

Abstract
Previous work established that increased expression of heat shock proteins (HSPs) in the vessel wall might evoke proinflammatory and autoimmune reactions in the pathogenesis of atherosclerosis. The present study was designed to further scrutinize the molecular mechanisms of HSP expression involving activation of heat shock transcription factors (HSFs) in atherosclerotic lesions in animal models. Severe atherosclerotic lesions developed in the aortas of rabbits 16 weeks after feeding a 0.2% cholesterol diet. When protein extracts from the aortas were subjected to Western blot analysis, the level of HSF1 in proteins from atherosclerotic lesions of hypercholesterolemic rabbits were significantly higher than those of normal vessels. Gel mobility shift assays revealed the formation of protein-heat shock element complexes containing HSF1 in protein extracts from atherosclerotic lesion. Furthermore, triglyceride-rich lipoprotein, oxidized-triglyceride-rich lipoprotein, low-density lipoprotein, and oxidized low-density lipoprotein did not activate HSF1 in cultured smooth muscle cells, whereas HSF1 was highly activated in cells treated with tumor necrosis factor-α. Interestingly, mechanical stretching of smooth muscle cells resulted in HSF1 translocation from the cytoplasm to the nucleus and hyperphosphorylation followed by increased HSP70 expression. Thus, our findings provide the first evidence that HSF1 is activated and highly expressed in atherosclerotic lesions and that cytokine stimulation and disturbed mechanical stress to the vessel wall may be responsible for such activation.
Atherosclerosis shares many similarities with chronic inflammatory and autoimmune disease. 1,2 In both conditions T lymphocytes and macrophages infiltrate into the lesions where they release several cytokines elaborating the lesion progression. 3 The presence of mast and dendritic cells and the aberrant expression of HLA class II antigen in endothelial cells and smooth muscle cells (SMCs) have been observed in atherosclerotic lesions. 4-6 Furthermore, previous studies indicated the involvement of the complement system, 7 whereas recent reports established the role of autoantibodies in atherogenesis. 8 These findings support the concept that inflammatory and autoimmune mechanisms are crucial in the pathogenesis of atherosclerosis.
Heat shock proteins (HSPs) are a family of conserved proteins that are expressed in almost all types of mammalian cells. 9 According to their molecular mass, they are divided into HSP90, HSP70, HSP60, and small HSPs. Although HSPs are believed to localize within the cell, accumulating evidence indicates that HSP60 is expressed on the surface of cells. 10,11 Recently, we 12 demonstrated that HSP60 is present in a soluble form in the circulation, which is positively correlated with the severity of atherosclerosis in a large population. Reports from several groups 13-16 have demonstrated that recombinant HSPs stimulate endothelial cells, macrophages, and SMCs; produce proinflammatory cytokines, eg, tumor necrosis factor (TNF)-α, interleukin (IL)-1, and IL-6; and express adhesion molecules, eg, E-selectin, ICAM-1, and VCAM-1. In the early 1990’s we 8 provided the first evidence that anti-HSP60 antibody levels are associated with atherosclerosis. This was confirmed by many other groups. 17-20 Interestingly, mucosal administration of HSP65 reduces atherosclerotic lesions in hyperlipidemic animals. Overexpression of host HSP60, HSP70, and HSP47 was demonstrated in atherosclerotic lesions co-existing with chlamydial HSP60. 21-24 However, it is unknown whether heat shock transcription factor (HSF) is activated or overexpressed in atherosclerotic lesions.
HSFs interact with a specific regulatory element, the heat shock element (HSE), present in the promoters of HSP genes. 25,26 In the cell, HSFs are constitutively present in a non-DNA-binding state; they are activated in response to various stresses to a DNA-binding form. This activation process seems to involve the oligomerization of HSF from a monomeric to a trimeric state and is associated with HSF hyperphosphorylation. 25,26 Mouse HSF1 deficiency alters cardiac redox homeostasis and increases mitochondrial oxidative damage. 27 Whereas most of our knowledge concerning HSF activation and expression comes from studies in cultured cells in vitro, no data exists concerning its activation in the disease situation in vivo. To explore the molecular mechanisms of HSP expression in atherosclerosis, the present study was designed to investigate HSF activation and expression in vivo. We demonstrated that HSF1 was highly expressed as well as activated in atherosclerotic lesions of hypercholesterolemic animals. We provide evidence that proinflammatory cytokines and mechanical stress are primarily responsible for HSF activation in SMCs.
Materials and Methods
Animal Models for Atherosclerosis
All animal experiments were performed according to protocols approved by the Institutional Committee for Use and Care of Laboratory Animals. The procedure for establishing a rabbit model of atherosclerosis was similar to that described previously. 28,29 In short, New Zealand White male rabbits received water ad libitum and were fed either a normal standard chow diet or a cholesterol-enriched diet (0.2% w/w) for 16 weeks, as described previously. 28,29 Animals were sacrificed by heart puncture under ketamine (25 mg/kg) and xylazine (5 to 10 mg/kg) anesthesia. Blood was collected for cholesterol assays. The aortas were carefully removed intact from the aortic arch to the iliac bifurcation, immediately put into cold phosphate-buffered saline (4°C), and prepared for histological analysis and protein extractions. For conventional histology, tissue fragments were fixed in 4% buffered formaldehyde (pH 7.2), embedded in paraffin, and sectioned for hematoxylin and eosin (H&E) staining. Serum cholesterol values were measured every 2 or 4 weeks using an enzymatic procedure (Sigma, St. Louis, MO).
