

Abstract
The traditional view on the role of serine proteases in tumor biology has changed with the recent discovery of a family of protease-activated receptors (PARs). In this study we explored the expression and functional role of the thrombin receptor PAR-1 in human colon cancer cells. Reverse transcriptase-polymerase chain reaction analysis showed that PAR-1 mRNAs are present in 11 of 14 human colon cancer cell lines tested but not in normal human colonic epithelial cells. This is in line with the immunolocalization of PAR-1 in human colon tumors and its absence in normal human colonic mucosa. The functional significance of the aberrant expression of PAR-1 in colon cancer cells was then investigated. We found that 1) a prompt increase in intracellular calcium concentration was observed on thrombin (10 nmol/L) or PAR-1 agonist AP1 (100 μmol/L) challenge of HT29 cells; 2) HT29 quiescent cells treated with thrombin (0.01 to 20 nmol/L) or AP1 (1 to 300 μmol/L) exhibited dramatic mitogenic responses (3.5-fold increase in cell number). Proliferative effects of thrombin or AP1 were also observed in other colon cancer cell lines expressing PAR-1. This effect was reversed by the MEK inhibitor PD98059 in consonance with the ability of thrombin or AP1 to induce phosphorylation of p42/p44 extracellular-regulated protein kinases. 3) PAR-1 activation by thrombin or AP1 led to a two-fold increase in cell motility of wounded HT29-D4. Our results demonstrate for the first time the aberrant expression of the functional thrombin receptor PAR-1 in colon cancers and its important involvement in cell proliferation and motility. Thrombin should now be considered as a growth factor for human colon cancer.
Progression from normal colonic mucosa to malignant tumor results from the accumulation of molecular genetic alterations triggering activation of oncogenes, inactivation of tumor-suppressor genes, and abnormalities in genes involved in DNA mismatch repair. 1,2 Moreover, several genes have been shown to be consistently up-regulated but not mutated. 1,3 This epigenetic mechanism involving genes encoding growth factors and their receptors also plays a critical role in tumor development, in particular in mucosal hyperproliferation. 3
In recent times, the traditional view on the role of proteases in tumor growth and progression has significantly changed. Indeed, besides their contribution to cancer progression by the degradation of extracellular matrix proteins 4,5 it is now clear that some proteases serve as signal molecules controlling cell functions through specific membrane receptors, the protease-activated receptors (PARs). This new class of receptors 6,7 belongs to the superfamily of the serpentine G protein-coupled receptors. These receptors are activated by serine proteases that cleave a single peptide bond in the N-terminal extracellular domain of the receptor resulting in the appearance of a new N-terminal domain that serves as a receptor agonist. 6,8 Four members of PARs are currently known. PAR-1, PAR-3, and PAR-4 are activated by thrombin 8-10 and PAR-2 is activated by trypsin-like enzymes but not by thrombin. 7,11 In addition to proteolytic activation, PAR-1, PAR-2, and PAR-4 can be also activated by synthetic peptides designed to mimic their tethered ligands. 6,7,12 PARs appear to couple primarily to Gq-mediated stimulation of inositol phosphate levels to activate target cells. PAR-1 appears also to signal via other effector systems by its ability to couple with multiple G proteins. 6,7 We recently showed that the serine protease trypsin is a very potent growth factor for human colon cancer cells through its interaction with the G protein-coupled receptor PAR-2. 13
The role of thrombin in colon cancer remains primarily unknown, although many observations reported a direct association between the thrombin pathway and colon tumor pathogenesis. First, clinical observations indicate that the coagulation system is activated in patients with colon cancer. 14,15 This activation of blood coagulation leads to generation of thrombin. 16 Second, elevation of the levels of thrombin/anti-thrombin complexes as well as thrombin fragments has been found in the blood of patients with colon cancer 14 in particular in the draining veins of colon tumors. 17 Third, it has been clearly demonstrated that colon cancer cells aggregate platelets by generation of thrombin activity. 18,19 Although activation of the thrombin pathway is probably a consequence of the disease, activation of coagulation during the disease processes could, in turn, contribute to pathogenesis of colon cancer. Thus it seems reasonable to explore whether thrombin has any direct influence on colon cancer cells.
In this context, the purpose of the present study was to determine thrombin receptor expression in human normal colon and in colon cancer cells in vivo and in vitro and to examine the potential involvement of thrombin in colon cancer cell proliferation and motility. We showed by reverse transcriptase-polymerase chain reaction (RT-PCR) the presence of PAR-1 mRNA in human colon cancer cell lines but not in normal colonic mucosa. In addition, we immunolocalized PAR-1 on the cell surface of cultured cells and in primary human colonic tumors. Finally, we provided evidence for a major role of PAR-1 activation in colon cancer cell proliferation and motility.
Materials and Methods
Reagents
The reagents were obtained from the following sources: activating peptide TFLLRNH2 (AP1) and SLIGKV (AP2), Neosystem (Strasbourg, France); highly purified α-thrombin (3000 U/mg), Kordia Laboratory Supplies (Leiden, The Netherlands); recombinant hirudin, Hoechst (Paris, France); trypsin (16,000 U/mg), goat anti-mouse IgG coupled to fluorescein and mitomycin C, Sigma Chemical Co. (St Louis, MO); Fura-2-AM, Molecular Probes (Leiden, Netherlands); Trizol reagent, Gibco-BRL (Cergy-Pontoise, France); PD98059, Calbiochem (San Diego, CA); 5-bromo-2′-deoxyuridine (BrdU) labeling kit, Boehringer Mannheim (Le Meylan, France). All other chemicals were from Bioprobe (Paris, France) or Interchim (Asnière, France).
Cell Culture
The human colon cancer cell lines HT29, SW480, HCT116, Caco-2, SW48, LoVo, LS174T, SW620, T84, HCT8, COLO-HSR, and COLO-205 were obtained from American Type Culture Collection (Rockville, MD), the HT29-Cl.19A cell line 20 was provided by Dr. C. Laboisse (INSERM U539, Nantes, France) and the HT29-D4 cell line 21 was from Dr. J. Marvaldi (Université d’Aix-Marseille, France). The main characteristics of the cell lines used in this study are described elsewhere. 22 Cells were routinely cultured in 25-cm2 plastic flasks (Costar, Cambridge, MA) and maintained at 37°C in humidified atmosphere of 5% CO2/air in Dulbecco’s modified Eagle’s medium (DMEM) (Life Technologies, Inc., Grand Island, NY) containing 4.5 g glucose/L, supplemented with 10% fetal calf serum (FCS). Caco-2 cells were grown in DMEM with 20% FCS and 1% nonessential amino acids. HCT-8 cells were grown in RPMI 1640 and T84 in 50% DMEM/50% Ham’s F12 supplemented with 10% FCS.
