

Abstract
Recognition of immune complexes in glomeruli by activator Fcγ receptors (FcγRI and FcγRIII) is an important step in the development of glomerulonephritis. The low-affinity receptor (FcγRIII) has previously been shown to be important in passive heterologous immune complex glomerulonephritis. However, most forms of human glomerulonephritis involve an active immune response, and the relative importance of FcγRI (high-affinity receptor) and FcγRIII in an active model of glomerulonephritis is not known. We have now studied accelerated nephrotoxic nephritis in FcγRIII−/− mice and FcγRI/III double-deficient mice, and compared them with matched wild-type controls and FcRγ chain-deficient (FcRγ−/−) mice. Mice were immunized against sheep IgG and injected with sheep anti-mouse glomerular basement membrane antibody 5 days later. Both FcγRI/III double-deficient mice and FcRγ−/− mice were strongly protected from renal injury. In contrast, FcγRIII−/− mice developed substantial nephritis, although there was a dose-dependent partial protection from glomerular crescents and thrombosis. Despite this histological protection from injury, the macrophage infiltrate was not reduced, implying a dissociation of macrophage accumulation from activation in the absence of activatory FcγRIII. Therefore, both FcγRI and FcγRIII play a role in this active model of glomerulonephritis, because both had to be deficient to protect markedly from disease.
Most forms of human glomerulonephritis are associated with deposition of immune complexes in glomeruli. Immune complexes containing IgG communicate with cells via Fcγ receptors, an important link between the innate and adaptive immune systems. Increasing evidence implicates Fcγ receptors in immune-mediated inflammation 1 and the recognition of immune complexes deposited in glomeruli by cellular activator Fcγ receptors is an important step in the development of glomerulonephritis. 2-5 Blockade of this pathway is, therefore, an attractive therapeutic target but design of rational therapy requires a better understanding of the exact role of the various types of Fcγ receptor.
Murine phagocytic effector cells express three different classes of IgG receptors, FcγRI, FcγRII, and FcγRIII. FcγRI and FcγRIII are hetero-oligomeric activatory receptors in which a ligand-binding α chain is associated with a signal-transducing γ-chain. The γ-chain is required for receptor assembly and triggering of effector functions including phagocytosis, antibody-dependent cytotoxicity, and release of inflammatory mediators. 6 These functions are mediated through an immunoreceptor tyrosine activation motif within the γ-chain. FcγRIII is a low-affinity activatory Fcγ receptor that interacts with IgG1, IgG2a, and IgG2b immune complexes and is widely expressed on circulating leukocytes, especially macrophages and polymorphs. 7,8 In contrast, FcγRI is a high-affinity activatory receptor that interacts predominantly with monomeric IgG2a. 9 The third receptor class for IgG, FcγRII is a single α-chain receptor and contains an immunoreceptor tyrosine inhibitory motif in the cytoplasmic domain. FcγRII has been shown to be a negative regulator of activatory Fcγ receptors I and III in immune complex-mediated inflammation. 10,11
Mice deficient in the FcRγ chain are protected from several forms of glomerulonephritis, including a spontaneous model of lupus nephritis and an active model of immune complex glomerulonephritis. 2-5 However, the FcRγ chain is the intracellular signaling subunit for both activator murine Fcγ receptors I and III as well as the high-affinity IgE receptor, 6 a collagen receptor on platelets, 12 the recently described paired immunoglobulin-like receptors, 13 and the natural killer receptor-P1. 14 Because FcRγ−/− mice lack several receptors, reduced glomerulonephritis in FcRγ−/− mice suggests, but does not conclusively prove, a role for activator Fcγ receptors in immune-mediated nephritis.
The development of mice lacking the α chain of specific Fcγ receptors has facilitated the study of the individual contributions of these receptors. 7,15,16 In a passive model of heterologous nephrotoxic nephritis, FcγRIII was found to be the most important receptor involved in neutrophil infiltration. 17 In this model, rabbit IgG directed against glomerular basement membrane was injected without preimmunization, leading to a glomerular neutrophil influx that resolved within 24 hours. This acute model depends on the interaction of the heterologous deposited glomerular antibody with Fcγ receptors, because the murine immune response does not develop this early in the disease. In contrast, most forms of human glomerulonephritis involve an autologous antibody response to exogenous or endogenous antigen with deposition of host immune complexes. We have therefore studied the model of accelerated nephrotoxic nephritis in which mice were preimmunized with sheep immunoglobulin and then given an injection of sheep anti-mouse glomerular basement membrane globulin. This binds to the glomerulus where it acts as a planted antigen to which mouse IgG becomes bound. This has been shown to be a T-cell-dependent model characterized by leukocyte infiltration, proteinuria, thrombosis of glomerular capillaries, glomerular crescent formation, and renal impairment. 18
We have used this model to elucidate the role of individual activator FcRγ receptors. Specifically we have studied the model in mice lacking FcγRIII or lacking both FcγRI/III and compared them with matched wild-type (WT) controls and mice lacking the common FcRγ chain.