Western Blot Analysis
The procedure used for protein extracts was similar to that described previously. 29 Briefly, the atherosclerotic intimal lesions and media were dissected on ice from the remaining adventitia with tweezers and scissors. Tissues were homogenized in a buffer (20 mmol Hepes, pH 7.4, 50 mmol/L β-glycerophosphate, 2 mmol/L EGTA, 1 mmol/L dithiothreitol, 1 mmol/L Na3VO4, 1% Triton X-100, 10% glycerol, 1 μg/ml leupeptin, 100 μmol/L phenylmethyl sulfonyl fluoride, and 1 μg/ml aprotinin), and proteins were extracted. A similar procedure was used for preparation of protein extracts from cultured SMCs (see below). Proteins (10 or 50 μg/lane) were separated by electrophoresis through a 7%, 10%, or 15% sodium dodecyl sulfate-polyacrylamide gel and transferred to a membrane. The membranes were processed with either rabbit anti-HSF1 antibodies 30,31 (1:2000 in dilution; a gift from Dr. R. I. Morimoto, University of Northwestern, Evanston, IL) or rat monoclonal antibody against HSF1 (1:200 in dilution; Santa Cruz Biotechnology, Santa Cruz, CA) or anti-phosphoERK1/2, -pan-ERK1/2 and HSP70 (1:200 in dilution; Santa Cruz Biotechnology), respectively, and specific antigen-antibody complexes were detected with the ECL Western blot detection kit (Amersham Co., Little Chalfont, UK).
Cell Cultures
Rat vascular SMCs were cultivated from arteries using the procedure as described previously. 32 In short, thoracic aortas of rats were removed, and intima and media were carefully dissected from the vessel, cut into pieces (∼1 mm3), and implanted onto a gelatin (0.02%)-coated plastic bottle (Falcon). The bottle was incubated up-side-down at 37°C in a humidified atmosphere of 95% air/5% CO2 for 3 hours, and then medium supplemented with 20% fetal calf serum, penicillin (100 U/ml), and streptomycin (100 μg/ml) was slowly added. Cells were passaged by treatment with 0.05% trypsin/0.02% ethylenediaminetetraacetic acid (EDTA) solution. Experiments were conducted on SMCs that had just achieved confluence.
Low-Density Lipoprotein (LDL) and Triglyceride-Rich Lipoprotein (TRLP) Isolation and Oxidation
Blood samples were obtained 2 hours after the ingestion of a fat-rich meal from normolipidemic males aged 27 ± 7 years. The patients did not suffer from any digestive or metabolic disease as verified by medical history. Blood was drawn under vacuum from a cubital vein into precooled vacutainer tubes (1 g/L EDTA-K3). Triacylglycerol-rich lipoproteins (TRLP) (d = 0.934 to 0.989) were obtained from plasma as described previously. 33 Remnant lipoproteins were obtained with columns packed with immunoaffinity gel containing anti-apoA-1 and anti-apoB100 (JI-H) monoclonal antibodies (Japan Immunoresearch Laboratories, Co., Ltd., Takasaki, Japan) conjugated to solid phase Sepharose-4B. Human serum was applied to the columns and then incubated for 20 minutes at room temperature. The unbound fraction containing apoE-enriched lipoproteins and albumin were eluted with 10 mmol/L of phosphate-buffered saline, pH 7.4. The remnant lipoprotein fraction was centrifuged at 100,000 × g on an ultracentrifuge Beckman L8-70M (Beckman Instruments, Palo Alto, CA) to remove albumin. LDL (d = 1.019 to 1.063 g/ml) was isolated by differential centrifugation using solid KBr to adjust the density as described previously. 34 remnant lipoproteins and LDL oxidation was performed by incubation of the lipoproteins (1 mg/ml) with 10 μmol/L of CuCl2 at 37°C for 18 hours. 35 Samples were immediately dialyzed against 10 mmol/L of phosphate saline buffer (pH 7.4) for 24 hours at 4°C. The extent of oxidation was assessed by measurement of TBARS (9.8 ± 1.3 nmol/mg).
Cyclic Strain Stress Treatment
SMCs were plated on silicone elastomer-bottomed culture plates (Flexcell Co., McKeesport, PA). Cells were subjected to mechanical stress with the Cyclic Stress Unit as described previously. 36 In short, SMCs achieving 90% confluence were serum-starved for 3 days, and subjected to mechanical stress. Vacuum (15 to 20 kPa) was repeatedly applied to the elastomer-bottomed plates via the base plate, which was placed in a humidified incubator with 5% CO2 at 37°C. Cyclic deformation (60 cycles/minute) and 15% elongation of elastomer-bottomed plates were used.
Gel Mobility Shift Assays
For atherosclerotic lesion analysis, whole protein extracts were used. For cultured cells, nuclear proteins were prepared. Subconfluent (80 to 90%) SMCs were treated with TRLP, LDL, cytokines, or mechanical stress for various time periods. For heat shock, cells were heated at 42°C for 30 minutes. The cells were washed and harvested with cold Tris-buffered saline (10 mmol/L Tris, 150 mmol/L NaCl, 1 mmol/L EDTA, pH 7.4). The cell pellet (2 to 3 × 106) was incubated with 400 μl of a cold buffer (10 mmol/L Hepes, 10 mmol/L KCl, 0.1 mmol/L EDTA, 0.1 mmol/L EGTA, 1 mmol/L DDT, 0.5 mmol/L phenylmethyl sulfonyl fluoride, pH 7.9) on ice for 15 minutes after pipetting. Then, 25 μl (10%) of Nonidet P-40 was added, vortexed for 10 seconds, and centrifuged for 30 seconds at 13,000 rpm. The nuclear pellet was incubated with 50 μl of cold buffer B (20 mmol/L Hepes, 0.4 mol/L NaCl, 20% glycerol, 1.5 mmol/L MgCl2, 1 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L dithiothreitol, 1 mmol/L phenylmethyl sulfonyl fluoride, 1 μg/ml leupeptin, 1 μg/ml aprotinin, pH 7.9) at 4°C for 15 minutes on a shaking platform with vigorous rocking. The suspension was centrifuged for 5 minutes at 4°C at 13,000 rpm. The supernatant was collected as nuclear proteins and protein concentration was determined with the Bio-Rad assay.
The procedure for gel mobility shift assays has been described previously. 37,38 In short, DNA binding was determined after incubation of 5 μg of nuclear protein extract with 10 fmol of an oligonucleotide containing HSE sequence from the Drosophila hsp70 promoter (5′-GCCTCGAATGTTCGCGAAGTTT-3′) labeled with 32P-dATP. Reaction buffer contained 10 mmol/L Hepes, (pH 7.9), 1 mmol/L dithiothreitol, 1 mmol/L EDTA, 80 mmol/L KCl, 4% Ficoll, and 1 μg poly(dIdC) (Pharmacia Biotech, Uppsala, Sweden) as a nonspecific competitor. Samples were electrophoresed through a 4% polyacrylamide gel in 0.5× TBE (1× TBE: 89 mmol/L Tris, 89 mmol/L boric acid, 20 mmol/L EDTA, pH 8.3), after which the gel was dried onto DE81 paper and exposed to autoradiographic film. Supershift assays were performed using antibodies generated against and specific to HSF1 and HSF2. The antibodies were added to samples after the initial binding reactions between protein extracts and oligonucleotides were allowed to take place.