Fresh normal human colons with no digestive disease were obtained from France-Transplant following French bioethical law. They were removed and immediately carried to our laboratory in an isothermic box on ice. Colonic epithelial cells were isolated using ethylenediaminetetraacetic acid as previously described 23 and stored in guanidium isothiocyanate at −70°C until isolation of total RNA.
Cell Culture and Proliferation Assay
Determination of cellular proliferation was accomplished by direct cell count. Cells, 5000 per well, were seeded in 96-cluster wells (Costar) in the appropriate medium and allowed to attach for 3 days. They were starved in serum-free medium for 48 hours. Then 200 μl of a fresh serum-free medium, with or without test substances was added. Ten percent FCS was used as a positive control. After 96 hours of culture, cells were detached from triplicate wells by trypsin/ethylenediaminetetraacetic acid and counted in a hemocytometer. Cell death was evaluated with trypan blue. No significant cell death was observed after treatment with up to 500 μmol/L of AP1 or 20 nmol/L of α-thrombin.
Cell Migration
The human colonic adenocarcinoma HT29-D4 cell line was used for these studies. Wounding assays were essentially performed according to Bürk 24 with slight modifications. Briefly, cells were seeded into 35-mm diameter 6-well plates at 1.5 × 105 cells/cm2 and allowed to grow to confluency. Cell monolayers were then grown in serum-free medium for 24 hours and wounded by pressing a sterilized razor blade onto the plate to cut the cell sheet and to mark on the plate a sharp visible demarcation at the wound edge. The wounded monolayers were then washed in DMEM to remove cell debris and incubated for 24 hours at 37°C in serum-free medium with or without test substances. To distinguish between enhanced restitution by migration and proliferation, HT29-D4 cells were treated with 2.5 μg/ml of mitomycin C for 2 hours before performing wounding assays and during the 24-hour incubation with test substances. Preliminary experiments showed that 2.5 μg/ml of mitomycin C inhibited cell proliferation without affecting cell viability. At the end of incubation, cells were rinsed with phosphate-buffered saline (PBS), fixed in 3% paraformaldehyde, and stained with May-Grünwald/Giemsa. Migration of cells was assessed in blinded manner in three microscopic fields, using an inverted Leica microscope, by numbering the cell nuclei observed across the wound borders. Results (average ± SD) are from three or four independent experiments.
RT-PCR
Total cellular RNA was extracted by the acid guanidinium-thiocyanate technique. Four μg of total RNA were reverse-transcribed using oligo (dT) primer. Twenty-five percent of the cDNA mixture was amplified using human PAR-1 sense primer 5′GCC AGA ATC AAA AGC AAC 3′ and anti-sense primer 5′ATG ATG TTT AGT GGG AGG3′. PAR-2 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) cDNAs were amplified as described elsewhere. 13 Each of the 30 cycles of amplification consisted of 94°C for 40 seconds, 50°C for 40 seconds, and 72°C for 40 seconds. Amplicons were separated by electrophoresis in a 2% agarose gel, stained with ethidium bromide, and viewed under ultraviolet illumination. They were purified by the Qiaquick PCR purification kit (Qiagen, Courtaboeuf, France) and sequenced.
Immunofluorescence Staining
Indirect immunofluorescence was performed on cells grown on glass coverslips. Cells were washed in PBS, incubated with PBS containing 1% bovine serum albumin for 15 minutes before application of the primary monoclonal anti-PAR-1 antibody (WEDE15) 25 diluted 1:100, for 2 hours at room temperature. Secondary goat anti-mouse IgG antibody coupled to fluorescein was applied for 45 minutes at room temperature. Then, cells were fixed in 2% paraformaldehyde and images were examined on a Leica photomicroscope.
Tissue Immunohistochemistry
Immunohistochemistry was performed on archival materials routinely fixed in 10% paraformaldehyde, including normal sigmoid mucosa from three control patients with symptoms suggestive of irritable bowel syndrome, taken as control patients, and 12 cases of colonic adenocarcinoma (Pathology Department of Bichat Hospital, Paris). Six adenocarcinomas were located in the right colon and six in the left colon. All tumoral tissue sections stained with hematoxylin and eosin showed both cancer and adjacent mucosa either subnormal or with low-grade dysplasia. Dewaxed sections were overlaid overnight with the WEDE15 antibody diluted 1:50 in PBS. Sections incubated in PBS without primary antibody served as controls. Specific binding was detected using the streptavidin-biotin-peroxidase method (Universal Immunostaining Kit; Immunotech, Marseille, France). Sections were counterstained with Mayer’s hematoxylin.
PAR-1 immunostaining was assessed by two independent observers with a semiquantitative method. First, the percentage of immunostained epithelial cancer cells was evaluated and second, the staining intensity was scored on a scale where − represents no staining; (±), discrete; ±, weak; +, moderate; ++, strong staining.
Fura-2/AM Loading and Intracellular Calcium Measurement
Intracellular calcium concentrations were measured using Fura-2/AM. HT29 cells (5 × 103/ml) were seeded onto the center of glass coverslips and cultured in DMEM for 5 days to 70 to 90% confluence. These coverslips were then loaded with 5 μmol/L of fura-2/AM in Na-Hepes-buffered saline (saline pH 7.4, containing 135 mmol/L NaCl, 4.6 mmol/L KCl, 1.2 mmol/L MgCl2, 11 mmol/L Hepes, 11 mmol/L glucose, and 1.5 mmol/L CaCl2) containing 0.01% pluronic acid for 45 to 60 minutes at 37°C. They were then washed in Na-Hepes buffer and placed at 37°C in a fluorimeter. The fluorescence was measured with a dual-wavelength excitation fluorimeter at 340 and 380 nm for excitation and 510 nm for emission.
Western Blot Analysis
For PAR-1 expression, membrane fractions were obtained by lysing cells in 5 mmol/L of Hepes containing a cocktail of protease inhibitors (Sigma Chemical Co., St. Louis, MO) for 30 minutes at 4°C followed by homogenization in a glass homogenizer. The lysates were centrifuged at 12,000 × g for 30 minutes at 4°C. The pellet containing the membrane fraction was aliquoted and stored at −80°C until use. Protein concentrations were measured by the Bio-Rad protein assay (Bio-Rad, Hercules, CA). One hundred μg of the pellet were tested with the WEDE15 antibody diluted 1:100 (Immunotech, Marseille, France).