Materials and Methods
Mice
FcRγ−/− mice were generated in the laboratory of Professor T. Saito, Chiba University, Chiba, Japan. 3 FcγRIII-deficient mice were generated as previously described. 7 FcRγ−/− mice were on a pure C57BL/6 background, and FcγRIII−/− mice were backcrossed to C57BL/6 for nine generations. Age- and sex-matched C57BL/6 controls were purchased from Harlan Ltd. (Bicester, UK). FcγRI/III−/− mice were on a hybrid C57BL/6/129/BALB/c background with a major contribution from C57BL/6 and were compared with a genetically matched control line derived from littermates. 15 All experiments were performed according to institutional guidelines.
Induction of Accelerated Nephrotoxic Nephritis
A sheep was immunized with murine glomerular lysate initially in complete and then incomplete Freund’s adjuvant at monthly intervals. A γ-globulin-enriched fraction of the serum was prepared by ammonium sulfate precipitation, after first heat-inactivating complement activity. The precipitate was then extensively dialyzed against phosphate-buffered saline (PBS), and finally sterile filtered before storing in aliquots at −70°C. The endotoxin content of the nephrotoxic serum measured using the BioWhittaker 1000-QCL LPS assay kit was less than 0.1 EU/ml (BioWhittaker, Walkersville, MD).
Mice were immunized intraperitoneally with 0.2 mg of sheep IgG in a 50:50 mix with complete Freund’s adjuvant (Sigma Chemical Co., St. Louis, MO). Five days later, the animals were injected with 5 or 10 mg of nephrotoxic serum via a tail vein. At time points from 2 hours to 26 days after nephrotoxic serum injection, mice were anesthetized with midazolam and fentanyl intraperitoneally and exsanguinated before harvesting the kidneys.
Histological Studies and Quantitative Immunofluorescence
Kidneys were fixed for 2 hours in Bouin’s solution, transferred to 70% ethanol, processed to paraffin, and stained with periodic acid-Schiff (PAS) reagent. Samples were assessed by an observer blinded to the experimental group. Glomerular thrombosis was assessed by scoring individual glomeruli for PAS-positive material as follows: grade 0, no PAS-positive material; grade 1, 0 to 25% of glomerular cross-section; grade 2, 25 to 50%; grade 3, 50 to 75%; grade 4, 75 to 100%. Fifty glomeruli were scored per point, and the mean glomerular thrombosis score was calculated for each mouse. Glomerular crescents were defined as glomeruli containing two or more layers of cells in Bowman’s space (50 glomeruli counted per point, converted to percentage crescents).
For immunofluorescence, kidneys were embedded in OCT (CellPath, Powys, UK), snap-frozen in isopentane cooled with liquid nitrogen, and stored at −70°C. Sections of 5 μm were fixed in acetone for 10 minutes. Fluorescein isothiocyanate-conjugated goat anti-mouse IgG (Fc-specific) and fluorescein isothiocyanate-conjugated monoclonal mouse anti-sheep IgG clone GT34 (Sigma Chemical Co.) were used. For quantitative immunofluorescence, blinded sections were examined at ×100 magnification using an Olympus BX4 fluorescence microscope (Olympus Optical, London, UK) and a Photonic Science Color Coolview camera (Photonic Science, Robertsbridge, UK). Samples from each experiment were measured together. The mean intensity of 20 glomeruli for each sample was recorded in arbitrary fluorescence units.
For immunoperoxidase staining for cell markers, kidneys were fixed for 4 hours in periodate lysine paraformaldehyde, washed overnight in 13% sucrose in PBS, and frozen in isopentane cooled with liquid nitrogen. Sections of 5 μm were stained for macrophages with FA11 (monoclonal rat anti-mouse CD68;, Serotec, Oxford, UK) in a three-layer immunoperoxidase technique as previously described. 19
Serum Creatinine, Albumin, and Albuminuria
Serum creatinine and albumin were measured in the Department of Chemical Pathology, Hammersmith Hospital. Serum albumin was analyzed using the bromocresol green method and serum creatinine was measured using an Olympus AU640 autoanalyzer. Individual 24-hour collections of urine were performed using metabolic cages, with free access to food and water. Albuminuria was assessed by radial immunodiffusion against a rabbit anti-mouse albumin as previously described. 19 Hemacombistix (Bayer Diagnostics, Newbury, UK) were used to assess proteinuria when metabolic cages were not available.
Measurement of the Murine Anti-Sheep IgG Immune Response
Serum mouse anti-sheep IgG levels were measured by enzyme-linked immunosorbent assay. 19 Briefly, enzyme-linked immunosorbent assay plates (Nunc Maxisorb; Fisher Scientific, Loughborough, UK) were coated overnight at 4°C with 100 μg/ml of sheep IgG (Sigma Chemical Co.). After blocking with 3% bovine serum albumin (ICN Biomedicals Inc., Aurora, OH) diluted serum samples were incubated for 1 hour at 37°C. For each experiment a range of serum dilutions was tested with a standard curve of a known positive sample. Goat anti-mouse IgG (Fc-specific) conjugated to alkaline phosphatase (Sigma Chemical Co.) was used as a secondary and the optical density at 405 nm was measured using p-nitrophenyl phosphate as the substrate (Sigma Chemical Co.).