Statistical Analysis
Statistical analyses were performed using the Mann-Whitney U-test. Results are given as mean ± SD. A P value <0.05 was considered significant.
Results
Atherosclerotic Lesions in Rabbit Model
Blood cholesterol levels in rabbits receiving a 0.2% cholesterol diet were significantly elevated and reached 500 mg/dl at 4 weeks, and ∼700 mg/dl at 16 weeks, whereas animals in the control group (chow diet) had blood cholesterol levels less than 100 mg/dl (Figure 1a) ▶ . Rabbit aortas from both groups were examined morphologically 16 weeks after chow or cholesterol feeding. H&E-stained sections from the vessel of a control animal revealed the normal intima containing a monolayer of endothelium as indicated by an arrow, and the media, major portion of the aorta (Figure 1b) ▶ . In cholesterol-fed rabbits, ∼60% of aortic intima was covered by atherosclerotic lesions, including fatty streaks and plaques. Aortic lesions of rabbits fed with a cholesterol-enriched diet were characterized by cell proliferation, foam cell accumulation, and lipid deposition in the intima (Figure 1c) ▶ . Atherosclerotic lesions displayed the great variation of cell types and distribution. Immunohistological staining demonstrated that lesions contained SMCs, macrophages, and T lymphocytes (data not shown).
Figure 1.

Blood cholesterol levels and atherosclerotic lesions in rabbits. a: The values of serum cholesterol were measured every 2 or 4 weeks using an enzymatic procedure. Note that serum cholesterol levels in rabbits fed a 0.2% cholesterol diet were significantly higher than in those receiving the chow diet. b and c: H&E-stained sections from animals receiving chow (b) or cholesterol-enriched diets (c) for 16 weeks. Arrows indicate internal elastic lamina.
Elevated Protein Levels and Activities of HSF1
Because of the increased intensity of HSP70 and HSP60 immunostaining in lesions, 22,24 it would be interesting to determine HSF1 protein levels in atherosclerotic lesions. Protein extracts from tissues of normal intima/media (I/M), intimal lesions (AS), and media (M) were analyzed by Western blot analysis. Abundant HSF1 proteins in atherosclerotic lesions (AS) were observed (Figure 2a) ▶ . HSF1 proteins in intimal lesions (AS) were significantly higher than intima/media (I/M) of control animals and media (M) of cholesterol-fed rabbits. HSF1 levels are normalized against actin (Figure 2a ▶ , bottom). The graphics in Figure 2 ▶ summarize data from five animals, implicating that HSF1 levels were fourfold to sixfold increased in atherosclerotic lesions. To determine whether a proportion of HSF1 in lesions was modified, a small amount of proteins (10 μg/lane) was loaded in 10% gel, which allows better separation of proteins with moderate changes in molecular weight. HSF1 bands were hardly seen in proteins of normal vessels, because HSF1 protein was too little in a small amount of protein extracts (10 μg/lane). Because of higher levels of HSF1 in lesions, two or three of the HSF1 bands were shifted to higher molecular weight species in protein extracts from atherosclerotic lesions (Figure 2b) ▶ , suggesting that HSF phosphorylation occurs in the lesions.
Figure 2.

Elevated HSF1 proteins in atherosclerotic lesions. Intima and media (I/M) from chow-fed rabbits and media (M) and atherosclerotic intimal lesions (AS) from cholesterol-fed rabbits (16-week diet) were dissected on ice from the remaining adventitia with tweezers and scissors. Tissues were frozen in liquid nitrogen and homogenized with a polytron homogenizer. a: Protein extracts (50 μg/lane) were separated on 7% sodium dodecyl sulfate-polyacrylamide gel, transferred onto membranes, and probed with the antibody against HSF1 (top) and anti-actin antibodies after stripping (bottom). Immunocomplexes were visualized by a Western blot detection kit. Each lane represents an individual animal. The molecular weight of HSF1 and actin is ∼90 and 39 kd, respectively. Graphic data show mean ± SD obtained from five rabbits per group. *, Significant difference from the intima/media, P < 0.001. b: Protein extracts (10 μg/lane) were separated on 10% sodium dodecyl sulfate-polyacrylamide gel, transferred onto membranes, and probed with the antibody against HSF1. Immunocomplexes were visualized by a Western blot detection kit.
To determine whether HSF1 is activated, gel mobility shift assays using protein extracts from the vessels were performed. As shown in Figure 3a ▶ , levels of protein-DNA-binding activity were increased in atherosclerotic proteins compared to normal vessels. The specificity of DNA binding was determined by a competition experiment using cold oligonucleotides. Figure 3b ▶ shows the results of a gel mobility shift assay in the presence or absence of either an unlabeled HSE, or nuclear factor (NF)-κB binding element. The increased binding activity in atherosclerotic proteins was specific for the HSE, as unlabeled HSE effectively competed for binding to the factor, whereas the NF-κB-binding element did not (Figure 3b) ▶ . There is evidence to indicate that HSF1 and HSF2 mediate the HSP induction in response to different stimuli, 39 therefore, it was of interest to determine which of these HSFs were involved. We examined the composition of the DNA-binding complexes using antibodies to HSF1 and HSF2. Addition of the anti-HSF1 antibody to the binding reaction resulted in a complete shift of the binding complexes to a slower migrating species (Figure 3c) ▶ , whereas the anti-HSF2 antibody had no effect (data not shown). The supershift pattern of antibody-HSF1-DNA complexes is similar to that observed in heat shock-treated cells, 30,38 ie, the complexes retarded within the well. These results indicate the presence of HSF1, but not HSF2, proteins in the HSE-binding complexes in protein extracts of atherosclerotic lesions.
Figure 3.