For extracellular-regulated protein kinase (ERK) phosphorylation assays, cells were grown in six-cluster wells (Costar) to 50% confluence and serum-deprived for 48 hours. Then quiescent cells were treated with test substances for various time periods. Cells were lysed with RIPA buffer (1× PBS, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate) containing protease inhibitor cocktail and 1 mmol/L sodium orthovanadate for 30 minutes at 4°C and lysates were centrifuged at 12,000 × g for 15 minutes. Equal amounts of extracts (50 μg) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane that was incubated first with Tris-buffered saline buffer (20 mmol/L Tris, 500 mmol/L NaCl) containing 5% (w/v) of low-fat milk and 0.1% (v/v) Tween 20 and then with phospho-specific antibodies to ERK1/2 diluted 1:2000 (New England Biolabs, Beverly, MA) overnight at 4°C. Then blots were washed and incubated with the anti-IgG-peroxidase-linked secondary antibody for 1 hour at room temperature before detection using the chemiluminescent detection kit (NEN Life Science, Paris, France) and exposure to X-ray films. The same membrane was reprobed with a polyclonal anti-ERK1/2 antibody (1:1000) (Santa Cruz Biotechnology, Santa Cruz, CA) that recognizes ERK1/2 regardless of its phosphorylation status, and served as loading control.
Statistical Analysis
All results are expressed as mean ± SD. Differences between data were tested by analysis of variance, followed by the Student’s t-test for unpaired data. A P value of <0.05 was regarded as statistically significant.
Results
Human Colon Cancer Cell Lines in Culture Express PAR-1
A large collection of human colon cancer cell lines was investigated for the expression of PAR-1 transcripts using RT-PCR. Expression of PAR-1 mRNA in confluent human colon cancer cells appears to be very frequent, eg,11 of 14 cell lines tested. As shown in the Figure 1A ▶ , a single band of the expected size (280 bp) was obtained with HT29, T84, CL19A, HCT8, SW480, LoVo, HCT116, SW48, Colo-HSR, and HT29-D4 cells but was undetectable in LS174T, SW620, and Colo205 cells (Figure 1A) ▶ . In addition, PAR-1 mRNA was expressed by both undifferentiated Caco-2 cells in exponential growth phase (Figure 1B) ▶ and by postconfluent Caco-2 cells (Figure 1, A and B) ▶ that exhibit enterocyte-like differentiation. 22 In contrast, PAR-1 mRNA is absent in epithelial cells isolated from normal human colon under experimental conditions in which a band was detected in HT29 cells (Figure 1C) ▶ . Interestingly, this expression profile is quite different from that of PAR-2, another member of the PAR family that is similarly expressed in normal and colonic cancer cells (Figure 1C) ▶ . 13 The authenticity of the amplified products in HT29 cells was verified by DNA sequence analysis, which revealed 100% identity with human PAR-1 cDNA sequence (EMBL:M62424).
Figure 1.

Constitutive expression of PAR-1 mRNA in various human colon cancer cell lines (A), undifferentiated exponentially growing (day 5) and differentiated postconfluent (day 20) Caco-2 cells (B), and HT29 cells (C) versus normal human colon epithelium. Total RNA extracted was reverse-transcribed and PCR-amplified with PAR-1 (A–C, top), GAPDH primers (A and B, bottom), or PAR-2 primers (C, bottom). A single PCR-amplified product of the exact predicted size (280 bp) for PAR-1 was visualized after electrophoresis in a 2% agarose gel. Note that in B, PAR-1 is present in both undifferentiated and differentiated Caco-2 cells and that in C, contrary to PAR-1, PAR-2 is equally expressed in normal and cancer cells.
To provide evidence for aberrant PAR-1 protein expression in cultured colon cancer cells, indirect immunofluorescence studies were performed on nonpermeabilized cells using the WEDE15 monoclonal antibody directed against an extracellular epitope of PAR-1. 25 Under such experimental conditions cell surface localization of PAR-1 can be readily demonstrated. We first investigated cell lines exhibiting expression of PAR-1 mRNA such as HT29, HT29-D4, and HCT116 and detected PAR-1 on the surface of these cells (Figure 2 ▶ , left). In contrast, no fluorescence could be detected on the surface of LS174T cells (Figure 2 ▶ , left) in which no PAR-1 mRNA was detectable (Figure 1A) ▶ . As shown in HT29 cells, the anti-PAR-1 antibody recognizes a 66-kd protein in immunoblotting experiments (Figure 2 ▶ , right). These data are consistent with the molecular weight of human PAR-1. 26
Figure 2.

Immunodetection of PAR-1 receptor in human colon cancer cells with WEDE15 antibody. Left: Immunofluorescence detection of PAR-1. Cells were fixed using 2% paraformaldehyde. PAR-1 protein is evident on the surface of HT29 cells (A), HT29-D4 cells (B), and HCT116 cells (C). No immunofluorescence was observed on LS174T cells (D). Right: Immunoblot of PAR-1 in HT29 cells. One band running at a size of ∼66 kd can be seen. Simultaneous detection with a control IgG does not reveal any immunoreactivity on HT29 lysate. Original magnification, ×400 (A–D).
PAR-1 Is also Aberrantly Expressed in Human Colon Tumors in Vivo
Because these results suggest an ectopic expression of PAR-1 in colon cancer, we investigated PAR-1 expression in human colonic adenocarcinomas versus normal colonic epithelium by immunohistochemistry. In normal human colonic mucosa from control patients without colonic cancer no staining was observed (Figure 3A) ▶ . In patients with colon cancer a discrete immunoreactivity in the normal mucosa, far from the neoplastic tissue, was detected (Figure 3B) ▶ . In contrast, this staining clearly increased in the low-grade dysplastic mucosa contiguous to the cancerous lesion, being seen mainly in the upper part of the crypts (Figure 3, C and D) ▶ . Eleven of 12 adenocarcinomas (91%) also expressed PAR-1 in the cancerous epithelium regardless of the site of the tumor within the colon (Table 1 ▶ and Figure 3E ▶ ), although the staining appeared variable in intensity and generally weaker than in adjacent dysplastic mucosa. No immunolabeling was seen when the primary antibody was omitted (not shown). We conclude that PAR-1 is aberrantly expressed in epithelial cells during colon carcinogenesis.
Figure 3.