Statistics
Results are expressed as mean ± SE. Nonparametric tests of significance were applied throughout. For comparing two groups, the Mann-Whitney U-test was used. For groups of three or more, the Kruskal-Wallis test was used, with Dunn’s multiple comparisons test (P values given are from Dunn’s test). GraphPad Prism (GraphPad Software, San Diego, CA) was used to analyze the data. Differences were considered significant when P was <0.05.
Results
FcRγ−/− Mice Were Strongly Protected from Nephritis, but FcγRIII−/− Mice Were Only Partially Protected in a Dose-Dependent Manner
Accelerated nephrotoxic nephritis was simultaneously induced in WT (n = 8), FcγRIII−/− (n = 8), and FcRγ−/− (n = 9) female mice, with 10 mg of nephrotoxic serum. All mice were sacrificed on day 7 after nephrotoxic serum injection, except for one WT mouse that was sacrificed on day 5 because of renal disease. WT and FcγRIII−/− mice developed glomerular thrombosis, glomerular crescents, and renal impairment whereas FcRγ−/− mice were completely protected (Figures 1 and 2 ▶ and Table 1 ▶ ). At this dose of nephrotoxic serum, there was no statistically significant difference between histological disease, renal impairment, or serum albumin between WT and FcγRIII−/− mice, suggesting the involvement of other FcRγ-associated receptors such as FcγRI in pathogenesis. Disease was then compared between WT (n = 9) and FcγRIII−/− (n = 12) mice using half the dose of nephrotoxic serum (5 mg) to day 8 after nephrotoxic serum injection. With this lower dose, partial protection of FcγRIII−/− mice from glomerular thrombosis and glomerular crescents was revealed (Figure 1 ▶ and Table 1 ▶ ). Despite less evidence of glomerular thrombosis or crescents, FcγRIII−/− mice had as many (experiment 1) or more (experiment 2) infiltrating macrophages than WT, indicating a dissociation of inflammatory cell infiltrate and the development of glomerular crescents and thrombosis.
Figure 1.
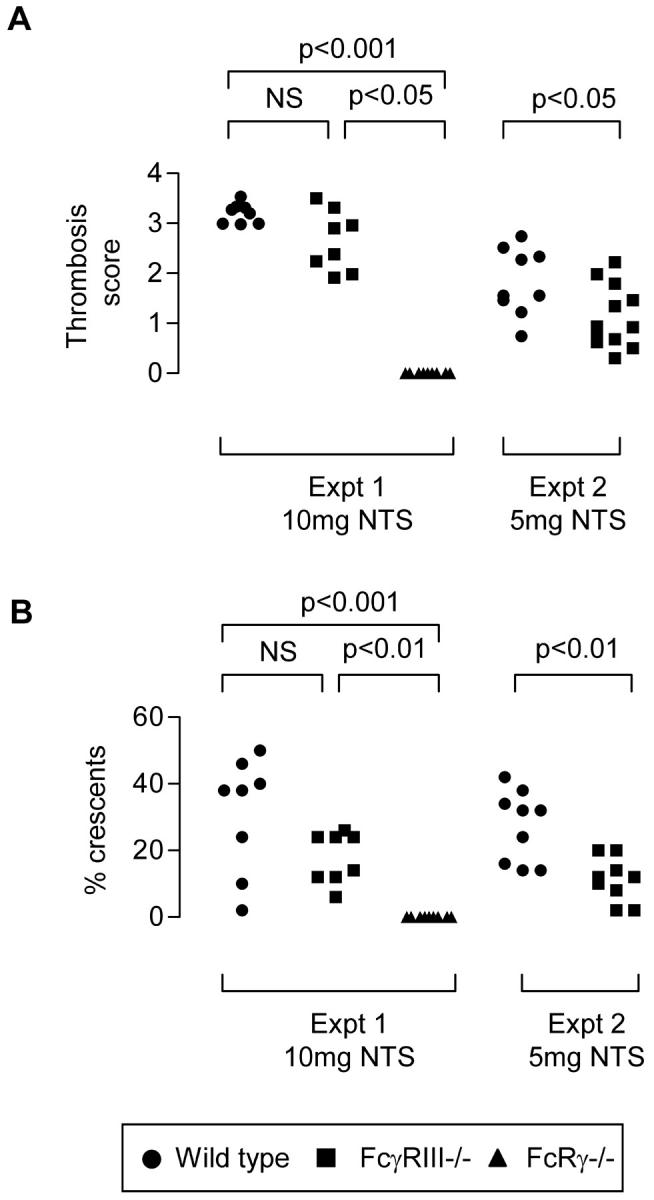
Glomerular thrombosis score (n = 50 glomeruli per point; A) and glomerular crescents (n = 50 glomeruli per point; B), day 7 after 10 mg of nephrotoxic serum (experiment 1) or 5 mg of nephrotoxic serum (experiment 2). FcγRIII−/− mice developed substantial disease that was only significantly attenuated compared with WT at the 5 mg dose. FcRγ−/− mice were strongly protected from disease.