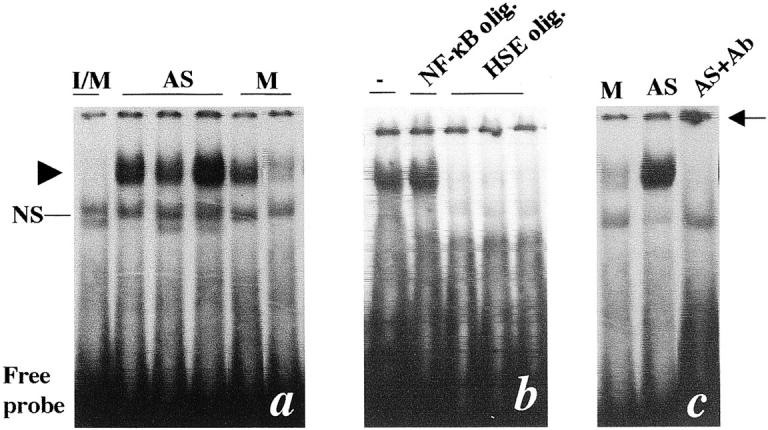
Gel mobility shift assays for protein extracts from atherosclerotic lesions. Intima and media (I/M) from chow-fed rabbits and media (M) and atherosclerotic intimal lesions (AS) from cholesterol-fed rabbits (16-week diet) were dissected and protein extracts were prepared as described in the Materials and Methods. a: Gel mobility shift assay performed using protein extracts (20 μg protein per lane) from I/M of control animals, atherosclerotic lesions (AS), or media (M). Arrowhead indicates protein-DNA-binding complexes; NS, nonspecific binding. b: Extracts obtained from atherosclerotic lesions were incubated with a radiolabeled oligonucleotide containing a HSF-binding site (HSE) with no addition (−), in the presence of unlabeled NF-κB, or unlabeled HSE, oligonucleotides (50:1). c: Protein extracts obtained from the media (M) or atherosclerotic lesions (AS) were incubated with a radiolabeled oligonucleotide containing a HSF-binding site in the presence of antibodies (1:20, Ab) specific to HSF1. Supershifted DNA-binding complexes indicative of HSF1 protein-containing complexes are indicated by the arrow. Each lane on the blot (a) represents an individual animal.
HSF1 Was Activated by TNF-α and Mechanical Stress, but Not Lipoproteins in SMCs
TRLP and LDL are main lipoproteins carrying and transporting triglyceride and cholesterol in hyperlipidemic animals. They can be oxidized in the arterial wall. 40 To test whether TRLP, oxidized TRLP, LDL, and oxidized LDL can induce HSF1 activation, vascular SMCs were stimulated with human TRLP, oxidized TRLP, LDL, and oxidized LDL, and the gel mobility shift assays were performed. As shown in Figure 4a ▶ , TRLP, oxidized TRLP, LDL, and oxidized LDL did not induce HSF-DNA binding in SMCs, whereas heat shock treatment did. This coincided with results obtained by Western blot analysis (Figure 4b) ▶ . To confirm that the treatment of SMCs with lipoproteins was effective, we determined the phosphorylation of MAP kinase ERK1/2 in SMCs. ERK1/2 was activated 10 minutes after treatment with all types of lipoproteins.
Figure 4.

TRLP, remnant lipoprotein, oxidized TRLP, LDL, and oxidized LDL do not activate HSF-DNA binding. a: Gel mobility shift assay for protein extracts from cultured SMCs. Rat SMCs were treated with TRLP, remnant lipoprotein, oxidized TRLP (100 μg/ml), LDL, and oxidized LDL (100 μg/ml) at 37°C for 1 hour. The cells were washed and harvested with cold Tris-buffered saline. Nuclear proteins were prepared as described in Materials and Methods and protein concentration was determined with a Bio-Rad assay. Protein extracts (5 μg per lane) were incubated with a radiolabeled HSE oligonucleotide on ice for 20 minutes. The gel mobility shift assay was performed in 4% gel. Arrowhead indicates specific HSE-binding complexes. b: Western blot analysis. SMCs were treated with TRLP, remnant lipoprotein, oxidized TRLP, LDL, and oxidized LDL (μg/ml) at 37°C for 12 hours. Protein extracts (50 μg/lane) were separated on 10% sodium dodecyl sulfate-polyacrylamide gel, transferred onto a membrane, and probed with the antibody against HSF1. Immunocomplexes were visualized by a Western blot detection kit. c: Western blot analysis for ERK1/2. Protein extracts from cultured SMCs treated with lipoproteins were separated on 15% sodium dodecyl sulfate-polyacrylamide gel, transferred onto a membrane, and probed with the antibody against phospho-ERK1/2 and pan-ERK1/2. Blots were stripped between antibody incubations. Immunocomplexes were visualized by a Western blot detection kit. Ctl−, negative control without treatment; CtL+, SMCs treated at 42°C for 30 minutes.
Because lipoproteins had no effects on HSF1 activation, other atherosclerotic risk factors have to be responsible for this event. TNF-α, a proinflammatory cytokine, is highly expressed in atherosclerotic lesions and may be involved in the development of atherosclerosis. 2,41 TNF-α was used to treat SMCs, and HSF activities were examined. The data shown in Figure 5 ▶ indicate that TNF-α treatment resulted in HSF1-DNA binding in SMCs, while H2O2 treatment did not stimulate HSF activation (Figure 5a) ▶ . There is evidence that altered hemodynamic (mechanical) stress plays a crucial role in the pathogenesis of atherosclerosis. 42 To study the effects of mechanical stress on HSF1 activation, an in vitro stretch model was used to mimic in vivo pathological conditions, eg, hypertension. Figure 5b ▶ shows data that indicates that mechanical stress markedly resulted in HSF1-DNA binding in SMCs. The increase in HSF activity in stretched SMCs was time-dependent (Figure 5b) ▶ . Furthermore, Western blot analysis revealed that HSF1 is translocated into the nuclei only in response to mechanical stress, because no protein was detectable in nuclear extracts from untreated SMCs (Figure 5c) ▶ . However, if whole cell proteins are used for Western blot analysis, HSF1 protein levels are similar between treated and untreated SMCs, but the band of stretched SMCs shifted to a slower species (Figure 5c ▶ , right). This HSF1 activation was followed by increased HSP70 production in mechanical stress-treated SMCs (Figure 5d) ▶ , suggesting that mechanical stress may play a crucial role in the activation of HSF1 and HSP expression.
