

Abstract
The structure of DNA breaks in early necrosis was analyzed and compared with apoptotic DNA degradation using in vivo and cell culture models. Early necrosis (1 hour after cell death) was produced in vivo by the freezing-thawing of rat thymus and in cell culture of Jurkat cells. Apoptosis was induced in the same cell types using dexamethasone for thymus and staurosporine for Jurkat cells. Selective detection of double-strand DNA breaks with blunt ends was performed by in situ ligation. Blunt-ended breaks bearing 5′ phosphates were detected in apoptotic but not in early necrotic cells. Pretreatment of apoptotic and necrotic tissue with Klenow enzyme with or without added dNTPs reduced all 3′ or 5′ overhangs to blunt ends. Subsequent in situ ligation with blunt-ended probes revealed no 3′ overhangs in necrotic cells. However double-strand cuts with 5′ overhangs were abundant in necrotic DNA. 5′ Overhangs were also detected in apoptotic cells. Presence of exclusively 5′ overhangs in early necrosis with absence of a variety of possible DNA ends, suggests the existence of a specific orderly mechanism of DNA degradation.
Discrimination between apoptotic and necrotic cell death is important for evaluation of the effects of anti-cancer drugs, as well as for study of the molecular biology of various diseases of immune system, liver, brain, and heart, in which cell death plays a prominent role. Apoptotic process is intimately connected to the orderly DNA fragmentation, in which the key apoptotic proteases (caspases) initiate specific DNA cleavage. 1,2 In necrosis, DNA is usually not cut in an orderly manner, which is explained by the activation of nonspecific lysosomal nucleases 3 and evidenced by a smear in electrophoresis. These differences in the structure of DNA damage are frequently used to discriminate between apoptosis and necrosis. However mechanistic boundaries between apoptosis and necrosis are becoming more and more vague. A recent report demonstrated the occurrence of the apoptotic ladder-type DNA fragmentation in the early necrosis of thymocytes, 4 and showed that DNA degradation in necrosis can also be systematic. We decided to investigate DNA damage in necrosis using criteria different from the detection of a ladder-type DNA fragmentation, because the nonrandom character of DNA damage can manifest as a preferential 3′→5′ or 5′→3′ exonuclease activity.
Disintegration of cellular membranes in necrosis and the infiltration of necrotic tissues by macrophages and other cell types eventually leads to the involvement of multiple enzymatic activities in DNA degradation creating random DNA cleavage. For this reason we focused on the early stages of DNA degradation in necrosis in vivo and in cell culture models.
We analyzed necrotic and apoptotic DNA degradation in situ in cells and tissues using a combination of direct labeling techniques such as terminal dUTP nick-end labeling (TUNEL), Klenow enzyme-based labeling, and a recently introduced in situ ligation. 5 The in situ ligation visualizes double-strand DNA breaks that are either blunt-ended or possess 3′ overhangs. When modified, it also permits the selective detection of both 3′→5′ and 5′→3′ exonuclease activities in situ via labeling of 3′ and 5′ overhangs of all lengths. The method relies on ligase-based attachment of double-stranded DNA probes with specific ends to the ends of DNA in tissue sections. The ligation of the probe occurs only when an exposed 5′ phosphate is present in cellular DNA. In its original form, the technique specifically detects blunt ends and short 3′ overhangs that are characteristic for DNA cleaved by the two major apoptotic nucleases, DNase I and caspase-activated deoxyribonuclease. 5-7 When Klenow enzyme pretreatment of cells is used before in situ ligation to either fill up 3′ overhangs or to cut 5′ overhangs, it reduces both to the blunt ends. This expands the utility of in situ ligation for the detection of all 3′ or 5′ overhangs.
We compared early necrosis (1 hour after instantaneous cell death induced by freezing-thawing) and apoptosis, which developed in rat thymus and in culture of Jurkat cells after 18 to 24 hours of treatment by dexamethasone or staurosporine. We demonstrate that blunt-ended and 3′-protruding DNA ends are present exclusively in apoptotic cells, whereas 5′ overhangs are generated in both apoptotic and early necrotic cells. The selective generation of 5′ overhangs in cells at the early stage of necrosis indicates a 3′→5′ exonuclease activity. Presence of 5′ overhangs in both apoptosis and necrosis suggests that the mechanism that generates this type of DNA damage cannot be fully explained by the activity of apoptosis-specific nucleases. We hypothesize that this damage could be attributed to the ubiquitous 3′→5′ proofreading exonuclease activity associated with cellular polymerases.
Materials and Methods
Apoptotic Cells
Jurkat cells (human leukemic T-cell lymphoblasts) were grown in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum, 50 U/ml penicillin, 50 μg/ml streptomycin, and 2 mmol/L glutamine at 37°C in humidified air atmosphere with 5% CO2. To induce apoptosis, the cells plated at 0.5 to 1 × 106 cells/ml in complete medium were treated with 1 μmol/L of staurosporine (Roche Molecular Biochemicals, Indianapolis, IN) a potent inhibitor of various protein kinases, for 18 hours at 37°C. The cells were washed in ice-cold phosphate-buffered saline (PBS), and fixed in ice-cold methanol.
Necrotic Cells
A cell culture dish with Jurkat cells was partially submerged into liquid nitrogen for 15 seconds. The application was repeated twice and 60 minutes later the cells were washed in ice-cold PBS and fixed in ice-cold methanol.
Apoptotic Tissue
Sprague-Dawley rats (150 g) were injected subcutaneously with 6 mg/kg of dexamethasone (Sigma, St. Louis, MO) dissolved in 30% dimethyl sulfoxide in water. Twenty-four hours later, the animals were sacrificed, and the thymus was removed and immediately fixed in 4% paraformaldehyde. After 18 hours in paraformaldehyde, the tissue was placed in 70% ethanol, and treated with graded alcohols to 100% ethanol, placed in chloroform overnight, and then embedded in paraffin.
Necrotic Tissue
Sprague-Dawley rats were anesthetized using 2.5% isoflurane and artificially ventilated. After thoracotomy, a cotton swab was placed into liquid nitrogen and then applied directly on the thymus for 3 seconds. The application was repeated twice, and the incision was closed. Sixty minutes later, the animal was sacrificed, and the thymus was removed and processed, as stated above.