Table 1.
Accelerated Nephrotoxic Nephritis in WT, FcγRIII−/−, and FcRγ−/− Mice
| WT control Controls | Experiment 1 | Experiment 2 | ||||
|---|---|---|---|---|---|---|
| WT | FcγRIII−/− | FcRγ−/− | WT | FcγRIII−/− | ||
| Dose of NTS (mg) | 0 | 10 | 10 | 10 | 5 | 5 |
| Days after NTS | N/A | 7* | 7 | 7 | 8 | 8 |
| Sex | m/f | f | f | f | f | f |
| Number of mice developing disease | 0/12 | 8/8 | 8/8 | 0/9 | 9/9 | 12/12 |
| % crescents | 0 ± 0 | 31 ± 6 | 18 ± 3 | 0 ± 0† | 27 ± 4 | 14 ± 2† |
| Glomerular thrombosis score | 0 ± 0 | 3.2 ± 0.1 | 2.7 ± 0.2 | 0 ± 0† | 1.8 ± 0.2 | 1.1 ± 0.2† |
| Macrophages/gcs | 0.1 ± 0.01 | 1.2 ± 0.2 | 1.7 ± 0.2 | 0.8 ± 0.4‡ | 1.0 ± 0.1 | 3.7 ± 0.7† |
| Serum creatinine (μmol/L) | 51 ± 2.0 | 166 ± 12 | 118 ± 18 | 50 ± 3† | 84 ± 11 | 69 ± 6 |
| Albuminuria(mg/24 hour) day 7 | 0.06 ± 0.001 | ND | ND | 7.3 ± 4 | 13 ± 2 | 14 ± 3 |
| Serum albumin (g/l) | 32 ± 1 | 19 ± 1 | 13 ± 0.8 | 30 ± 1‡ | 16 ± 1 | 13 ± 1 |
| Serum anti-sheep IgG (relative OD) | 0.09 | 0.88 ± 0.1 | 0.7 ± 0.1 | 0.86 ± 0.1 | 0.7 ± 0.1 | 1 ± 0.1† |
*One WT mouse was sacrificed at day 5.
†P <0.05 compared with WT and FcγRIII−/− (experiment 1) or WT (experiment 2).
‡P <0.05 compared with FcγRIII−/−, NS compared with WT.
N/A, not applicable; ND, albuminuria was not measured for these mice.
FcγRIII−/− Mice Were Not Protected from Early Neutrophil Infiltrate in Accelerated Nephrotoxic Nephritis
Before the development of glomerular thrombosis, there was an influx of neutrophils that peaked in the first 2 hours after nephrotoxic serum injection. The influx of neutrophils was compared between WT, FcγRIII−/−, andFcRγ−/− preimmunized mice 2 hours and 24 hours after 10 mg of nephrotoxic serum. WT and FcγRIII−/− mice both developed neutrophil infiltrates, but FcRγ−/− mice were protected (Figure 3A) ▶ . WT and FcγRIII−/− mice developed albuminuria in the first 24 hours after disease induction, whereas FcRγ−/− mice were completely protected (P < 0.05) (Figure 3B) ▶ . These results are in contrast to results previously obtained with the heterologous model. 17 To confirm those results using our model, 10 mg of nephrotoxic serum was injected without preimmunization and the glomerular neutrophil influx was quantitated at 2 hours. In this heterologous model, FcγRIII−/− mice were protected (P < 0.01) (Figure 3C) ▶ .
Figure 3.

A and B: Preimmunized mice were injected with 10 mg of nephrotoxic serum (NTS) and sacrificed after 2 or 24 hours. C: Naïve mice were injected with 10 mg of NTS and sacrificed after 2 hours. A: Glomerular neutrophil infiltration 2 and 24 hours after NTS (50 glomeruli counted per point). gcs, glomerular cross-section. B: Albuminuria in the first 24 hours after NTS injection. C: Glomerular neutrophil infiltration at 2 hours after NTS to nonimmunized mice. Nonimmunized FcγRIII−/− mice were protected from neutrophil infiltration, but preimmunized FcγRIII−/− mice were not protected.
Immune Responses of FcRγ−/− and FcγRIII−/− Mice
The immune response was assessed by quantitative immunofluorescence of deposited glomerular murine immunoglobulins at 2 hours and 24 hours after induction of disease and the end of the experiments (day 7). There was no significant difference between the groups. Data at 24 hours is shown in Figure 4 ▶ . The circulating mouse anti-sheep IgG antibody response was measured at the end of the experiments. The circulating anti-sheep IgG levels of FcγRIII−/− and FcRγ−/− mice measured by enzyme-linked immunosorbent assay were as strong as the WT mice (Table 1) ▶ . In addition, the binding of sheep IgG to the glomerular basement membrane was assessed by quantitative immunofluorescence for each experiment. No significant differences were found (data are shown for the 24-hour time point in Figure 4B ▶ ).