Figure 5.
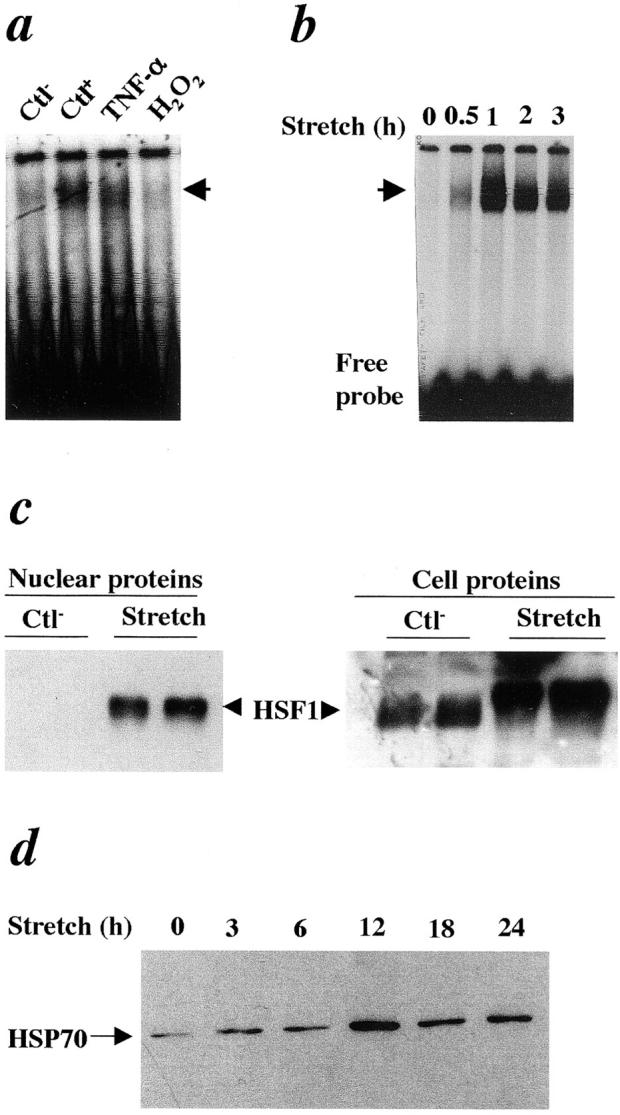
TNF-α- and mechanical stretch-activated HSF1 in SMCs. a: Gel mobility shift assay of protein extracts from SMCs treated with TNF-α (50 ng/ml) or H2O2 (200 μmol/L) at 37°C for 1 hour. b: Gel mobility shift assay of protein extracts from SMCs treated with mechanical stretch (1 Hz) for the indicated times. c: Western blot analysis for HSF1 using nuclear proteins (left) and cell proteins (right) of SMCs treated with mechanical stretch (1 Hz) 37°C for 1 hour. d: Western blot analysis for HSP70 of SMCs treated with mechanical stretch (1 Hz) 37°C for the indicated times. Ctl−, negative control without treatment; CtL+, SMCs treated at 42°C for 30 minutes.
Discussion
Atherosclerosis is an inflammatory- and immune-mediated disease, in which HSPs were repeatedly implicated to be crucial. 43 Several families of HSPs, including HSP70, HSP60, HSP47, and chlamydial HSP60, have been demonstrated to be expressed at high levels in atherosclerotic lesions. 21-24 These highly expressed HSPs in lesions could promote disease progression via evoking proinflammatory and autoimmune responses. 43 Mucosal administration of HSP65 reduces atherosclerotic lesions in animal models. 44 In the present study, we provide the first evidence of increased HSF1 activities in atherosclerotic lesions in vivo, which coincide with high levels of HSF1 proteins. These observations could be important to understand the molecular mechanisms of HSP expression in atherogenesis.
At present, four different HSFs have been identified, ie, HSF1, HSF2, HSF3, and HSF4. 45 HSFs are transcription products of four different genes. HSF1, HSF2, and HSF4 have been identified in human tissues. 45 HSF3 has only been described in chickens, in which it is involved in the tissue development. In response to stress stimuli, HSF1 is activated and translocated from cytoplasm into the nucleus in cultured cells. 46 We found increased HSF1, but not HSF2, in atherosclerotic lesions that was mainly localized in the nuclei, and the molecular weight of HSF1 from lesional extracts was larger than that of normal vessels. This indicated that HSF1 in lesions was phosphorylated (modified) and activated. Usually, HSF1 proteins in cultured cells do not markedly change in response to stress, eg, heat shock or mechanical stress (Figure 5c) ▶ . Surprisingly, we demonstrated that protein levels of HSF1 in atherosclerotic lesions were much higher compared to normal vessels, suggesting different mechanisms of HSF1 activation and regulation of HSP expression in vivo from in vitro cultured cells.
It is believed that LDL and oxidized LDL are important in the development of atherosclerosis. 40 Oxidized LDL possesses several proatherogenic properties, including interactions with several receptors, leading to the engorgement of cells with lipids, inhibition of endothelium-dependent vascular relaxation, cytotoxicity to proliferatingcells, and stimulation of chemoattractant secretion. 40,47 Oxidized LDL also triggers in vitro HSP60 expression in monocytes/macrophages, 48 and HSP70 in human endothelial cells 49 and SMCs. 50 Furthermore, TRLPs may be a key factor in the development of atherosclerosis. 51 Our data demonstrated that TRLP, oxidized TRLP, LDL, and oxidized LDL do not activate HSF1 in cultured SMCs, indicating that HSP expression induced by oxidized LDL was not regulated at a transcriptional level and that lipoproteins may not directly stimulate HSF1 activation in atherosclerotic lesions in vivo. However, oxidized LDL and oxidized TRLP may exert their role in HSF1 activation in vivo via stimulating cells producing a panel of cytokines 52 that result in HSF1 activation. In fact, it has been found that TNF-α is present in high concentrations in atherosclerotic lesions. 2,41 We demonstrate that TNF-α can activate HSF1 in SMCs, supporting the role of cytokines in HSF1 activation in atherosclerotic lesions.