Probes for in Situ Ligation
Two oligonucleotide probes were used: Probe 1 forms blunt-ended hairpin. It was used for in situ ligation and detected DNA breaks with no protruding ends. Biotin was incorporated into the probe by chemical insertion of biotin-triethylene glycol phosphoramidite (Glen Research, Sterling, VA) directly into the oligonucleotide backbone (Serologicals, Gaithersburg, MD). The sequence of the probe was: 5′ GCG CTA GAC C5*G GTC TAG CGC 3′. 5* represents biotin-triethylene glycol spacer (Glenn Research). Probe 2 forms single 3′ dA overhang. It was used in one series only to detect DNA breaks with single 3′ dT overhangs present in apoptotic cells in dexamethasone-treated thymus (Figure 1A) ▶ . The sequence of the probe was: 5′ GCG CTA GAC C5*G GTC TAG CGC A 3′. 5* represents biotin-triethylene glycol spacer (Glenn Research). Probes were detected in reaction with streptavidin-fluorescein isothiocyanate conjugate (probe 1) or streptavidin-Texas Red conjugate (probe 2) (Vector Laboratories, Burlingame, CA) (see In Situ Ligation).
Figure 1.
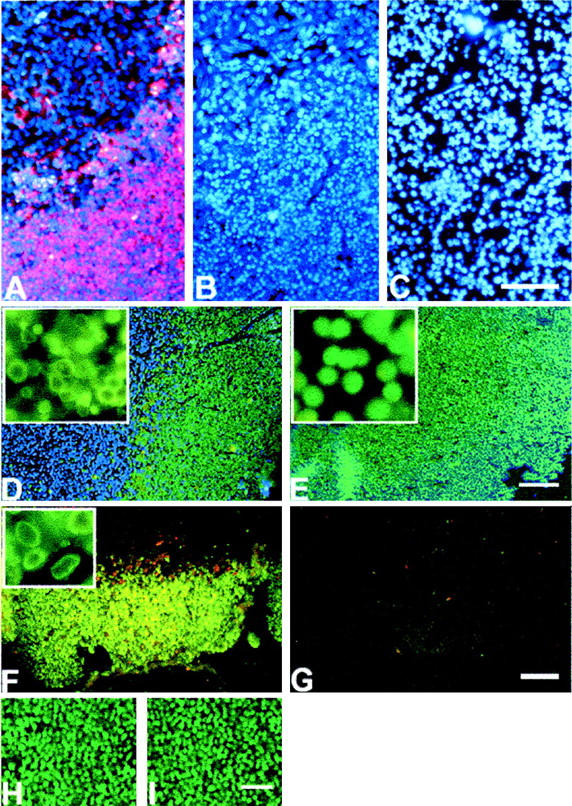
A–C: Comparison of apoptotic (A, B) and necrotic (C) thymus stained by DAPI (blue fluorescence) and by in situ ligation using oligonucleotide probes with single dA overhangs (red fluorescence). A and C are dual-stained images, B is a single-stained (DAPI) image and is provided for the easy comparison of nuclear morphology with C. D and E: Comparison of apoptotic and necrotic thymus stained by TUNEL. D: Apoptotic thymus (green fluorescence, TUNEL staining; blue fluorescence, DAPI staining). E: Necrotic thymus (green fluorescence, TUNEL staining; blue fluorescence, DAPI staining). Insets show high-magnification images of apoptotic and necrotic nuclei labeled by TUNEL (side of the inset, 80 μm). Strong positive staining is present in both cases. However necrotic thymus is uniformly positive with the loss of the characteristic pattern of cell death seen in glucocorticoid-induced apoptosis. F and G: Comparison of apoptotic and necrotic thymus using in situ ligation with blunt-ended probes detecting double-strand DNA breaks bearing 5′ phosphates. F: Apoptotic thymus, blunt ends detection. G: Necrotic thymus, blunt ends detection. Note that in situ ligation does not detect double-strand DNA breaks bearing 5′ phosphates in necrotic thymus. H and I: DNA nicks relegation in necrotic thymus using T4 DNA ligase. H: Necrotic thymus TUNEL-stained before T4 ligase pretreatment; I: the same thymus after T4 ligase pretreatment. DNA nicks do not contribute to the strong positive staining of necrotic thymus by TUNEL. No change in TUNEL signal intensity occurred after treatment of necrotic tissue with T4 DNA ligase to seal DNA nicks. Apoptosis was induced in the thymus by an intraperitoneal injection of 6 mg/kg of dexamethasone. Necrosis was induced in the thymus by freezing with liquid nitrogen. Scale bars: 200 μm (C, E); 300 μm (G); 150 μm (I).
In Situ Ligation
Tissue Section
In situ ligation using the hairpin-shaped oligo probe was performed on 6-μm-thick tissue sections. Sections were deparaffinized with xylene, rehydrated in graded alcohol concentrations, briefly washed in water, and then treated with proteinase K (50 μg/ml) in PBS for 15 minutes. Sections were rinsed with water, and then 80 μl of the ligation buffer without the probe was applied to each section for 15 minutes, to ensure even saturation of the section with ligation buffer before addition of the enzyme and the probe. The ligation buffer contained 66 mmol/L of Tris-HCl, pH 7.5, 5 mmol/L MgCl2, 0.1 mmol/L dithioerythritol, 1 mmol/L ATP, and 15% polyethylene glycol (8000 MW; Sigma). The buffer was aspirated, and the full ligation mix containing the ligation buffer with the hairpin probe (35 μg/ml) and T4 DNA ligase (250 U/ml, Roche Molecular Biochemicals) was applied to the sections. In a mock control reaction solution, an equal volume of 50% glycerol in water was substituted for T4 DNA ligase. Sections were covered with glass coverslips, and placed in a humidified box for 16 hours at room temperature (23°C). They were then washed in water three times for 20 minutes. Streptavidin-fluorescein isothiocyanate conjugate (for blunt-ended oligonucleotide probe 1) or streptavidin-Texas Red conjugate (for single 3′ dA overhang oligonucleotide probe 2) (Vector Laboratories) were added to the sections at 4 μg/ml in 50 mmol/L sodium bicarbonate, 15 mmol/L sodium chloride, pH 8.2, for 45 minutes. Sections were washed in the same buffer three times for 30 minutes, and then in water for 20 minutes. The sections were then counterstained with the DNA-binding dye 4,6-diamidino-2-phenylindole (DAPI) (1 μg/ml), mounted in Vectashield (Vector Laboratories), and observed via fluorescence microscopy.