Figure 4.
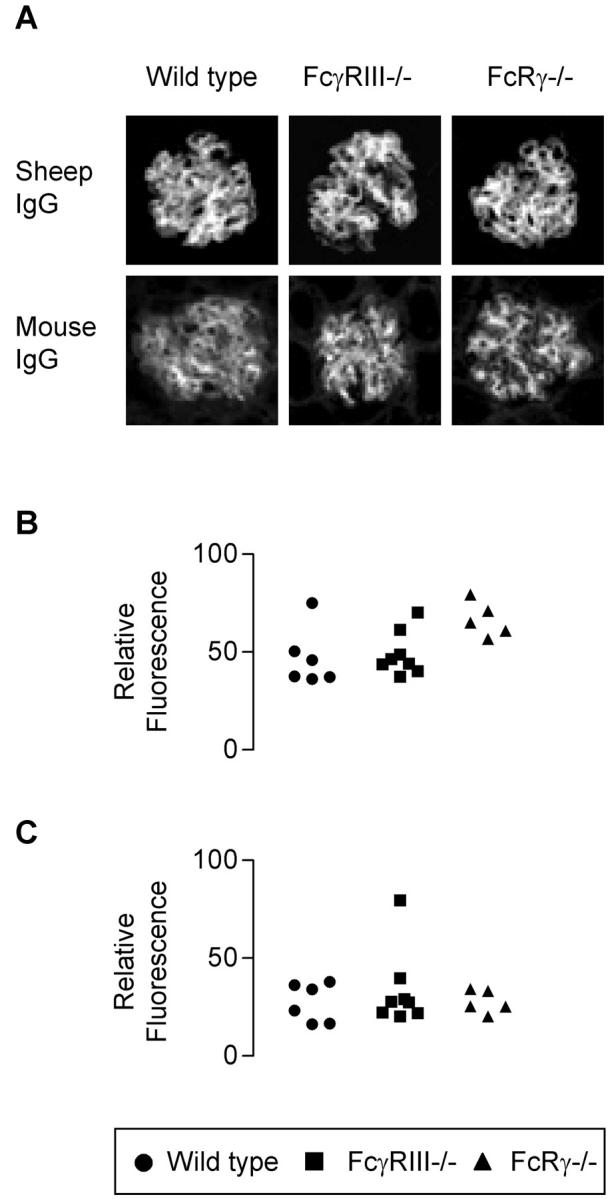
A: Representative glomeruli showing immunofluorescence for glomerular sheep and mouse IgG 24 hours after nephrotoxic serum injection. B: Quantitative immunofluorescence for glomerular sheep IgG. C: Mouse IgG deposition 24 hours after nephrotoxic serum injection. There was no significant difference between the groups.
FcRγ−/− Mice Developed Albuminuria and a Macrophage Influx in a Dose-Dependent Manner
Although FcRγ−/− mice were completely protected from glomerular thrombosis and glomerular crescents in our model, it was noted that after 10 mg of nephrotoxic serum a proportion of the mice developed a macrophage infiltrate and proteinuria at day 7 (Table 1) ▶ . This phenomenon was studied in more detail by measuring albuminuria at weekly intervals in preimmunized FcRγ−/− mice injected with 10 mg or 5 mg of nephrotoxic serum (Figure 5) ▶ . FcRγ−/− mice injected with 10 mg of nephrotoxic serum (especially females) had albuminuria at day 7 after nephrotoxic serum that later resolved, whereas those injected with 5 mg of nephrotoxic serum did not develop albuminuria at any time. It is not clear why the females had a greater propensity to develop albuminuria than the males, but it may relate to their smaller size.
Figure 5.

Weekly albuminuria measurements in FcRγ−/− preimmunized mice injected with 5 mg or 10 mg of nephrotoxic serum. Mice injected with 10 mg of nephrotoxic serum, especially females, had albuminuria at day 7 that resolved by day 26. Horizontal line represents the mean 24-hour albuminuria of a group of normal C57BL/6 mice.
FcγRI/III Double-Deficient Mice Were Protected from Accelerated Nephrotoxic Nephritis
As stated in the introduction, FcRγ−/− mice lack several receptors that require the FcRγ chain for a signaling subunit. These receptors include both activator Fcγ receptors, as well as several others such as the high-affinity IgE receptor (FcεRI). To confirm that protection of FcRγ−/− mice in this model was because of the lack of FcγRI and III, accelerated nephrotoxic nephritis was induced in male mice lacking both FcγRI and III (FcγRI/III−/−) (n = 5) and WT strain-matched controls (n = 6) using 10 mg of nephrotoxic serum. Mice were sacrificed at day 8 after nephrotoxic serum injection. FcγRI/III−/− mice were substantially protected from glomerular thrombosis (score, 0.5 ± 0.3) compared with WT (score, 2.4 ± 0.2) (P < 0.005). FcγRI/III−/− mice were also protected from glomerular crescents (percent crescents, 0.4 ± 0.1%) compared with WT (percent crescents, 5 ± 1.1%) (P < 0.005; Figure 6, A and B ▶ ). Representative histology is shown in Figure 7 ▶ . There was no significant difference in the macrophage infiltrate or proteinuria as assessed by urine dipstick at day 8, but this is consistent with previous findings with FcRγ−/− animals (Figure 6C) ▶ . Quantitation of deposited glomerular IgG showed no difference in deposited glomerular mouse or sheep IgG between the groups at day 8 after nephrotoxic serum. The enzyme-linked immunosorbent assay for total mouse IgG against sheep IgG showed higher levels in the FcγRI/III−/− mice at day 8 than WT animals.