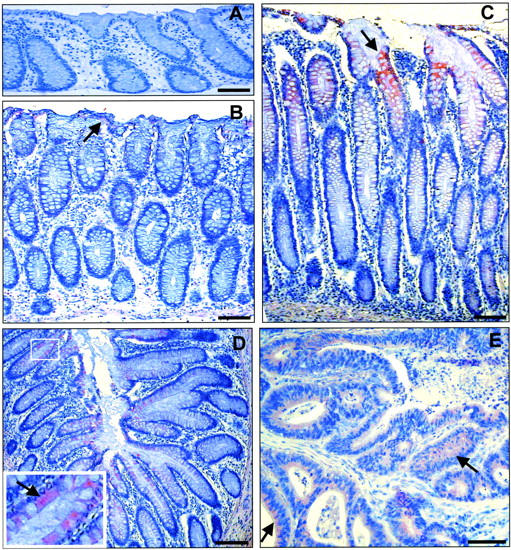
Representative immunostaining for the thrombin receptor PAR-1 with WEDE15 (A–E) in paraffin sections of normal colonic mucosa and colonic tissues from patients with adenocarcinoma. A: PAR-1 immunoreactivity is absent in normal mucosa from control patients. B and C: Two sections from a colonic mucosa distant from a right colon adenocarcinoma. B: Normal mucosa 4 cm from the lesion shows very discrete immunoreactivity on the surface of the epithelium (arrow). C: In the low-grade dysplastic colonic mucosa contiguous to the cancerous lesion, clear immunoreactivity is present in the epithelium of the upper part of the crypt (arrow). D: In the low-grade dysplastic colonic mucosa contiguous to a left colon adenocarcinoma from another patient, similar immunoreactivity as in C is seen. Arrow in the enlarged area in D shows positive immunoreactivity in the colonocytes except in the goblet cells. E: Partial view of the adenocarcinoma contiguous to the dysplastic colonic mucosa seen in D. PAR-1 immunoreactivity in the cancerous epithelial cords is uniformly present but weaker than in the dysplastic mucosa (arrows). Scale bars, 100 μm.
Table 1.
Estimation of PAR-1 Expression in Colon Carcinoma Epithelial Cells
| Cases | Dukes stage | Tumor site | Percent of stained cells, % | Staining intensity |
|---|---|---|---|---|
| 1 | B | Right colon | 90 | + to ++ |
| 2 | B | Cecum | 80 | ± to ++ |
| 3 | B | Sigmoid | 50 | ± to ++ |
| 4 | B | Sigmoid | 100 | ± |
| 5 | C | Right colon | 0 | − |
| 6 | B | Sigmoid | 70 | ± to ++ |
| 7 | B | Cecum | 100 | + to ++ |
| 8 | B | Left colon | 100 | ± to ++ |
| 9 | B | Right colon | 100 | ± to ++ |
| 10 | B | Left colon | 40 | ± |
| 11 | A | Sigmoid | 100 | ± |
| 12 | B | Right colon | 50 | (±) |
Staining intensity was often variable from place to place and scored as: −, negative; (±), discrete; ±, weak; +, moderate; ++, strong.
PAR-1 Is Functionally Active in Human Colon Cancer Cells
To characterize the functionality of PAR-1 in colon cancer cells, we first studied the effect of PAR-1 activators on intracellular calcium mobilization in HT29 cells (Figure 4) ▶ . Thrombin and the AP1 peptide, a selective PAR-1 agonist, 27 were used as activators. As shown in Figure 4A ▶ , thrombin (10 nmol/L) produced an increase in cytosolic calcium transients. The peptide AP1 also elicited a rapid rise of intracellular calcium (Figure 4B) ▶ . After a first challenge with AP1 the calcium response to thrombin was almost completely abolished. This suggests that the thrombin receptor has been desensitized by the selective PAR-1 agonist and that thrombin signals calcium in HT29 cells mainly via PAR-1. As expected, responses to thrombin were blocked by hirudin, a thrombin inhibitor, 28 (Figure 4C) ▶ whereas this inhibitor did not affect the response to AP1 (Figure 4D) ▶ . In HT29 cells PAR-1 is co-expressed with PAR-2. 13 Indeed, after a first challenge with thrombin or AP1, a further challenge with PAR-2 activators (trypsin or AP2), produced a robust calcium response (Figure 4, A and B) ▶ . Calcium mobilization was also observed on stimulation with thrombin and AP1 in the other tested cell lines HT29-D4, Cl19A, HCT116, and LoVo, which exhibit detectable PAR-1 mRNA or protein (Figure 1A ▶ and Figure 2 ▶ ). We also performed experiments in which HT29 cells were assayed in Ca2+-free medium on exposure to AP1 (100 μmol/L) or thrombin (10 nmol/L). The calcium responses to AP1 or thrombin (not shown) were unaffected indicating that Ca2+ derives from intracellular pools.
Figure 4.

Effect of α-thrombin and AP1 on calcium signaling. HT29 cells were loaded with Fura-2/AM and assayed in medium containing Ca2+. A: Thrombin (1 U/ml) followed by a second challenge with trypsin (10 nmol/L). B: The cells were challenged first with the activating peptide AP1 (100 μmol/L) followed by a second challenge with thrombin (1 U/ml) and a third challenge with the activating peptide for PAR-2, AP2 (100 μmol/L). C: HT29 cells were challenged with thrombin (1 U/ml) pretreated with its inhibitor, hirudin (10 U/ml), then with AP1 (100 μmol/L). Note that cells are still responsive to a second challenge with AP1. D: HT29 cells were challenged with AP1 (100 μmol/L) preincubated with hirudin. All of the compounds were given at the arrows. These results are representative of three others.
Thrombin Acting through PAR-1 Is a Growth Factor for Human Colon Cancer Cells
Thrombin is known to induce proliferation of endothelial cells and a few other cell types. 6,7 The finding of PAR-1 expression in human colon cancer cells prompted us to evaluate the effect of thrombin on cell growth. This was first investigated in HT29 cells. In the proliferation assay conditions used, thrombin elicited a dramatic increase in cell number. Indeed, a 333% augmentation of cell number greater than basal was observed on challenge of cells with 20 nmol/L of thrombin. Thrombin is active in the range of concentrations between 0.1 and 20 nmol/L (Figure 5) ▶ with half-maximal response obtained for ∼3 nmol/L of thrombin. At these doses, cells showed morphology similar to that of untreated cells. At higher concentrations of thrombin an important detachment of HT29 cells from plastic wells occurred and the assay was no longer reliable. The selective PAR-1-activating peptide AP1 also elicited a dose-dependent mitogenic response in HT29 cells (Figure 5) ▶ , a half-maximal response being obtained with 50 μmol/L of AP1. Maximal response to AP1 (300 μmol/L) was similar to that induced by 20 nmol/L of thrombin (Figure 5) ▶ . Unlike thrombin, AP1 did not induce HT29 cell detachment regardless of peptide concentration used (not shown). Maximal responses elicited by thrombin (20 nmol/L) or AP1 (300 μmol/L) were very important in HT29 cells because they represented as much as 40% of the maximal mitogenic response induced by 10% serum, which reached a maximum of ∼10-fold more than serum-starved controls (not shown). When the specific thrombin inhibitor hirudin was preincubated with thrombin, a profound inhibition of thrombin-induced growth effect was observed (Figure 5) ▶ . The mitogenic effects of thrombin and AP1 have been extended to HCT116, HT29-D4, Cl.19A, and LoVo cell lines. 29 Figure 6 ▶ shows the mitogenic responses to thrombin (10 nmol/L), AP1 (100 μmol/L), and FCS (10%). As expected, neither thrombin nor AP1 had a significant effect on proliferation of LS174T cells that do not express PAR-1 at the cell surface (see above).