For Klenow enzyme pretreatment, to fill in 5′ overhangs, a 20-μl solution containing: 70 mmol/L Tris-HCl, pH 7.5, 70 mmol/L MgCl2, 10 mmol/L dithiothreitol, 2.5 mmol/L of dATP, dGTP, dCTP, dTTP, and 5 U of Klenow enzyme (Roche Molecular Biochemicals) was added to sections after proteinase K treatment and washing. Sections were incubated at 37°C for 30 minutes, then washed in water two times for 10 minutes each, and were labeled by in situ ligation using blunt-ended probes.
To remove 3′ overhangs using the Klenow enzyme, dNTPs were omitted from the reaction mixture. Sections were incubated with the reaction mixture for 30 minutes at 37°C, and after washing were labeled by in situ ligation using blunt-ended probes. Klenow enzyme does not destroy blunt ends generated as a result of trimming of 3′ DNA overhangs in tissue sections. We verified this by treating 6-μm-thick sections of paraformaldehyde-fixed normal bovine adrenal with AluI restriction endonuclease (30 U/section, 8 hours, 37°C), producing massive amounts of blunt-ended DNA cuts. These sections were then treated with Klenow enzyme without added dNTPs, and no change in signal occurred in in situ ligation with blunt-ended probes. Measurements taken by the digital MicroMAX camera system containing Peltier cooled RTE/CCD-1300-Y/HS (Princeton Instruments, Inc, Trenton, NJ) before Klenow enzyme treatment without added dNTPs and measurements taken 30 minutes and 1 hour after addition of enzyme (5 U/section, 37°C) were the same (results not shown).
Cultured Cells
To remove 3′ overhangs using the Klenow enzyme, cells were treated as described for tissue sections (see Tissue Sections), except for proteinase K treatment that was substituted by a 15-minute wash with PBS.
To fill up overhangs using labeled Texas Red dUTP, a 20-μl solution containing 70 mmol/L Tris-HCl, pH 7.5, 70 mmol/L MgCl2, 10 mmol/L dithiothreitol, 2.5 mmol/L of dATP, dGTP, dCTP, 1.75 mmol/L dTTP, and 0.75 mmol/L of Texas Red dUTP (Molecular Probes, Eugene, OR) and 5 U of Klenow enzyme (Roche Molecular Biochemicals) was added to cells after 15 minutes of washing in PBS. The cells were incubated at 37°C for 30 minutes, and washed in water two times for 10 minutes each.
The signal was acquired using Olympus IX-70 fluorescent microscope equipped with a MicroMax digital camera system (Princeton Instruments, Inc.) containing an RTE/CCD-1300-Y/HS array cooled by a Peltier device. Background fluorescence because of nonspecific binding of Texas Red dUTP and streptavidin-fluorescein isothiocyanate to cells was measured in control slides containing cells incubated in reaction solutions without added enzymes (Klenow fragment and T4 DNA ligase), and subtracted from experimental slide images using MetaMorph 4.1 program (Advanced Scientific, Inc., Fort Lauderdale, FL).
TUNEL
TUNEL staining for detection of free 3′OH groups in cellular DNA in tissue sections was performed using the ApoTaq Fluorescein kit for indirect immunofluorescence (Serologicals), using the standard technique recommended by the manufacturer. Terminal transferase-based detection in cell culture was performed using the incorporation of biotin-16 dUTP as described elsewhere. 8 The signal was detected using a streptavidin-fluorescein-isothiocyanate conjugate.
Results
Evaluation of Apoptotic and Early Necrotic Models
In our experiments, we used rat thymus 24 hours after administration of glucocorticoid, a model for apoptosis well established previously 9 and used by us before. 5,10,11 Within 24 hours such intervention results in massive cell death of the cortical lymphocytes, whereas the core areas remain primarily unaffected. 5 Staining of tissue sections by in situ ligation or by TUNEL produces a characteristic pattern of doughnut-shaped apoptotic areas with uniform cell death and central unstained zones with no dead cells. 5,10 Control untreated rat thymus also has small amounts of apoptotic cells in cortical areas, which is consistent with the observation that ∼1% of thymic cells show features of cell death in postnatal animals. 12 We have previously visualized these cells using both in situ ligation and TUNEL. 5 For direct comparison with apo-ptotic thymus, we used necrotic thymus where nonprogrammed cell death was induced by repeated freezing-thawing of thymic tissue in vivo in an anesthetized animal. Such intervention is frequently used to induce fast and massive necrosis in cryotherapy of many tissues. 13-15
We verified thymic apoptosis using in situ ligation with oligo probes with single 3′ nucleotide extension a well-established test for this system. 5,10,11 Figure 1A ▶ demonstrates the results of the staining and shows characteristic clustering of dying cells in the cortical area of thymus (red fluorescence) with unaffected central part (blue fluorescence). Necrotic thymus was completely negative (Figure 1C) ▶ , which corresponds with the previously reported results that oligo probe with 3′ single nucleotide overhang does not react with necrotic tissue. 5,10 The comparison of the nuclei in apoptosis and necrosis using the fluorescent dye DAPI revealed differences in their morphology (Figure 1, B and C) ▶ . The nuclei in cortical apoptotic areas were condensed whereas necrotic nuclei in the same areas were bigger because of intracellular edema and swelling.
Detection of Multiple Free 3′ OH Groups in DNA of Necrotic Cells
Sections of apoptotic and necrotic thymus, consecutive to those used in in situ ligation, were used in experiments with TUNEL (Figure 1, D and E) ▶ . TUNEL detects 3′ OH groups at the ends of single-strand and double-strand DNA cuts.