Figure 6.

FcγRI/III−/− mice and WT controls at day 8 after induction of nephritis. A: Glomerular thrombosis score (n = 50 glomeruli scored per point). B: Percent glomerular crescents (n = 50 glomeruli). C: Macrophages per glomerular cross-section (n = 50 glomeruli per point). gcs, glomerular cross-section.
Figure 7.

Representative PAS-stained glomeruli from WT control (A) and FcγRI/III−/− (B) mice at day 8 after induction of nephritis. The WT mice had thrombosis of glomerular capillaries and occasional crescent formation, whereas the FcγRI/III−/− mice were protected.
Discussion
The results presented here confirm that FcRγ−/− mice were protected from accelerated nephrotoxic nephritis, and demonstrate that this was as a result of deficiency of FcγRI and FcγRIII. Both FcγRI and FcγRIII appeared to play a role, because both needed to be deficient to protect markedly from disease. Interestingly, in this active model, early neutrophil influx was not entirely dependent on FcγRIII, as was the case with the passive model of heterologous glomerulonephritis. 17 These results demonstrate that under different conditions, the relative importance of FcγRI and FcγRIII can be changed, which is an important consideration if a therapeutic approach of receptor blockade was to be attempted.
To exclude an effect of Fcγ receptors on the priming of the immune response we carefully examined the systemic immune response to sheep IgG and deposition of immunoglobulin within glomeruli. There were no differences in either of these parameters in the Fcγ receptor-deficient mice, implying that the role of activator Fcγ receptors in this model is in the intraglomerular response to deposited IgG.
In a previous study we examined the relative importance of FcRγ chain on intrinsic renal cells and circulating leukocytes by studying mice made chimeric by bone marrow transplantation. 19 Mice lacking the FcRγ chain on bone marrow-derived cells were completely protected from nephrotoxic nephritis whereas absence of FcRγ on intrinsic renal cells had no effect. The results of the present experiments demonstrate that both FcγRI and FcγRIII on circulating leukocytes are involved.
There are at least two reasons why FcγRI may play a role in the accelerated model of glomerulonephritis in addition to FcγRIII. FcγRI, much like the high-affinity IgE receptor of mast cells, circulates loaded with monomeric immunoglobulin. 20 Activation occurs on cross-linking of bound antibodies with a target antigen. FcγRI is more likely to be involved when high titers of specific antibody are present to compete with bound monomeric IgG, which might occur more often in an active disease model with high levels of circulating specific antibody. Secondly, accelerated nephrotoxic nephritis is dependent on the production of interferon-γ. 21 Interferon-γ is one of the most potent cytokines involved in the up-regulation of FcγRI on macrophages. 22 Local interferon-γ production may therefore cause an increase in macrophage FcγRI expression and explain why FcγRI has a relatively important role in this model.
It is interesting to compare our results with those in other models of inflammation. FcγRIII alone was shown to be important in several forms of immune complex-mediated inflammation, such as the reverse passive Arthus reaction, autoimmune hemolytic anemia, and heterologous glomerulonephritis. 7,23,24 However, in other models both FcγRI and FcγRIII have been shown to play a role. 24,25 FcγRI may play a more important role in models and genetic backgrounds in which T cells are involved and up-regulate expression of FcγRI on infiltrating leukocytes, whereas FcγRIII may be relatively more important in passive models.
Macrophages are important effectors of injury in glomerulonephritis. Our results demonstrate that it is possible to uncouple glomerular immunoglobulin deposition and macrophage infiltration from glomerular injury manifested by thrombosis and crescent formation. Indeed, there were more macrophages in the glomeruli of FcγRIII−/− mice than controls. This implies that the mechanisms of macrophage accumulation in immune complex glomerulonephritis are different from those that prime macrophages for injury and that macrophage activation for injury is critically dependent on Fcγ receptors. Fcγ receptor-deficient macrophages have markedly impaired antibody-dependent cellular cytotoxicity and phagocytosis. 6,7,15,16 Interfering with signaling from activator Fc receptors might provide one mechanism to reduce renal injury in established glomerulonephritis and even perhaps allow macrophages to develop a reparative phenotype.