Figure 5.

Concentration-dependent stimulation of HT29 cell proliferation by thrombin or AP1. Quiescent cells were grown in serum-free medium with the indicated concentration of thrombin (filled circles), AP1 (open circles), or thrombin and hirudin (10 U/ml) (filled triangle). After 96 hours, cells from triplicate wells were counted for each condition. Data are means ± SD of three different experiments.
Figure 6.

Ability of PAR-1 ligands (AP1, thrombin) to induce cell proliferation in different human colon cancer cell lines. Quiescent cells were grown for 96 hours in serum-free medium without or with 100 μmol/L of AP1, 10 nmol/L of thrombin, or 10% FCS. Cells from triplicate wells were counted for each condition. Data are means ± SD. ***, P < 0.0001; **, P < 0.001, thrombin-, AP1-, or 10% FCS-treated cells versus control cells.
The ERK cascade plays a critical role in the regulation of cell proliferation. 30 In an attempt to document the mechanism whereby thrombin promotes colon cancer cell proliferation, we tested the MEK inhibitor PD98059 (Figure 7) ▶ . Pretreatment of HT29 cells with increasing concentrations of PD98059 resulted in a dose-dependent decrease in cell number induced by thrombin (10 nmol/L) or AP1 (100 μmol/L) (Figure 7) ▶ . At the maximal dose of inhibitor tested (25 μmol/L), the proliferative effects of thrombin or AP1 were reversed by 70 to 80% (Figure 7) ▶ . Cell counts from control cells treated with PD98059 alone were comparable to those of diluent-treated cells (Figure 7) ▶ . In addition, trypan blue staining of cells treated with 25 μmol/L of PD98059 revealed 99.9% viability, suggesting that PD98059 is not toxic to HT29 cells. These results suggest that cell proliferation induced by thrombin or AP1 involves the phosphorylation of ERKs in HT29 cells. This was demonstrated because addition of thrombin (10 nmol/L) or AP1 (100 μmol/L) to quiescent HT29 cells induced a rapid and significant phosphorylation of p42/p44 (Figure 8A) ▶ . The maximum of phosphorylation for the two agonists was reached within 5 to 10 minutes and stayed above the basal level for the remaining time tested. An immunoblot using anti-ERK1/2 was performed at each time of activation to ensure equal loading of total ERK on the gel (Figure 8A) ▶ . In addition, the thrombin- or AP1-induced phosphorylation of ERK1/2 is abrogated in the presence of the MEK inhibitor PD98059 (Figure 8B) ▶ . All these results clearly show that thrombin acts as a growth factor for colon cancer cells through the activation of PAR-1 and via the MAPK pathway.
Figure 7.

Dose-dependent inhibition of thrombin-induced cell proliferation by the MEK inhibitor PD98059. Quiescent HT29 cells were grown for 96 hours in serum-free medium without (control) or with 100 μmol/L of AP1 or 10 nmol/L of thrombin in the presence of the indicated concentrations of MEK inhibitor (PD98056). Data are means ± SD of three separate experiments.
Figure 8.

Activation of p42/p44 MAPK by thrombin and AP1 and effect of MEK inhibitor (PD98059) on thrombin- and AP1-induced phosphorylation. A: Immunoblot with phospho-specific p42/p44 MAPK antibodies on HT29 cell lysate treated with or without thrombin (10 nmol/L) or AP1 (100 μmol/L) for the indicated time periods (shown in minutes). B: Phosphorylated form of p42/p44 MAPK in HT29 cells pretreated with 20 μmol/L of PD98059 and then with or without thrombin (10 nmol/L) or AP1 (100 μmol/L) addition for 5 minutes. To confirm equal protein loading, the membranes were stripped and blotted with p42/p44 antibodies.
PAR-1 Activators also Affect Colon Cancer Cell Motility
To be invasive, cancer cells undergo a sequence of events initiated by cell migration from their origin sites. Therefore we investigated the potential role of thrombin on colon cancer motility by using the in vitro wounded HT29-D4 model. 31 When the HT29-D4 monolayer was wounded and incubated for 24 hours in serum-free medium alone, very few cells crossed the wound (Figure 9) ▶ . Addition of thrombin (5 nmol/L) induced a significant enhancement of colon cancer cell migration into the denuded area. Similar results were obtained with the PAR-1 agonist AP1 (Figure 9) ▶ , which induced a dose-dependent stimulation of cell migration (Figure 10A) ▶ . Interestingly, thrombin was as active as insulin (Figure 10B) ▶ , a well-known stimulator of HT29-D4 cell motility. 31 In our experimental conditions few nuclei showed BrdU labeling confirming that mitomycin C did inhibit proliferation (see Materials and Methods). This indicated that the enhancement of cell migration by thrombin or AP1 was not the result of an effect on cell proliferation. These results show that PAR-1 activation is not only involved in proliferation but also in migration of colon cancer cells.
Figure 9.
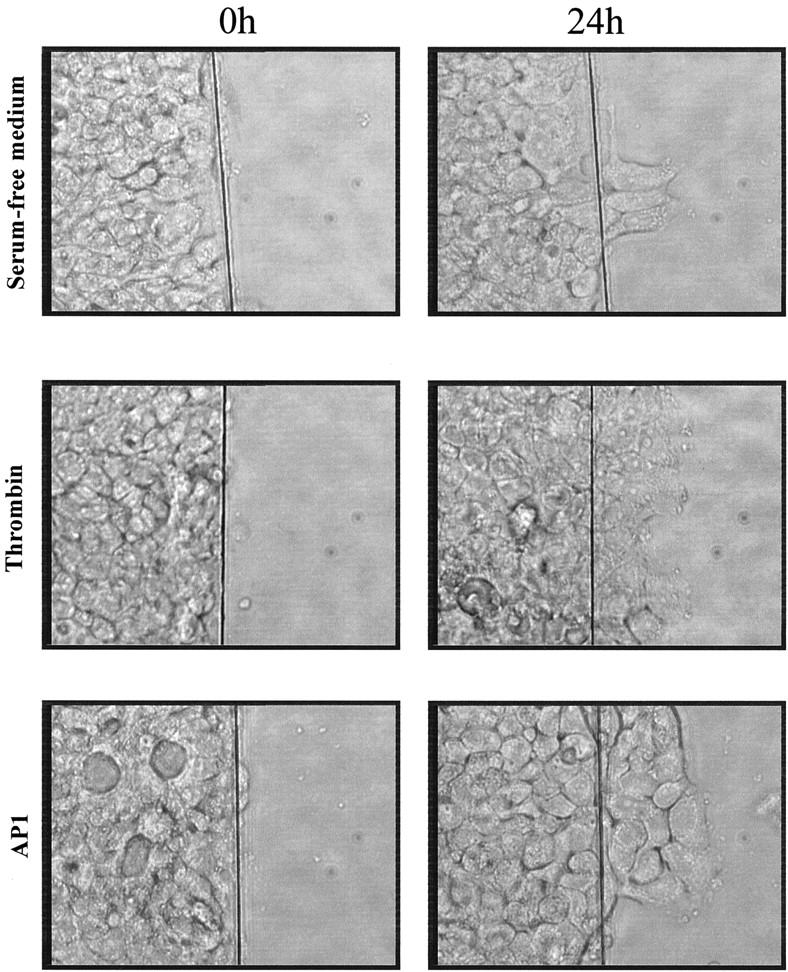
Effect of thrombin on migration of HT29-D4 cells. Wounded monolayers were cultured for 24 hours in serum-free medium in the absence or presence of 5 nmol/L of thrombin or 300 μmol/L of AP1. This result is representative of four independent experiments.