Multiple breaks with terminal 3′ hydroxyls were detected in both apoptotic and necrotic cells in thymus (Figure 1, D and E) ▶ . However in apoptosis, the pattern of TUNEL labeling was identical to the ligase-based staining shown in Figure 1A ▶ and confined to the cortical regions undergoing uniform extensive cell death (Figure 1D ▶ , green fluorescence). The characteristic apoptotic ring-type chromatin aggregation on the nuclear membrane was also clearly observed (Figure 1D ▶ , inset). In necrotic thymus, TUNEL staining revealed DNA breaks with terminal 3′ OH evenly distributed throughout the section and not confined to the cortical regions as in apoptosis (Figure 1E) ▶ . The nuclear morphology was also different demonstrating a solid, uniform nuclear staining without a characteristic for apoptosis ring-type DNA condensation (Figure 1E ▶ , inset).
Presence of Blunt-Ended DNA Breaks in Apoptotic but Not Early Necrotic Cells
In situ ligation with blunt-ended probes detected blunt-ended breaks in apoptotic, but not early necrotic cells (Figure 1, F and G) ▶ . The nuclei labeled by in situ ligation concentrated in the core areas of dexamethasone-treated thymus and had apoptotic morphology containing blunt-ended breaks in the areas of chromatin condensation on the nuclear membrane (Figure 1F ▶ , inset). Early necrotic tissue was completely negative showing complete absence of blunt-ended breaks at this stage of necrosis (Figure 1G) ▶ . The breaks detected by the technique in apoptotic cells are 5′ phosphorylated and can be attributed to the activity of specific apoptotic nucleases, such as caspase-activated deoxyribonuclease or DNase I-like endonucleases. Both groups of enzymes in their action on cellular chromatin were shown to produce primarily blunt-ended 5′ phosphorylated DNA breaks. 6,7,16,17
Estimation of the Types of DNA Damage Labeled by TUNEL in Early Necrosis
We performed additional series of experiments in an attempt to characterize what types of DNA breaks were labeled by terminal transferase in necrotic thymus. Terminal transferase is capable of labeling several types of DNA breaks, which include DNA nicks, 3′ and 5′ overhangs, and blunt ends. 18,19 We have already shown that blunt ends were not present in necrotic cells at the time point investigated. The next series investigated nicks as possible candidates for necrotic DNA damage.
Resealing of DNA Nicks Using T4 DNA Ligase Does Not Reduce the TUNEL Signal in Necrotic Tissue
To determine whether nicks were responsible for the positive TUNEL signal seen in early necrosis, we used T4 DNA ligase to repair them. T4 DNA ligase is known to efficiently reseal DNA nicks of a 3′OH to 5′ PO4 configuration, 20 which could be generated by DNase I-like endonuclease. 21 In our experiment, we compared consecutive sections of necrotic tissue with and without preincubation with T4 ligase. Such treatment should reseal nicks in DNA in the sections. However, a 24-hour preincubation of necrotic tissue with ligase did not reduce the fluorescent TUNEL signal (Figure 1, H and I) ▶ , which indicates that DNA nicks were not responsible for the TUNEL staining we observed in necrotic thymocytes.
Elsewhere we demonstrated that double-strand DNA breaks with single nucleotide 3′ overhangs are very scarce in necrosis, although they are abundant in apoptosis 5,10 (Figure 1, A and C) ▶ . Although we confirmed this data in our present experiments but possibility still remained that longer 3′ overhangs can be more characteristic for necrotic cells. The other remaining types of DNA cleavage were 5′ overhangs. We tested the presence of both candidate types of DNA cleavage in our experiments with necrotic thymus model and in cell culture model of necrosis.
Detection of 5′ but Not 3′ Overhangs in Necrotic Thymus
To investigate whether 3′ or 5′ extensions were responsible for the TUNEL labeling of early necrotic tissue, we performed experiments using a combination of in situ ligation with pretreatment of the cells with Klenow enzyme. Enzymatic pretreatment, depending on its conditions, reduced either 3′ or both 3′ and 5′ overhangs to blunt ends. After each type of overhang is converted to a blunt end, they can be detected by in situ ligation, using a blunt-ended probe.
To detect 3′ overhangs of various lengths, we treated sections of necrotic thymus with Klenow enzyme in the absence of added triphosphates. Because of the 3′→5′ exonuclease activity of the Klenow enzyme, which is 400% higher for single-stranded than for double-stranded DNA, this reduced 3′ overhangs present in cellular DNA to blunt ends. 22 Although excessive pretreatment with Klenow enzyme could in theory result in more advanced DNA degradation with generation of 5′ overhangs from blunt-ended DNA, we confirmed (see Materials and Methods) that in our experimental conditions, this type of reaction does not occur. This may be because of extensive crosslinking existing between strands of double-stranded DNA as a result of paraformaldehyde fixation. Because no blunt ends were initially present in our model of necrosis, the 3′ overhangs reduced to blunt ends by Klenow enzyme pretreatment are the only possible source of blunt ends under these conditions. The in situ ligation procedure was used to detect the newly generated blunt ends originating from 3′ overhangs.
To detect both 5′ and 3′ overhangs of various lengths, we treated sections of necrotic thymus with Klenow polymerase in the presence of all four nucleotides. Because of the 5′→3′ polymerase activity of the Klenow enzyme, this filled in all 5′ overhangs in cellular DNA producing blunt ends. 23 In addition, because of 3′→5′ exonuclease activity of the enzyme 3′ overhangs, if present, should be also reduced to blunt ends. Because no blunt ends were initially present in necrotic tissue, presence of either of 3′ or 5′ overhangs would result in generation of blunt ends detectable by in situ ligation with blunt-ended probes.