Despite the lack of FcγRI and FcγRIII, some of the FcRγ−/− mice developed albuminuria, peaking at day 7 after nephrotoxic serum injection, which later resolved. Non-FcRγ-dependent pathways such as complement, direct antibody-mediated injury to podocytes, or T cell-mediated mechanisms may be involved. Angiotensin type 1 receptor blockade ameliorated the macrophage influx and proteinuria in FcRγ−/− mice in nephrotoxic nephritis. 4 However, despite mild proteinuria and macrophage influx both FcRγ and FcγRI/III double-deficient mice were almost completely protected from crescents that are the most destructive form of glomerular injury.
In summary, both FcRγ−/− mice and FcγRI/III−/− mice were substantially protected from glomerular thrombosis and crescent formation during murine nephrotoxic nephritis, a model of anti-glomerular basement membrane antibody nephritis. Unlike heterologous glomerulonephritis, in which FcγRIII is responsible for neutrophil infiltration, in this active model of glomerulonephritis both FcγRI and FcγRIII play a role in injury. Blockade of activator Fcγ receptors may therefore be an attractive therapeutic target in established immune complex glomerulonephritis, but our results show that it may be necessary to target both FcγRI and FcγRIII.
Figure 2.

Representative PAS-stained glomeruli from WT (A), FcγRIII−/− (B), and FcRγ−/− (C) kidneys at day 7 after 10 mg of nephrotoxic serum (NTS) injection to preimmunized mice. A and B show hypercellularity, glomerular crescent formation, and thrombosis; C shows mild hypercellularity only.
Acknowledgments
We thank Professor T. Saito for the FcRγ−/− mice; Mr. John Meek (Department of Chemical Pathology, Hammersmith Hospital) for performing creatinine and albumin analyses; Mrs. Margarita Lewis for preparing tissue for histology; Dr. Marina Botto for assistance in maintenance of the mouse colonies; and Dr. Joseph Boyle for helpful comments on the manuscript.
Footnotes
Address reprint requests to Dr. H. Terence Cook, Department of Histopathology, Imperial College School of Medicine, Hammersmith Hospital, Du Cane Rd., London, UK W12 0NN. E-mail: t.cook@ic.ac.uk.
Supported by Wellcome Trust grant O54838 and a Wellcome Trust Research Training Fellowship (to R. T.).
References
- 1.Ravetch JV, Bolland S: IgG Fc receptors. Annu Rev Immunol 2001, 19:275-290 [DOI] [PubMed] [Google Scholar]
- 2.Clynes R, Dumitru C, Ravetch JV: Uncoupling of immune complex formation and kidney damage in autoimmune glomerulonephritis. Science 1998, 279:1052-1054 [DOI] [PubMed] [Google Scholar]
- 3.Park SY, Ueda S, Ohno H, Hamano Y, Tanaka M, Shiratori T, Yamazaki T, Arase H, Arase N, Karasawa A, Sato S, Ledermann B, Kondo Y, Okumura K, Ra C, Saito T: Resistance of Fc receptor-deficient mice to fatal glomerulonephritis. J Clin Invest 1998, 102:1229-1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suzuki Y, Shirato I, Okumura K, Ravetch JV, Takai T, Tomino Y, Ra C: Distinct contribution of Fc receptors and angiotensin II-dependent pathways in anti-GBM glomerulonephritis. Kidney Int 1998, 54:1166-1174 [DOI] [PubMed] [Google Scholar]
- 5.Wakayama H, Hasegawa Y, Kawabe T, Hara T, Matsuo S, Mizuno M, Takai T, Kikutani H, Shimokata K: Abolition of anti-glomerular basement membrane antibody-mediated glomerulonephritis in FcRgamma-deficient mice. Eur J Immunol 2000, 30:1182-1190 [DOI] [PubMed] [Google Scholar]
- 6.Takai T, Li M, Sylvestre D, Clynes R, Ravetch JV: FcR gamma chain deletion results in pleiotrophic effector cell defects. Cell 1994, 76:519-529 [DOI] [PubMed] [Google Scholar]
- 7.Hazenbos WL, Gessner JE, Hofhuis FM, Kuipers H, Meyer D, Heijnen IA, Schmidt RE, Sandor M, Capel PJ, Daeron M, van de Winkel JG, Verbeek JS: Impaired IgG-dependent anaphylaxis and Arthus reaction in Fc gamma RIII (CD16) deficient mice. Immunity 1996, 5:181-188 [DOI] [PubMed] [Google Scholar]
- 8.Hazenbos WL, Heijnen IA, Meyer D, Hofhuis FM, Renardel de Lavalette CR, Schmidt RE, Capel PJ, van de Winkel JG, Gessner JE, van den Berg TK, Verbeek JS: Murine IgG1 complexes trigger immune effector functions predominantly via Fc gamma RIII (CD16). J Immunol 1998, 161:3026-3032 [PubMed] [Google Scholar]
- 9.Gavin AL, Leiter EH, Hogarth PM: Mouse FcgammaRI: identification and functional characterization of five new alleles. Immunogenetics 2000, 51:206-211 [DOI] [PubMed] [Google Scholar]
