrated by quantitative in situ hybridization histochemistry. Eur Heart J 1991, 12:S121-S123 [DOI] [PubMed] [Google Scholar]
- 46.Klingel K, Rieger P, Mall G, Selinka HC, Huber M, Kandolf R: Visualization of enteroviral replication in myocardial tissue by ultrastructural in situ hybridization: identification of target cells and cytopathic effects. Lab Invest 1998, 78:1227-1237 [PubMed] [Google Scholar]
- 47.Huber SA, Mortensen A, Moulton G: Modulation of cytokine expression by CD4+ T cells during coxsackievirus B3 infections of BALB/c mice initiated by cells expressing the gamma delta + T-cell receptor. J Virol 1996, 70:3039-3044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Opavsky MA, Penninger J, Aitken K, Wen WH, Dawood F, Mak T, Liu P: Susceptibility to myocarditis is dependent on the response of alpha/beta T lymphocytes to coxsackieviral infection. Circ Res 1999, 85:551-558 [DOI] [PubMed] [Google Scholar]
- 49.Kimura T, Griffin DE: The role of CD8(+) T cells and major histocompatibility complex class I expression in the central nervous system of mice infected with neurovirulent Sindbis virus. J Virol 2000, 74:6117-6125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guidotti LG, Ishikawa T, Hobbs MV, Matzke B, Schreiber R, Chisari FV: Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity 1996, 4:25-36 [DOI] [PubMed] [Google Scholar]
- 51.Seko Y, Takahashi N, Yagita H, Okumura K, Yazaki Y: Expression of cytokine mRNAs in murine hearts with acute myocarditis caused by coxsackievirus B3. J Pathol 1997, 183:105-108 [DOI] [PubMed] [Google Scholar]
- 52.Young HA, Hardy KJ: Role of interferon-gamma in immune cell regulation. J Leukoc Biol 1995, 58:373-381 [PubMed] [Google Scholar]
- 53.Leipner C, Grun K, Borchers M, Stelzner A: The outcome of coxsackievirus B3-(CVB3-) induced myocarditis is influenced by the cellular immune status. Herz 2000, 25:245-248 [DOI] [PubMed] [Google Scholar]
- 54.Das G, Sheridan S, Janeway CA, Jr: The source of early IFN-gamma that plays a role in Th1 priming. J Immunol 2001, 167:2004-2010 [DOI] [PubMed] [Google Scholar]
- 55.Mailliard RB, Egawa S, Cai Q, Kalinska A, Bykovskaya SN, Lotze MT, Kapsenberg ML, Storkus WJ, Kalinski P: Complementary dendritic cell-activating function of CD8+ and CD4+ T cells: helper role of CD8+ T cells in the development of T helper type 1 responses. J Exp Med 2002, 195:473-483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Peterson KE, Stromnes I, Messer R, Hasenkrug K, Chesebro B: Novel role of CD8(+) T cells and major histocompatibility complex class I genes in the generation of protective CD4(+) Th1 responses during retrovirus infection in mice. J Virol 2002, 76:7942-7948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vikingsson A, Pederson K, Müller D: Altered kinetics of CD4+ T cell proliferation and interferon-gamma production in the absence of CD8+ T lymphocytes in virus-infected beta2-microglobulin-deficient mice. Cell Immunol 1996, 173:261-268 [DOI] [PubMed] [Google Scholar]
- 58.Henke A, Zell R, Ehrlich G, Stelzner A: Expression of immunoregulatory cytokines by recombinant coxsackievirus B3 variants confers protection against virus-caused myocarditis. J Virol 2001, 75:8187-8194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Horwitz MS, Krahl T, Fine C, Lee J, Sarvetnick N: Protection from lethal coxsackievirus-induced pancreatitis by expression of gamma interferon. J Virol 1999, 73:1756-1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Spriggs MK, Koller BH, Sato T, Morrissey PJ, Fanslow WC, Smithies O, Voice RF, Widmer MB, Maliszewski CR: Beta 2-microglobulin-, CD8+ T-cell-deficient mice survive inoculation with high doses of vaccinia virus and exhibit altered IgG responses. Proc Natl Acad Sci USA 1992, 89:6070-6074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Franco MA, Greenberg HB: Role of B cells and cytotoxic T lymphocytes in clearance of and immunity to rotavirus infection in mice. J Virol 1995, 69:7800-7806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Taylor SF, Cottey RJ, Zander DS, Bender BS: Influenza infection of beta 2-microglobulin-deficient (beta 2m−/−) mice reveals a loss of CD4+ T cell functions with aging. J Immunol 1997, 159:3453-3459 [PubMed] [Google Scholar]
- 63.Christianson GJ, Brooks W, Vekasi S, Manolfi EA, Niles J, Roopenian SL, Roths JB, Rothlein R, Roopenian DC: Beta 2-microglobulin-deficient mice are protected from hypergammaglobulinemia and have defective antibody responses because of increased IgG catabolism. J Immunol 1997, 159:4781-4792 [PubMed] [Google Scholar]
- 64.Weller AH, Hall M, Huber SA: Polyclonal immunoglobulin therapy protects against cardiac damage in experimental coxsackievirus-induced myocarditis. Eur Heart J 1992, 13:115-119 [DOI] [PubMed] [Google Scholar]
- 65.Bryant D, Becker L, Richardson J, Shelton J, Franco F, Peshock R, Thompson M, Giroir B: Cardiac failure in transgenic mice with myocardial expression of tumor necrosis factor-alpha. Circulation 1998, 97:1375-1381 [DOI] [PubMed] [Google Scholar]