Results of the experiments are shown in Figure 2, A and B ▶ . Klenow enzyme treatment without added nucleotides did not generate any blunt ends in necrotic thymus (Figure 2B) ▶ . Furthermore, we have previously shown that blunt ends were not detected in necrotic thymus before Klenow enzyme treatment (Figure 1G) ▶ . This indicates that no 3′ overhangs were present in early necrosis. The data correlates with our other results, demonstrating that short 3′ overhangs were not detected in necrosis by in situ ligation of oligonucleotides with single base 3′ extensions. 5,10 In contrast, treatment of the sections with a reaction mixture containing both Klenow enzyme and nucleotides resulted in the generation of new blunt ends created by filled up 5′ overhangs (Figure 2A) ▶ . These results indicate that 5′ overhangs were already present in DNA of necrotic cells at detectable concentrations 1 hour after the onset of necrosis. Presence of 5′ overhangs and absence of blunt ends and 3′ overhangs in early necrosis indicates a nonrandom character of DNA damage, which could not be mediated by nonspecific lysosomal nucleases. The lysosome-mediated massive destruction of DNA in necrosis is thought to be a late rather than an early event. 24 Besides, lysosomal nucleases should produce a random mixture of different breaks, not limited to 5′ overhangs. 25,26
Figure 2.
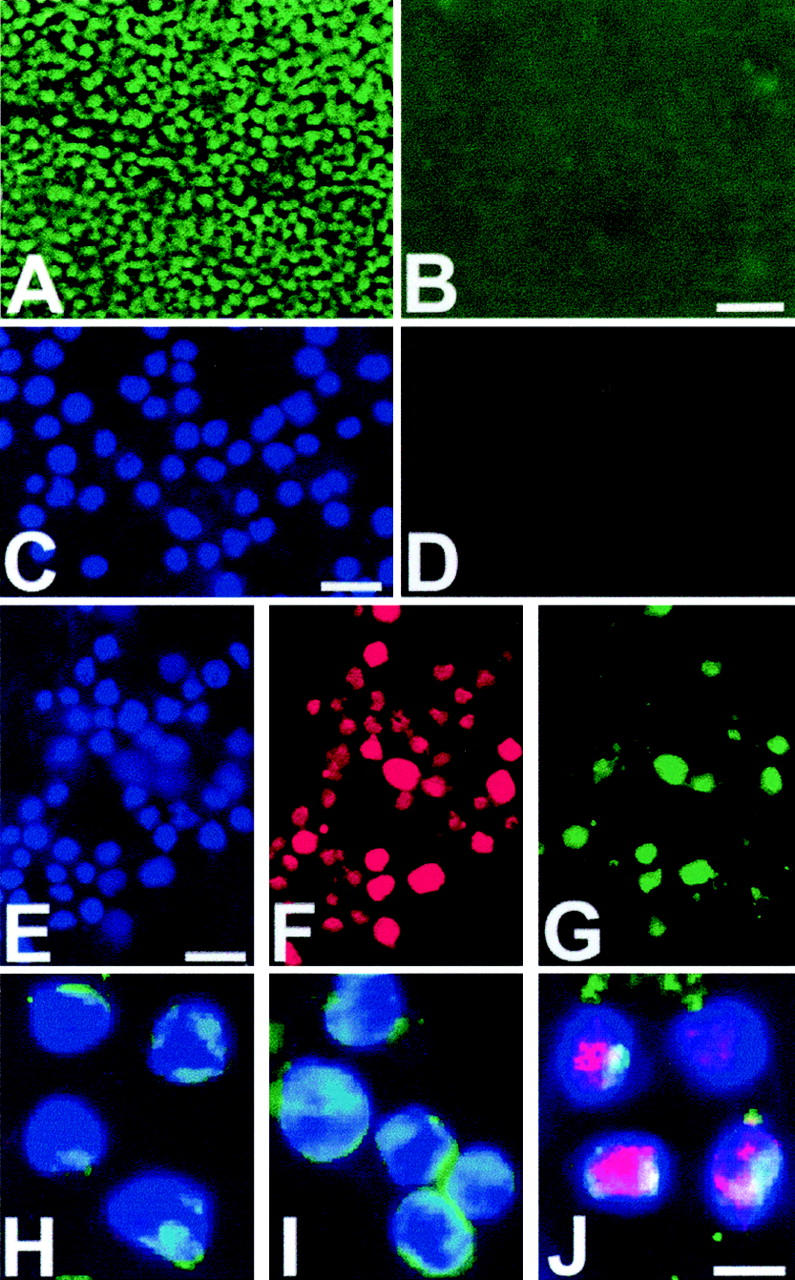
Detection of 5′ overhangs but not 3′ overhangs in necrotic and apoptotic thymus and Jurkat cells in culture. A: Detection of 5′overhangs: necrotic thymus is labeled by in situ ligation (green fluorescence) with the use of a blunt-ended probe after treatment with Klenow enzyme with addition of dNTPs. B: Necrotic thymus labeled by in situ ligation with the use of a blunt-ended probe after treatment with Klenow enzyme without addition of dNTPs; no 3′overhangs detected. C: DAPI-stained necrotic Jurkat cells. D: Same cells labeled by in situ ligation with the use of a blunt-ended probe after treatment with Klenow enzyme without addition of dNTPs; no 3′overhangs detected in necrotic Jurkat cells. E, F, G: Detection of 5′overhangs. G: Necrotic Jurkat cells are labeled by in situ ligation (green fluorescence) with the use of a blunt-ended probe after treatment with Klenow enzyme with addition of dNTPs. F: Necrotic Jurkat cells are labeled by filling-in reaction using Klenow enzyme and Texas Red dUTP (red fluorescence). E: DAPI staining (blue fluorescence). H, I, J: Apoptotic thymocytes: detection of blunt ends (green fluorescence) (H), 3′overhangs (green fluorescence) (I), and 5′ overhangs (pink) (J). DAPI staining (blue fluorescence). Apoptosis was induced in Jurkat cells by the addition of staurosporine. Apoptosis was induced in the thymus by an intraperitoneal injection of 6 mg/kg dexamethasone. Necrosis was induced in the thymus by freezing with liquid nitrogen. Scale bars: 50 μm (B); 30 μm (C, E); 10 μm (J).