In vivo, the vessel wall is exposed to two main hemodynamic forces or biomechanical stress: shear stress, the dragging frictional force created by blood flow, and mechanical stretch, a cyclic strain stress created by blood pressure. 42,53 Shear stress stimulates endothelial cells to release nitric oxide 54 and prostacyclin, 55 resulting in vessel relaxation and protection of vascular cells, whereas SMCs are stimulated by stretch or cyclic strain stress. 56 In humans, atherosclerotic lesions occur preferably at bifurcations and curvatures 57 where hemodynamic force is disturbed, ie, lower shear stress and higher mechanical stretch. 58 Therefore, mechanical stretch could be a crucial factor in the pathogenesis of atherosclerosis. Using an in vitro mechanical stress model, we provide evidence that mechanical forces evoke rapid activation of HSF1 in SMCs, followed by elevated HSP70 protein levels. Interestingly, SMC lines stably expressing dominant-negative rac (rac N17) abolished HSP protein production and HSF1 activation induced by mechanical forces, whereas a significant reduction of HSF1 activities was seen in ras N17 transfected-cell lines. 59 Therefore, mechanical stretch-induced HSF1 activation was regulated by rac/ras GTP-binding proteins 59 that may be primarily responsible for HSF1 activation seen in atherosclerotic lesions.
It is believed that HSFs are constitutively present in a non-DNA-binding state; they are activated in response to various stresses to a DNA-binding form. This activation process appears to involve the oligomerization of HSF from a monomeric to a trimeric state, but not de novo HSF protein production. 25,26 Surprisingly, we found that HSF1 protein levels in atherosclerotic lesions are much higher than normal vessels, indicating increased HSF1 protein production. How HSF1 induction is regulated, at the present, is unknown. We postulate that a special microenvironment of atherosclerotic lesions may require a large amount of HSF1 to maintain a high level of constitutively expressed HSPs. Supporting this hypothesis is the observation that SMCs have a similar amount of HSF1 proteins between treated and untreated controls, suggesting the importance of in vivo microenvironment of the vessel wall in regulation of HSF1 production. Thus, one should be cautious with the interpretation of in vitro data to in vivo situations.
In summary, risk factors for atherosclerosis, such as biomechanical stress and cytokines induced by hypercholesterolemia, directly stimulate cells of the arterial wall to express high levels of HSF1, which leads to increased expression of HSPs. Pathologically, overstimulation by the risk factors results in cell death, which releases intracellular HSPs into intercellular spaces to form soluble HSPs that lead to proinflammatory and autoimmune responses. Both of these contribute to the development of atherosclerosis. Our findings of HSF1 activation in vivo and in vitro could help us to understand better the molecular mechanisms of HSP involvement in atherogenesis.
Acknowledgments
We thank Immunoresearch Laboratories Co. Ltd., Takasaki, Japan, for providing us antibodies against TRLP; Dr. M. Mayr for help in setting gel mobility shift assays; and Mr. Zhang for excellent technical assistance.
Footnotes
Address reprint requests to Qingbo Xu, M.D., Ph.D., Department of Cardiological Sciences, St. George’s Hospital Medical School, Cranmer Terrance, London SW17 0RE, UK. E-mail: q.xu@sghms.ac.uk.
Supported in part by the Oak Foundation and The Royal Society, UK.
References
- 1.Ross R: Atherosclerosis—an inflammatory disease. N Engl J Med 1999, 340:115-126 [DOI] [PubMed] [Google Scholar]
- 2.Hansson GK, Libby P, Schonbeck U, Yan ZQ: Innate and adaptive immunity in the pathogenesis of atherosclerosis. Circ Res 2002, 91:281-291 [DOI] [PubMed] [Google Scholar]
- 3.Hansson GK: Immune mechanisms in atherosclerosis. Arterioscler Thromb Vasc Biol 2001, 21:1876-1890 [DOI] [PubMed] [Google Scholar]
- 4.Metzler B, Xu Q: The role of mast cells in atherosclerosis. Int Arch Allergy Immunol 1997, 114:10-14 [DOI] [PubMed] [Google Scholar]
- 5.Bobryshev YV, Lord RS, Rainer S, Jamal OS, Munro VF: Vascular dendritic cells and atherosclerosis. Pathol Res Pract 1996, 192:462-467 [PubMed] [Google Scholar]
- 6.Xu Q, Oberhuber G, Gruschwitz M, Wick G: Immunology of atherosclerosis: cellular composition and major histocompatibility complex class II antigen expression in aortic intima, fatty streaks, and atherosclerotic plaques in young and aged human specimens. Clin Immunol Immunopathol 1990, 56:344-359 [DOI] [PubMed] [Google Scholar]
- 7.Torzewski J, Bowyer DE, Waltenberger J, Fitzsimmons C: Processes in atherogenesis: complement activation. Atherosclerosis 1997, 132:131-138 [DOI] [PubMed] [Google Scholar]
- 8.Xu Q, Willeit J, Marosi M, Kleindienst R, Oberhollenzer F, Kiechl S, Stulnig T, Luef G, Wick G: Association of serum antibodies to heat-shock protein 65 with carotid atherosclerosis. Lancet 1993, 341:255-259 [DOI] [PubMed] [Google Scholar]
- 9.Morimoto RI: Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev 1998, 12:3788-3796 [DOI] [PubMed] [Google Scholar]
- 10.Xu Q, Schett G, Seitz CS, Hu Y, Gupta RS, Wick G: Surface staining and cytotoxic activity of heat-shock protein 60 antibody in stressed aortic endothelial cells. Circ Res 1994, 75:1078-1085 [DOI] [PubMed] [Google Scholar]