Figure 10.

Effect of thrombin and AP1 on migration of HT29-D4 cells. The migrating cells across the wound were fixed, stained, and counted. A: Wounded monolayers were cultured for 24 hours in serum-free medium in the presence of various concentration of AP1. B: Wounded monolayers were cultured for 24 hours in serum-free medium in the absence or presence of thrombin (5 nmol/L) or insulin (50 nmol/L) taken as a positive control. Results are means ± SD of three (A) and four (B) independent experiments. ***, P < 0.001; **, P < 0.01; *, P < 0.05, insulin-, thrombin- or AP1-treated cells versus control cells.
Discussion
We report here that the thrombin receptor subtype PAR-1 is ectopically expressed in human colon cancer cells. To our knowledge, this is the first evidence for thrombin receptors in colon cancer. In contrast, PAR-1 is clearly not present in epithelial cells of normal human colon. In colon cancer cells, thrombin appears to promote efficient cell growth and migration. Therefore, thrombin that does not exert a direct control of normal epithelial function in human colon becomes a potent growth factor for colon cancer owing to the aberrant expression of its receptor PAR-1.
This study shows that PAR-1 is expressed in many colon tumors tested in vivo as well as colon cancer cell lines in culture. In vivo, we have demonstrated by immunohistochemistry that PAR-1 is present in the low-grade dysplastic mucosa and cancer mucosal tissues but is expressed at a very low level, if any, in the adjacent normal mucosa. Normal mucosa from control colonic tissues does not stain at all with the PAR-1 antibody. This is in line with the failure of RT-PCR to detect any PAR-1 mRNA in isolated cells from normal human colon. With regard to colon tumor specimens they were only investigated by immunohistochemistry techniques and not used for RT-PCR and immunoblotting because they contain blood vessel cells that are a major source of PAR-1. 6,7 The presence of PAR-1 in human colon cancer was further demonstrated by RT-PCR, immunoblotting, and immunofluorescence analysis of cultured cell lines. Measurement of calcium transients showed that PAR-1 is functional in colon cancer cell lines. Interestingly, the expression of PAR-1 does not correlate with the differentiation state of colon cancer cell lines. Indeed PAR-1 expression was found in undifferentiated cells such as HT29, SW480, HCT8, LoVo, and HCT116 22 and polarized enterocyte-like cells such as CL19A and Caco-2. 20,22 The absence of PAR-1 in some cell lines is unclear. In this context, it is worth noting that LS174T, COLO205, and SW620, which do not express PAR-1 mRNAs, show no aberration of chromosome 5 (American Type Culture Collection: CCL-188, CCL-222, and CCL-227, respectively) on which PAR-1 gene is localized. 29 Another explanation to the absence of PAR-1 may be the existence of a down-regulation of PAR-1 gene by diffusable signals in some cancer cells. Further studies on PAR-1 regulation in colon cancer cells in vitro is needed.
Altogether our data demonstrate that functional PAR-1 is aberrantly expressed in human colon cancer cells. The mechanism whereby the PAR-1 gene is switched on in colon cancer is unknown but ectopic expression of PAR-1 can probably be extended to other epithelial cancers. Indeed, PAR-1 has been reported to be highly expressed in breast carcinoma, 32 pancreatic adenocarcinoma, 33 laryngeal carcinoma, 34 renal carcinoma, 35 lung adenocarcinoma, gastric carcinoma, 36 and oral squamous cell carcinoma. 37 Again, the expression of PAR-1 appears to be very low or absent in normal epithelial tissues from which these cancers arise. Thus it may be suggested that malignant transformation of epithelial cancers, including colon cancer, is associated with the induction of the PAR-1 gene. Because PAR-1-mediated signaling can cause cell transformation, 38 the aberrant expression of PAR-1 in colon cancers and more generally epithelial cancers can be considered as an important event in epithelial tumorigenesis.
The recent cloning of PARs showed the existence of two other thrombin receptors PAR-3 and PAR-4 in addition to PAR-1. Although, PAR-3 and PAR-4 mRNAs can be detected by RT-PCR in some colon cancer cells (unpublished data), several lines of evidence indicate that the effects of thrombin in human colon cancer cells are mediated by functional PAR-1 but probably not by PAR-3 or PAR-4: 1) the PAR-1 agonist AP1 27,39 mimics all of the effects of thrombin on intracellular calcium transients, cell proliferation, and cell motility whereas AP1 does not activate other PARs; 27 2) an initial challenge of HT29 cells with AP1 desensitizes the effect of thrombin on calcium transients; 3) PAR-4 is relatively insensitive to thrombin and may function as a low-affinity receptor, 7,10,40 and this contrasts with the high potency of thrombin in promoting HT29 colon cancer cell growth with an EC50 of 3 nmol/L; 4) PAR-3 itself may not be a fully functional receptor but rather may play a role as a tethering protein for thrombin. 7,41
In this study, we show for the first time that thrombin and its agonist, AP1, are very potent stimulators of human colon cancer cell proliferation, their effects representing up to 40% of the maximal mitogenic response induced by 10% FCS. The proliferative effect is 1) related to the proteolytic activity of thrombin because it is completely abrogated by hirudin, a specific thrombin inhibitor; 28 2) observed at low thrombin concentrations with an EC50 of 3 nmol/L; 3) PAR-1-mediated because it is mimicked by the specific PAR-1 agonist AP1. 27,39 In this context, thrombin should be considered as an important growth factor for colon cancer cells. It is as efficient as or even more potent than known growth factors for colonic cancer cells such as transforming growth factor-α, 42 insulin-like growth factors, 43 hepatocyte growth factor, 44 neurotensin, 45 or glycine-extended gastrin. 46 As extensively documented in the literature 6,7 thrombin stimulates MAP kinases in its target cells. We have pharmacological evidence demonstrating that the activation of MAP kinases does participate in the thrombin-mediated events leading to an increase in cell proliferation of colon cancer cells. This is based on the findings that the specific inhibitor of MAP kinase PD98059 abolishes the thrombin-promoting effect on colon cancer cell growth (Figure 8) ▶ . Thrombin has been also proposed to modulate cell adhesion and invasion in some tumors. 47-49 In this study, we show that thrombin induces cell migration in the in vitro wounded HT29-D4 model. This is in line with its ability to enhance adhesion and migration in SW480 colon cancer cells. 50 We further demonstrated that the motogenic effect of thrombin is PAR-1-mediated because it is mimicked by AP1 (Figures 9 and 10) ▶ . Therefore, thrombin combines proliferative and motogenic effects in colon cancer cells.