Detection of 5′ but Not 3′ Overhangs in Necrotic Jurkat Cells
We repeated detection of blunt ends, 3′ and 5′ overhangs in a cell culture model of nonprogrammed cell death using Jurkat cells (the immortalized T cell line) (see Materials and Methods). This cell culture model is useful for the analysis of apoptotic and necrotic DNA cleavage because it excludes the exogenous effects produced by leukocytes.
Results of these experiments are illustrated by Figure 2; C to G ▶ . To increase signal in these series a fluorescent nucleotide, Texas Red-dUTP, was added to the nucleotide mix (see Materials and Methods). This permitted direct visualization of the newly synthesized DNA strands (red fluorescence) in addition to a subsequent detection of the newly generated blunt ends by the blunt-ended hairpin oligo probes (green fluorescence).
Klenow enzyme pretreatment without added nucleotides did not generate any blunt-ended DNA breaks in necrotic Jurkat cells (Figure 2, C and D) ▶ . A very small number of solitary cells in the initial cell culture was undergoing apoptosis and was labeled by a blunt-ended probe (not shown). Similar to our experiments with thymic tissue, we did not detect blunt-ended DNA breaks in necrotic Jurkat cells before Klenow enzyme pretreatment (not shown). The negative signal after pretreatment indicated also the absence of 3′ overhangs in necrotic Jurkat cells (Figure 2D) ▶ .
To detect both 5′ and 3′overhangs of various lengths, we treated necrotic Jurkat cells with Klenow polymerase in the presence of all four nucleotides. This filled in all 5′ overhangs in cellular DNA producing blunt ends. 23 This was visualized by both incorporation of the fluorescent Texas-dUTP and by in situ ligation to the newly generated blunt ends. Results of this experiment are illustrated by Figure 2; E, F, and G ▶ . The images show that most Jurkat cells in freeze-thaw cell culture were stronger labeled by Texas Red dUTP than by in situ ligation. Both signals were more concentrated in the internal nuclear areas or were evenly distributed throughout the nuclei (Figure 2; E, F, and G) ▶ . The intensity of labeling significantly varied from cell to cell but invariably the majority of the labeled cells had much stronger red fluorescence (filling in 5′ overhangs) and relatively weak green fluorescent signal (in situ ligation to blunt ends). The much weaker in situ ligation signal is explained by the fact that positioning red and green fluorophores in close proximity creates fluorescence resonance energy transfer that quenches green fluorescence and increases red fluorophore output. 27 Additionally, the green fluorescence is initially weaker than red because the hairpin probes place a single fluorophore at the end of each detectable break, whereas the filling in reaction could incorporate multiple fluorophores into the newly synthesized DNA. Despite the variability of signal between different cells in culture in case of 5′ overhangs detection (Figure 2; E, F, and G) ▶ the signal is much stronger than in 3′ overhangs detection (Figure 2, C and D) ▶ .
Detection of Both 5′ and 3′ Overhangs in Apoptotic Jurkat Cells
We labeled of 5′ and 3′ overhangs in the culture of apoptotic staurosporine-treated Jurkat cells using the same approach as in the case of necrotic cells. The results of the experiments are illustrated by Figure 2; H, I, and J ▶ . Multiple blunt ends were initially present in apoptotic cells and were detected before Klenow enzyme pretreatment (Figure 2H) ▶ . The signal significantly increased after pretreatment with Klenow enzyme without dNTPs (Figure 2I) ▶ indicating detection of 3′overhangs of various lengths. In both cases (blunt ends and 3′overhangs) labeling occurred in the areas of apoptotic chromatin condensation on the nuclear membrane. 5′ Overhangs were also detected in apoptotic cells (Figure 2J) ▶ . Unlike blunt ends and 3′ overhangs, they concentrated primarily in the central areas of the nuclei (Figure 2; H, I, and J) ▶ .
Discussion
Our data demonstrates that DNA cleavage in early necrosis is characterized by selective generation of 5′ overhangs, but not 3′ overhangs or blunt ends. Instead, all three types of breaks can be detected in apoptotic cell death, indicating a considerably wider variety of DNA breaks created in this process.
The source of 5′ overhangs is open to speculation. This type of cleavage is too selective to warrant a nonenzymatic explanation. Because no new enzymes are synthesized in necrosis, the creation of these overhangs can only be attributed to the pre-existing enzymatic activity.
Endonucleases are unlikely candidates for early necrotic DNA cleavage. These enzymes can initiate DNA cleavage within the DNA molecule generating 1 to 4 nucleotides long 3′ and 5′ overhangs. 6,7,16,17 Very short single base 3′ and 5′ overhangs are characteristic for DNase I-like apoptotic nuclease and caspase activated deoxyribonuclease (DFF40) cleavage, respectively. 5,7 However, mechanisms of action of all these enzymes preclude them from exclusive generation of long 5′ overhangs without simultaneous production of blunt ends and 3′ overhangs. 6,7,16,17
The sole presence of 5′ overhangs indicates participation of 3′→5′ exonucleolytic activity. The 3′→5′ exonuclease would selectively remove nucleotides of one DNA strand starting with a pre-existing nick, creating a single strand gap. Two strands will separate when a nick is encountered on the opposite strand. Any 3′ overhangs will be rapidly eliminated, leaving only long 5′ overhangs. The two major sources of 3′→5′ exonuclease activity constitutively present in cells are lysosomal and proofreading exonucleases.
Freezing-thawing could destabilize lysosomal membranes releasing lysosomal enzymes, which would rapidly disintegrate DNA. However, if DNA is digested by a mixture of lysosomal nucleases a variety of different types of DNA breaks and not only 5′ overhangs should be present. Furthermore the same types of breaks are present in apoptosis, in which lysosomes are not affected.
Exonucleolytic proofreading is another possible candidate for early necrotic DNA damage. Both polymerases δ and ε have specific 3′→5′ exonuclease activities 28 that can selectively produce 5′ overhangs. Polymerase α 29 have also been associated with a 3′→5′ exonuclease, which was later purified from thymus cells. 30 The polymerase complexes responsible for the replication of nuclear DNA are sited in the midzonal nucleoplasmic regions, apart from peripherally located blocks of condensed chromatin. 31,32
It is not possible to discern at this stage if nuclear polymerases or the other enzymes play a role in early necrotic DNA degradation, described in this article. In addition, we cannot exclude that several sources could contribute to 3′→5′ exonucleolytic activity. It also not clear whether the same mechanism is responsible for creation of 5′ overhangs in apoptotic cells.