- 11.Khan IU, Wallin R, Gupta RS, Kammer GM: Protein kinase A-catalyzed phosphorylation of heat shock protein 60 chaperone regulates its attachment to histone 2B in the T lymphocyte plasma membrane. Proc Natl Acad Sci USA 1998, 95:10425-10430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu Q, Schett G, Perschinka H, Mayr M, Egger G, Oberhollenzer F, Willeit J, Kiechl S, Wick G: Serum soluble heat shock protein 60 is elevated in subjects with atherosclerosis in a general population. Circulation 2000, 102:14-20 [DOI] [PubMed] [Google Scholar]
- 13.Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, Stevenson MA, Calderwood SK: Novel signal transduction pathway utilized by extracellular HSP70. Role of Toll-like receptor (TLR) 2 and TLR4. J Biol Chem 2002, 277:15028-15034 [DOI] [PubMed] [Google Scholar]
- 14.Chen W, Syldath U, Bellmann K, Burkart V, Kolb H: Human 60-kDa heat-shock protein: a danger signal to the innate immune system. J Immunol 1999, 162:3212-3219 [PubMed] [Google Scholar]
- 15.Kol A, Bourcier T, Lichtman AH, Libby P: Chlamydial and human heat shock protein 60s activate human vascular endothelium, smooth muscle cells, and macrophages. J Clin Invest 1999, 103:571-577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohashi K, Burkart V, Flohe S, Kolb H: Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J Immunol 2000, 164:558-561 [DOI] [PubMed] [Google Scholar]
- 17.Frostegard J, Lemne C, Andersson B, van der Zee R, Kiessling R, de Faire U: Association of serum antibodies to heat-shock protein 65 with borderline hypertension. Hypertension 1997, 29:40-44 [DOI] [PubMed] [Google Scholar]
- 18.Gromadzka G, Zielinska J, Ryglewicz D, Fiszer U, Czlonkowska A: Elevated levels of anti-heat shock protein antibodies in patients with cerebral ischemia. Cerebrovasc Dis 2001, 12:235-239 [DOI] [PubMed] [Google Scholar]
- 19.Zhu J, Quyyumi AA, Rott D, Csako G, Wu H, Halcox J, Epstein SE: Antibodies to human heat-shock protein 60 are associated with the presence and severity of coronary artery disease: evidence for an autoimmune component of atherogenesis. Circulation 2001, 103:1071-1075 [DOI] [PubMed] [Google Scholar]
- 20.Huittinen T, Leinonen M, Tenkanen L, Manttari M, Virkkunen H, Pitkanen T, Wahlstrom E, Palosuo T, Manninen V, Saikku P: Autoimmunity to human heat shock protein 60, Chlamydia pneumoniae infection, and inflammation in predicting coronary risk. Arterioscler Thromb Vasc Biol 2002, 22:431-437 [DOI] [PubMed] [Google Scholar]
- 21.Berberian PA, Myers W, Tytell M, Challa V, Bond MG: Immunohistochemical localization of heat shock protein-70 in normal-appearing and atherosclerotic specimens of human arteries. Am J Pathol 1990, 136:71-80 [PMC free article] [PubMed] [Google Scholar]
- 22.Kleindienst R, Xu Q, Willeit J, Waldenberger FR, Weimann S, Wick G: Immunology of atherosclerosis. Demonstration of heat shock protein 60 expression and T lymphocytes bearing alpha/beta or gamma/delta receptor in human atherosclerotic lesions. Am J Pathol 1993, 142:1927-1937 [PMC free article] [PubMed] [Google Scholar]
- 23.Rocnik E, Chow LH, Pickering JG: Heat shock protein 47 is expressed in fibrous regions of human atheroma and is regulated by growth factors and oxidized low-density lipoprotein. Circulation 2000, 101:1229-1233 [DOI] [PubMed] [Google Scholar]
- 24.Kanwar RK, Kanwar JR, Wang D, Ormrod DJ, Krissansen GW: Temporal expression of heat shock proteins 60 and 70 at lesion-prone sites during atherogenesis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol 2001, 21:1991-1997 [DOI] [PubMed] [Google Scholar]
- 25.Wu C: Heat shock transcription factors: structure and regulation. Annu Rev Cell Dev Biol 1995, 11:441-469 [DOI] [PubMed] [Google Scholar]
- 26.Pirkkala L, Nykanen P, Sistonen L: Roles of the heat shock transcription factors in regulation of the heat shock response and beyond. EMBO J 2001, 15:1118-1131 [DOI] [PubMed] [Google Scholar]
- 27.Yan LJ, Christians ES, Liu L, Xiao X, Sohal RS, Benjamin IJ: Mouse heat shock transcription factor 1 deficiency alters cardiac redox homeostasis and increases mitochondrial oxidative damage. EMBO J 2002, 21:5164-5172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu Y, Dietrich H, Metzler B, Wick G, Xu Q: Hyperexpression and activation of extracellular signal-regulated kinases (ERK1/2) in atherosclerotic lesions of cholesterol-fed rabbits. Arterioscler Thromb Vasc Biol 2000, 20:18-26 [DOI] [PubMed] [Google Scholar]
- 29.Metzler B, Hu Y, Dietrich H, Xu Q: Increased expression and activation of stress-activated protein kinases/c-Jun NH(2)-terminal protein kinases in atherosclerotic lesions coincide with p53. Am J Pathol 2000, 156:1875-1886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sistonen L, Sarge KD, Morimoto RI: Human heat shock factors 1 and 2 are differentially activated and can synergistically induce hsp70 gene transcription. Mol Cell Biol 1994, 14:2087-2099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sarge KD, Murphy SP, Morimoto RI: Activation of heat shock gene transcription by heat shock factor 1 involves oligomerization, acquisition of DNA-binding activity, and nuclear localization and can occur in the absence of stress. Mol Cell Biol 1993, 13:1392-1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu Y, Zou Y, Dietrich H, Wick G, Xu Q: Inhibition of neointima hyperplasia of mouse vein grafts by locally applied suramin. Circulation 1999, 100:861-868 [DOI] [PubMed] [Google Scholar]