The proliferative and motogenic effects of thrombin on colon cancer cells raise the question of the endogenous sources of thrombin that could activate the PAR-1 receptor in colon tumors in vivo. Thrombin generation is probably the direct result of the overactivation of the coagulation system, a widely described abnormality in various cancer patients 51 including those with colon cancers. 14,15 Thus, blood is probably a major provider of thrombin to colon cancer. However, thrombin activation of PAR-1 in transformed colon epithelial cells requires that thrombin reaches the extracellular environment of colonic tumors. It may be suggested that thrombin can cross locally the leaky vessels 52 that are formed during neo-angiogenesis associated with the development of colon tumors. 53 Although one study using hirudin binding did not suggest the presence of thrombin in colon cancer tissues, 54 two other reports 18,19 demonstrated that colon adenocarcinoma cells produce thrombin-like activity as assessed by platelet aggregation assay. Therefore a second important source of thrombin for colon cancer cells could be the tumor itself suggesting that thrombin may be also involved in an autocrine/paracrine loop within the colonic tumor resulting in cancer cell growth and motility.
In conclusion, our results demonstrate for the first time the aberrant expression of the functional thrombin receptor PAR-1 in colon cancer cells and its involvement in thrombin-induced effects on proliferation and cell motility. On the basis of our results and other results reporting that PAR-1 can function as an oncoprotein that transforms fibroblasts in vitro, 38 we speculate that PAR-1 and its physiological activator, thrombin, should now be considered as crucial contributors to the development of human colon cancer.
Acknowledgments
We thank Dr. Francine Walker (Pathology Department of Bichat Hospital, Paris) for providing the specimens for theses studies.
Footnotes
Address reprint requests to Dalila Darmoul, Ph.D., Neuroendocrinologie et Biologie Cellulaire Digestives, INSERM U410, Faculté de Médecine Xavier Bichat 75018 Paris, France. E-mail: darmoul@bichat.inserm.fr.
References
- 1.Chung DC: The genetic basis of colorectal cancer: insights into critical pathways of tumorigenesis. Gastroenterology 2000, 119:854-865 [DOI] [PubMed] [Google Scholar]
- 2.Kinzler KW, Vogelstein B: Lessons from hereditary colorectal cancer. Cell 1996, 87:159-170 [DOI] [PubMed] [Google Scholar]
- 3.Favoni RE, de Cupis A: The role of polypeptide growth factors in human carcinomas: new targets for a novel pharmacological approach. Pharmacol Rev 2000, 52:179-206 [PubMed] [Google Scholar]
- 4.Mignatti P, Rifkin DB: Biology and biochemistry of proteinases in tumor invasion. Physiol Rev 1993, 73:161-195 [DOI] [PubMed] [Google Scholar]
- 5.McCawley LJ, Matrisian LM: Matrix metalloproteinases: multifunctional contributors to tumor progression. Mol Med Today 2000, 6:149-156 [DOI] [PubMed] [Google Scholar]
- 6.Dery O, Corvera CU, Steinhoff M, Bunnett NW: Proteinase-activated receptors: novel mechanisms of signaling by serine proteases. Am J Physiol 1998, 274:C1429-C1452 [DOI] [PubMed] [Google Scholar]
- 7.Macfarlane SR, Seatter MJ, Kanke T, Hunter GD, Plevin R: Proteinase-activated receptors. Pharmacol Rev 2001, 53:245-282 [PubMed] [Google Scholar]
- 8.Vu TK, Hung DT, Wheaton VI, Coughlin SR: Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 1991, 64:1057-1068 [DOI] [PubMed] [Google Scholar]
- 9.Ishihara H, Connolly AJ, Zeng D, Kahn ML, Zheng YW, Timmons C, Tram T, Coughlin SR: Protease-activated receptor 3 is a second thrombin receptor in humans. Nature 1997, 386:502-506 [DOI] [PubMed] [Google Scholar]
- 10.Xu WF, Andersen H, Whitmore TE, Presnell SR, Yee DP, Ching A, Gilbert T, Davie EW, Foster DC: Cloning and characterization of human protease-activated receptor 4. Proc Natl Acad Sci USA 1998, 95:6642-6646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nystedt S, Emilsson K, Wahlestedt C, Sundelin J: Molecular cloning of a potential proteinase activated receptor. Proc Natl Acad Sci USA 1994, 91:9208-9212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hollenberg MD, Compton SJ: International Union of Pharmacology. XXVIII proteinase-activated receptors. Pharmacol Rev 2002, 54:203-217 [DOI] [PubMed] [Google Scholar]
- 13.Darmoul D, Marie JC, Devaud H, Gratio V, Laburthe M: Initiation of human colon cancer cell proliferation by trypsin acting at protease-activated receptor-2. Br J Cancer 2001, 85:772-779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iversen LH, Okholm M, Thorlacius-Ussing O: Pre- and postoperative state of coagulation and fibrinolysis in plasma of patients with benign and malignant colorectal disease—a preliminary study. Thromb Haemost 1996, 76:523-528 [PubMed] [Google Scholar]
- 15.Modrau II, Iversen LL, Thorlacius-Ussing OO: Hemostatic alterations in patients with benign and malignant colorectal disease during major abdominal surgery. Thromb Res 2001, 104:309-315 [DOI] [PubMed] [Google Scholar]
- 16.Kakkar AK, DeRuvo N, Chinswangwatanakul V, Tebbutt S, Williamson RC: Extrinsic-pathway activation in cancer with high factor VIIa and tissue factor. Lancet 1995, 346:1004-1005 [DOI] [PubMed] [Google Scholar]
- 17.Garcia-Avello A, Galindo-Alvarez J, Martinez-Molina E, Cesar-Perez J, Navarro JL: Coagulative system activation and fibrinolytic system inhibition activities arise from tumoral draining vein in colon carcinoma. Thromb Res 2001, 104:421-425 [DOI] [PubMed] [Google Scholar]