The blunt-end detection by in situ ligation presented in this report refers only to 5′ phosphorylated breaks, because the breaks bearing 5′ OH groups, such as those produced by DNase II-like nucleases cannot be detected by ligation. 33 However the Klenow enzyme filling-in reaction labels both types irrespective of the 5′ functional group. The stronger signal obtained with this type of labeling compared to the simultaneous detection of the newly created blunt ends as a result of this reaction might indicate the presence of breaks with 5′OH groups. Such results are not surprising because 5′ groups are rapidly exchanged by cellular kinases and phosphatases. More detailed analysis of this phenomenon was hampered by the absence of an in situ technique allowing simultaneous detection of both 5′ OH and 5′ PO4 DNA breaks. The first such technique was recently introduced by us 34 and will be used in future analysis of this phenomenon.
We obtained a much more uniform signal visualizing 5′ overhangs in thymic tissue staining (Figure 2A) ▶ compared to cell cultures (Figure 2; E to G) ▶ . One probable explanation is in the differences in cell and tissue fixation, as we have used a crosslinking paraformaldehyde in tissue section experiments and noncrosslinking ethanol in cell culture experiments. The cut DNA fragments, which contain overhangs, could be lost in cell processing and washing unless they were cross linked to the cellular matrix, as in the case of paraformaldehyde fixation. Previously the short chromatin fragments produced in apoptotic DNA degradation were shown to be very loosely attached to the matrix. 35 In such a case, the choice of the fixative can be critical in reducing background and obtaining uniform strong signal.
Previous attempts to discriminate between apoptotic and necrotic cell death were made with the assumption of a more orderly character of DNA cleavage in programmed cell death compared to necrotic DNA degradation. In fact, we and the other investigators have demonstrated that various apoptotic executioner nucleases selectively produce specific types of DNA breaks. 5,7 Necrotic DNA degradation is expected to contain a random mixture of all possible configurations of DNA breaks generated by multiple nonspecific lysosomal nucleases. 3 Although this is a probable scenario for later necrosis, our data presented in this report demonstrates that this is not the case at the early stages of nonprogrammed cell death.
Our data correlates with results indicating that DNA polymerase I-based labeling that preferentially detects 5′ overhangs, 36 efficiently mark early necrotic cells. 37 TUNEL, which in addition to 5′ overhangs can label 3′ overhangs and blunt ends, was shown to label both apoptotic and necrotic cells. 37 Although it remains to be seen how selective this type of DNA break is for early necrosis, it is important to note that this finding suggests that necrosis (at least its freezing-thawing model) like apoptosis, is a nonrandom event, with specific nucleolytic activity in its early stages.
At later times of necrosis development we and the other investigators have detected blunt-ended breaks in necrotic Wilms’ tumor, and in placenta. 5,38 The generation of blunt-ended DNA breaks at later times in necrosis, with a complete absence of such breaks in early time periods, indicates involvement of additional mechanisms in late necrotic DNA degradation.
Footnotes
Address reprint requests to Dr. Vladimir V. Didenko, Department of Neurosurgery, Baylor College of Medicine, 2002 Holcombe Blvd., Bldg. 109, Rm. 204, Houston, TX 77030. E-mail: vdidenko@bcm.tmc.edu.
Supported by the National Cancer Institute, National Institutes of Health (grant R01 CA78912 to D. S. B.), the Texas Higher Education Coordinating Board (grant 004949-054 to D. S. B., V. V. D.), the DeBakey Medical Foundation (to V. V. D.), the Baylor College of Medicine (to V. V. D.), the Taub Foundation, the Henry J. N. Taub Fund for Neurosurgical Research, the George A. Robinson IV Foundation, the Blanche Greene Estate Fund of the Pauline Sterne Wolff Memorial Foundation, and the Seigo Arai and Koppelman Funds of the Neurological Research Foundation.
References
- 1.Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S: A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 1998, 391:43-50 [DOI] [PubMed] [Google Scholar]
- 2.Sakahira H, Enari M, Nagata S: Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature 1998, 391:96-99 [DOI] [PubMed] [Google Scholar]
- 3.DeDuve C, Wattiaux R: Functions of lysosomes. Annu Rev Physiol 1966, 28:435-492 [DOI] [PubMed] [Google Scholar]
- 4.Dong Z, Saikumar P, Weinberg JM, Venkatachalam MA: Internucleosomal DNA cleavage triggered by plasma membrane damage during necrotic cell death. Involvement of serine but not cysteine proteases. Am J Pathol 1997, 151:1205-1213 [PMC free article] [PubMed] [Google Scholar]
- 5.Didenko VV, Hornsby PJ: Presence of double-strand breaks with single-base overhangs in cells undergoing apoptosis but not necrosis. J Cell Biol 1996, 135:1369-1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lutter LC: Deoxyribonuclease I produces staggered cuts in the DNA of chromatin. J Mol Biol 1977, 117:53-69 [DOI] [PubMed] [Google Scholar]
- 7.Widlak P, Li P, Wang X, Garrard WT: Cleavage preferences of the apoptotic endonuclease DFF40 (caspase-activated DNase or nuclease) on naked DNA and chromatin substrates. J Biol Chem 2000, 275:8226-8232 [DOI] [PubMed] [Google Scholar]