- 33.Abia R, Pacheco YM, Perona JS, Montero E, Muriana FJ, Ruiz-Gutierrez V: The metabolic availability of dietary triacylglycerols from two high oleic oils during the postprandial period does not depend on the amount of oleic acid ingested by healthy men. J Nutr 2001, 131:59-65 [DOI] [PubMed] [Google Scholar]
- 34.Jurgens G, Xu QB, Huber LA, Bock G, Howanietz H, Wick G, Traill KN: Promotion of lymphocyte growth by high density lipoproteins (HDL). Physiological significance of the HDL binding site. J Biol Chem 1989, 264:8549-8556 [PubMed] [Google Scholar]
- 35.Amberger A, Maczek C, Jurgens G, Michaelis D, Schett G, Trieb K, Eberl T, Jindal S, Xu Q, Wick G: Co-expression of ICAM-1, VCAM-1, ELAM-1 and Hsp60 in human arterial and venous endothelial cells in response to cytokines and oxidized low-density lipoproteins. Cell Stress Chaperones 1997, 2:94-103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li C, Hu Y, Mayr M, Xu Q: Cyclic strain stress-induced mitogen-activated protein kinase (MAPK) phosphatase 1 expression in vascular smooth muscle cells is regulated by Ras/Rac-MAPK pathways. J Biol Chem 1999, 274:25273-25280 [DOI] [PubMed] [Google Scholar]
- 37.Xu Q, Ganju L, Fawcett TW, Holbrook NJ: Vasopressin-induced heat shock protein expression in renal tubular cells. Lab Invest 1996, 74:178-187 [PubMed] [Google Scholar]
- 38.Xu Q, Hu Y, Kleindienst R, Wick G: Nitric oxide induces heat-shock protein 70 expression in vascular smooth muscle cells via activation of heat shock factor 1. J Clin Invest 1997, 100:1089-1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morimoto RI: Cells in stress: transcriptional activation of heat shock genes. Science 1993, 259:1409-1410 [DOI] [PubMed] [Google Scholar]
- 40.Witztum JL, Steinberg D: The oxidative modification hypothesis of atherosclerosis: does it hold for humans? Trends Cardiovasc Med 2001, 11:93-102 [DOI] [PubMed] [Google Scholar]
- 41.Libby P, Sukhova G, Lee RT, Galis ZS: Cytokines regulate vascular functions related to stability of the atherosclerotic plaque. J Cardiovasc Pharmacol 1995, 25(Suppl 2):S9-S12 [DOI] [PubMed] [Google Scholar]
- 42.Xu Q: Biomechanical-stress-induced signaling and gene expression in the development of arteriosclerosis. Trends Cardiovasc Med 2000, 10:35-41 [DOI] [PubMed] [Google Scholar]
- 43.Xu Q: Role of heat shock proteins in atherosclerosis. Arterioscler Thromb Vasc Biol 2002, 22:1547-1559 [DOI] [PubMed] [Google Scholar]
- 44.Maron R, Sukhova G, Faria AM, Hoffmann E, Mach F, Libby P, Weiner HL: Mucosal administration of heat shock protein-65 decreases atherosclerosis and inflammation in aortic arch of low-density lipoprotein receptor-deficient mice. Circulation 2002, 106:1708-1715 [DOI] [PubMed] [Google Scholar]
- 45.Snoeckx LH, Cornelussen RN, Van Nieuwenhoven FA, Reneman RS, Van Der Vusse GJ: Heat shock proteins and cardiovascular pathophysiology. Physiol Rev 2001, 81:1461-1497 [DOI] [PubMed] [Google Scholar]
- 46.Craig EA, Weissman JS, Horwich AL: Heat shock proteins and molecular chaperones: mediators of protein conformation and turnover in the cell. Cell 1994, 78:365-372 [DOI] [PubMed] [Google Scholar]
- 47.Steinberg D, Witztum JL: Lipoproteins and atherogenesis. Current concepts. JAMA 1990, 264:3047-3052 [PubMed] [Google Scholar]
- 48.Frostegard J, Kjellman B, Gidlund M, Andersson B, Jindal S, Kiessling R: Induction of heat shock protein in monocytic cells by oxidized low density lipoprotein. Atherosclerosis 1996, 121:93-103 [DOI] [PubMed] [Google Scholar]
- 49.Zhu W, Roma P, Pellegatta F, Catapano AL: Oxidized-LDL induce the expression of heat shock protein 70 in human endothelial cells. Biochem Biophys Res Commun 1994, 200:389-394 [DOI] [PubMed] [Google Scholar]
- 50.Zhu WM, Roma P, Pirillo A, Pellegatta F, Catapano AL: Oxidized LDL induce hsp70 expression in human smooth muscle cells. FEBS Lett 1995, 372:1-5 [DOI] [PubMed] [Google Scholar]
- 51.Ginsberg HN: New perspectives on atherogenesis: role of abnormal triglyceride-rich lipoprotein metabolism. Circulation 2002, 106:2137-2142 [DOI] [PubMed] [Google Scholar]
- 52.Niemann-Jonsson A, Dimayuga P, Jovinge S, Calara F, Ares MP, Fredrikson GN, Nilsson J: Accumulation of LDL in rat arteries is associated with activation of tumor necrosis factor-alpha expression. Arterioscler Thromb Vasc Biol 2000, 20:2205-2211 [DOI] [PubMed] [Google Scholar]
- 53.Davies PF: Flow-mediated endothelial mechanotransduction. Physiol Rev 1995, 75:519-560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rubanyi GM, Romero JC, Vanhoutte PM: Flow-induced release of endothelium-derived relaxing factor. Am J Physiol 1986, 250:H1145-H1149 [DOI] [PubMed] [Google Scholar]
- 55.Bhagyalakshmi A, Frangos JA: Mechanism of shear-induced prostacyclin production in endothelial cells. Biochem Biophys Res Commun 1989, 158:31-37 [DOI] [PubMed] [Google Scholar]
- 56.Vinters HV, Berliner JA: The blood vessel wall as an insulin target tissue. Diabete Metab 1987, 13:294-300 [PubMed] [Google Scholar]
- 57.Agarwal ML, Taylor WR, Chernov MV, Chernova OB, Stark GR: The p53 network. J Biol Chem 1998, 273:1-4 [DOI] [PubMed] [Google Scholar]
- 58.Hansen RS, Braithwaite AW: The growth-inhibitory function of p53 is separable from transactivation, apoptosis and suppression of transformation by E1a and Ras. Oncogene 1996, 13:995-1007 [PubMed] [Google Scholar]
- 59.Xu Q, Schett G, Li C, Hu Y, Wick G: Mechanical stress-induced heat shock protein 70 expression in vascular smooth muscle cells is regulated by Rac and Ras small G proteins but not mitogen-activated protein kinases. Circ Res 2000, 86:1122-1128 [DOI] [PubMed] [Google Scholar]