- 10.Takai T, Ono M, Hikida M, Ohmori H, Ravetch JV: Augmented humoral and anaphylactic responses in Fc gamma RII-deficient mice. Nature 1996, 379:346-349 [DOI] [PubMed] [Google Scholar]
- 11.Clynes R, Maizes JS, Guinamard R, Ono M, Takai T, Ravetch JV: Modulation of immune complex-induced inflammation in vivo by the coordinate expression of activation and inhibitory Fc receptors. J Exp Med 1999, 189:179-185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nieswandt B, Bergmeier W, Schulte V, Rackebrandt K, Gessner JE, Zirngibl H: Expression and function of the mouse collagen receptor glycoprotein VI is strictly dependent on its association with the FcRgamma chain. J Biol Chem 2000, 275:23998-24002 [DOI] [PubMed] [Google Scholar]
- 13.Kubagawa H, Chen CC, Ho LH, Shimada TS, Gartland L, Mashburn C, Uehara T, Ravetch JV, Cooper MD: Biochemical nature and cellular distribution of the paired immunoglobulin-like receptors, PIR-A and PIR-B. J Exp Med 1999, 189:309-318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arase N, Arase H, Park SY, Ohno H, Ra C, Saito T: Association with FcRgamma is essential for activation signal through NKR-P1 (CD161) in natural killer (NK) cells and NK1.1+ T cells. J Exp Med 1997, 186:1957-1963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ioan-Facsinay A, de Kimpe SJ, Hellwig SM, van Lent PL, Hofhuis FM, van Ojik HH, Sedlik C, da Silveira SA, Gerber J, de Jong YF, Roozendaal R, Aarden LA, van den Berg WB, Saito T, Mosser D, Amigorena S, Izui S, van Ommen GJ, van Vugt M, van de Winkel JG, Verbeek JS: FcgammaRI (CD64) contributes substantially to severity of arthritis, hypersensitivity responses, and protection from bacterial infection. Immunity 2002, 16:391-402 [DOI] [PubMed] [Google Scholar]
- 16.Barnes N, Gavin AL, Tan PS, Mottram P, Koentgen F, Hogarth PM: FcgammaRI-deficient mice show multiple alterations to inflammatory and immune responses. Immunity 2002, 16:379-389 [DOI] [PubMed] [Google Scholar]
- 17.Coxon A, Cullere X, Knight S, Sethi S, Wakelin MW, Stavrakis G, Luscinskas FW, Mayadas TN: Fc gamma RIII mediates neutrophil recruitment to immune complexes. a mechanism for neutrophil accumulation in immune-mediated inflammation. Immunity 2001, 14:693-704 [DOI] [PubMed] [Google Scholar]
- 18.Huang XR, Tipping PG, Apostolopoulos J, Oettinger C, D’Souza M, Milton G, Holdsworth SR: Mechanisms of T cell-induced glomerular injury in anti-glomerular basement membrane (GBM) glomerulonephritis in rats. Clin Exp Immunol 1997, 109:134-142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tarzi RM, Davies KA, Robson MG, Fossati-Jimack L, Saito T, Walport MJ, Cook HT: Nephrotoxic nephritis is mediated by Fcgamma receptors on circulating leukocytes and not intrinsic renal cells. Kidney Int 2002, 62:2087-2096 [DOI] [PubMed] [Google Scholar]
- 20.Unkeless JC, Eisen HN: Binding of monomeric immunoglobulins to Fc receptors of mouse macrophages. J Exp Med 1975, 142:1520-1533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kitching AR, Holdsworth SR, Tipping PG: IFN-gamma mediates crescent formation and cell-mediated immune injury in murine glomerulonephritis. J Am Soc Nephrol 1999, 10:752-759 [DOI] [PubMed] [Google Scholar]
- 22.Sivo J, Politis AD, Vogel SN: Differential effects of interferon-gamma and glucocorticoids on Fc gamma R gene expression in murine macrophages. J Leukoc Biol 1993, 54:451-457 [DOI] [PubMed] [Google Scholar]
- 23.Sylvestre DL, Ravetch JV: A dominant role for mast cell Fc receptors in the Arthus reaction. Immunity 1996, 5:387-390 [DOI] [PubMed] [Google Scholar]
- 24.Ji H, Ohmura K, Mahmood U, Lee DM, Hofhuis FM, Boackle SA, Takahashi K, Holers VM, Walport M, Gerard C, Ezekowitz A, Carroll MC, Brenner M, Weissleder R, Verbeek JS, Duchatelle V, Degott C, Benoist C, Mathis D: Arthritis critically dependent on innate immune system players. Immunity 2002, 16:157-168 [DOI] [PubMed] [Google Scholar]
- 25.van Lent PL, Nabbe K, Blom AB, Holthuysen AE, Sloetjes A, van de Putte LB, Verbeek S, van den Berg WB: Role of activatory Fc gamma RI and Fc gamma RIII and inhibitory Fc gamma RII in inflammation and cartilage destruction during experimental antigen-induced arthritis. Am J Pathol 2001, 159:2309-2320 [DOI] [PMC free article] [PubMed] [Google Scholar]