- 8.Gavrieli Y, Sherman Y, Ben-Sasson SA: Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol 1992, 119:493-501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wyllie AH: Glucocorticoid-induced thymocytes apoptosis is associated with endogenous endonuclease activation. Nature 1980, 284:555-556 [DOI] [PubMed] [Google Scholar]
- 10.Didenko VV, Tunstead JR, Hornsby PJ: Biotin-labeled hairpin oligonucleotides: probes to detect double-strand breaks in DNA in apoptotic cells. Am J Pathol 1998, 152:897-902 [PMC free article] [PubMed] [Google Scholar]
- 11.Didenko VV, Boudreaux DJ, Baskin DS: Substantial background reduction in ligase-based apoptosis detection using newly designed hairpin oligonucleotide probes. BioTechniques 1999, 27:1130-1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Metcalf D: The thymus and lymphopoiesis. Good RA Gabrielson AE eds. Thymus in Immunobiology. Structure, Function, and Role in Disease. 1964:pp 150-182 Harper and Row, New York
- 13.Tacke J: Thermal therapies in interventional MR imaging. Cryotherapy Neuroimaging Clin N Am 2001, 11:759-765 [PubMed] [Google Scholar]
- 14.Conzemius MG, Brown TD, Zhang Y, Robinson RA: A new animal model of femoral head osteonecrosis: one that progresses to human-like mechanical failure. J Orthop Res 2002, 20:303-309 [DOI] [PubMed] [Google Scholar]
- 15.Gazzaniga S, Bravo A, Goldszmid SR, Maschi F, Martinelli J, Mordoh J, Wainstok R: Inflammatory changes after cryosurgery-induced necrosis in human melanoma xenografted in nude mice. J Invest Dermatol 2001, 116:664-671 [DOI] [PubMed] [Google Scholar]
- 16.Lutter LC: Precise location of DNase I cutting sites in the nucleosome core determined by high resolution gel electrophoresis. Nucleic Acids Res 1979, 6:41-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sollner-Webb B, Melchior W, Jr, Felsenfeld G: DNase I, DNase II and staphylococcal nuclease cut at different, yet symmetrically located, sites in the nucleosome core. Cell 1978, 14:611-627 [DOI] [PubMed] [Google Scholar]
- 18.Grosse F, Manns A: Terminal deoxyribonucleotidyl transferase. Burrell MM eds. Enzymes of Molecular Biology. 1993:pp 95-105 Humana Press, Totowa [DOI] [PubMed]
- 19.Smith A, Haaf T: DNA nicks and increased sensitivity of DNA to fluorescence in situ end labeling during functional spermiogenesis. BioTechniques 1998, 25:496-502 [DOI] [PubMed] [Google Scholar]
- 20.Maunders MJ: DNA and RNA ligases. Burrell MM eds. Enzymes of Molecular Biology. 1993:pp 213-230 Humana Press, Totowa [DOI] [PubMed]
- 21.Weir F: Deoxyribonuclease I and II. Burrell MM eds. Enzymes of Molecular Biology. 1993:pp 7-16 Humana Press, Totowa
- 22.Eun H-M: Enzymology Primer for Recombinant DNA Technology. 1996:p 361 Academic Press, San Diego
- 23.Maunders MJ: DNA polymerases. Burrell MM eds. Enzymes of Molecular Biology. 1993:pp 17-25 Humana Press, Totowa
- 24.Majno G, Joris I: Apoptosis, oncosis and necrosis. An overview of cell death. Am J Pathol 1995, 146:3-15 [PMC free article] [PubMed] [Google Scholar]
- 25.Holtzman E: Lysosomes. 1989:p 2 Plenum Press, New York
- 26.Van Dyck JM, Wattiaux R: Distribution intracellulaire de l’exonuclease acide dans le foie de rat. Eur J Biochem 1968, 7:13-20 [DOI] [PubMed] [Google Scholar]
- 27.Didenko VV: DNA probes using fluorescence resonance energy transfer (FRET): designs and applications. BioTechniques 2001, 31:1106-1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goodman MF, Bloom LB: Proofreading exonucleases: error correcting during DNA replication. Linn SM Lloyd RS Roberts RJ eds. Nucleases. 1993:pp 235-262 Cold Spring Harbor Laboratory Press, New York
- 29.Wang TS: Eukaryotic DNA polymerases. Annu Rev Biochem 1991, 60:513-552 [DOI] [PubMed] [Google Scholar]
- 30.Bialek G, Grosse F: An error-correcting proofreading exonuclease-polymerase that copurifies with DNA-polymerase-α-primase. J Biol Chem 1993, 268:6024-6033 [PubMed] [Google Scholar]
- 31.Bensch KG, Tanaka S, Hu SZ, Wang TS, Korn D: Intracellular localization of human DNA polymerase alpha with monoclonal antibodies. J Biol Chem 1982, 257:8391-8396 [PubMed] [Google Scholar]
- 32.Fry M, Loeb LA: Intracellular localization of nuclear polymerases. Animal Cell DNA Polymerases. 1986:pp 109-110 CRC Press, Boca Raton
- 33.Maunders MJ: MM Burrell eds. DNA and RNA ligases (EC 6.5.1.1, EC 6.5.1.2, and EC 6.5.1.3). Enzymes of Molecular Biology. 1993:pp 213-230 Humana Press, Totowa [DOI] [PubMed]
- 34.Didenko VV, Ngo H, Baskin DS: In situ detection of double-strand DNA breaks with terminal 5′OH groups. Didenko VV eds. In Situ Detection of DNA Damage: Methods and Protocols. 2002:pp 153-159 Humana Press, Totowa [DOI] [PubMed]
- 35.Arends MJ, Morris RG, Wyllie AH: Apoptosis. The role of the endonuclease. Am J Pathol 1990, 136:593-608 [PMC free article] [PubMed] [Google Scholar]
- 36.Eun H-M: Enzymology Primer for Recombinant DNA Technology. 1996:p 352 Academic Press, San Diego
- 37.Gold R, Schmied M, Giegerich G, Breitschopf H, Hartung HP, Toyka KV, Lassmann H: Differentiation between cellular apoptosis and necrosis by the combined use of in situ tailing and nick translation techniques. Lab Invest 1994, 71:219-225 [PubMed] [Google Scholar]
- 38.Al-Lamki RS, Skepper JN, Loke YW, King A, Burton GJ: Apoptosis in the early human placental bed and its discrimination from necrosis using the in-situ DNA ligation technique. Hum Reprod 1998, 13:3511-3519 [DOI] [PubMed] [Google Scholar]