- 18.Pearlstein E, Ambrogio C, Gasic G, Karpatkin S: Inhibition of the platelet-aggregating activity of two human adenocarcinomas of the colon and an anaplastic murine tumor with a specific thrombin inhibitor, dansylarginine N-(3-ethyl-1, 5-pentanediyl)amide. Cancer Res 1981, 41:4535-4539 [PubMed] [Google Scholar]
- 19.Chiang HS, Swaim MW, Huang TF: Characterization of platelet aggregation induced by human colon adenocarcinoma cells and its inhibition by snake venom peptides, trigramin and rhodostomin. Br J Haematol 1994, 87:325-331 [DOI] [PubMed] [Google Scholar]
- 20.Augeron C, Laboisse CL: Emergence of permanently differentiated cell clones in a human colonic cancer cell line in culture after treatment with sodium butyrate. Cancer Res 1984, 44:3961-3969 [PubMed] [Google Scholar]
- 21.Fantini J, Abadie B, Tirard A, Remy L, Ripert JP, el Battari A, Marvaldi J: Spontaneous and induced dome formation by two clonal cell populations derived from a human adenocarcinoma cell line, HT29. J Cell Sci 1986, 83:235-249 [DOI] [PubMed] [Google Scholar]
- 22.Zweibaum A, Laburthe M, Grasset E, Louvard D: The gastrointestinal system: intestinal absorption and secretion. Field M Frizzell RA eds. Handbook of Physiology. 1991, :pp 223-255 American Physiological Society, Bethesda [Google Scholar]
- 23.Salomon R, Couvineau A, Rouyer-Fessard C, Voisin T, Lavallee D, Blais A, Darmoul D, Laburthe M: Characterization of a common VIP-PACAP receptor in human small intestinal epithelium. Am J Physiol 1993, 264:E294-E300 [DOI] [PubMed] [Google Scholar]
- 24.Burk RR: A factor from a transformed cell line that affects cell migration. Proc Natl Acad Sci USA 1973, 70:369-372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoxie JA, Ahuja M, Belmonte E, Pizarro S, Parton R, Brass LF: Internalization and recycling of activated thrombin receptors. J Biol Chem 1993, 268:13756-13763 [PubMed] [Google Scholar]
- 26.Wojtukiewicz MZ, Tang DG, Ben-Josef E, Renaud C, Walz DA, Honn KV: Solid tumor cells express functional “tethered ligand” thrombin receptor. Cancer Res 1995, 55:698-704 [PubMed] [Google Scholar]
- 27.Kawabata A, Saifeddine M, Al-Ani B, Leblond L, Hollenberg MD: Evaluation of proteinase-activated receptor-1 (PAR1) agonists and antagonists using a cultured cell receptor desensitization assay: activation of PAR2 by PAR1-targeted ligands. J Pharmacol Exp Ther 1999, 288:358-370 [PubMed] [Google Scholar]
- 28.Liu LW, Vu TK, Esmon CT, Coughlin SR: The region of the thrombin receptor resembling hirudin binds to thrombin and alters enzyme specificity. J Biol Chem 1991, 266:16977-16980 [PubMed] [Google Scholar]
- 29.Schmidt VA, Vitale E, Bahou WF: Genomic cloning and characterization of the human thrombin receptor gene. Structural similarity to the proteinase activated receptor-2 gene. J Biol Chem 1996, 271:9307-9312 [PubMed] [Google Scholar]
- 30.Schramek H: MAP kinases: from intracellular signals to physiology and disease. News Physiol Sci 2002, 17:62-67 [DOI] [PubMed] [Google Scholar]
- 31.Andre F, Rigot V, Remacle-Bonnet M, Luis J, Pommier G, Marvaldi J: Protein kinases C-gamma and -delta are involved in insulin-like growth factor I-induced migration of colonic epithelial cells. Gastroenterology 1999, 116:64-77 [DOI] [PubMed] [Google Scholar]
- 32.Even-Ram S, Uziely B, Cohen P, Grisaru-Granovsky S, Maoz M, Ginzburg Y, Reich R, Vlodavsky I, Bar-Shavit R: Thrombin receptor overexpression in malignant and physiological invasion processes. Nat Med 1998, 4:909-914 [DOI] [PubMed] [Google Scholar]
- 33.Rudroff C, Seibold S, Kaufmann R, Zetina CC, Reise K, Schafer U, Schneider A, Brockmann M, Scheele J, Neugebauer EA: Expression of the thrombin receptor PAR-1 correlates with tumour cell differentiation of pancreatic adenocarcinoma in vitro. Clin Exp Metastasis 2002, 19:181-189 [DOI] [PubMed] [Google Scholar]
- 34.Kaufmann R, Schafberg H, Rudroff C, Nowak G: Thrombin receptor activation results in calcium signaling and protein kinase C-dependent stimulation of DNA synthesis in HEp-2g laryngeal carcinoma cells. Cancer 1997, 80:2068-2074 [DOI] [PubMed] [Google Scholar]
- 35.Kaufmann R, Junker U, Junker K, Nuske K, Ranke C, Zieger M, Scheele J: The serine proteinase thrombin promotes migration of human renal carcinoma cells by a PKA-dependent mechanism. Cancer Lett 2002, 180:183-190 [DOI] [PubMed] [Google Scholar]
- 36.D’Andrea MR, Derian CK, Santulli RJ, Andrade-Gordon P: Differential expression of protease-activated receptors-1 and -2 in stromal fibroblasts of normal, benign, and malignant human tissues. Am J Pathol 2001, 158:2031-2041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Y, Gilcrease MZ, Henderson Y, Yuan XH, Clayman GL, Chen Z: Expression of protease-activated receptor 1 in oral squamous cell carcinoma. Cancer Lett 2001, 169:173-180 [DOI] [PubMed] [Google Scholar]
- 38.Martin CB, Mahon GM, Klinger MB, Kay RJ, Symons M, Der CJ, Whitehead IP: The thrombin receptor, PAR-1, causes transformation by activation of Rho-mediated signaling pathways. Oncogene 2001, 20:1953-1963 [DOI] [PubMed] [Google Scholar]
- 39.Hollenberg MD, Saifeddine M, al-Ani B, Kawabata A: Proteinase-activated receptors: structural requirements for activity, receptor cross-reactivity, and receptor selectivity of receptor-activating peptides. Can J Physiol Pharmacol 1997, 75:832-841 [PubMed] [Google Scholar]
- 40.Kahn ML, Zheng YW, Huang W, Bigornia V, Zeng D, Moff S, Farese RV, Jr, Tam C, Coughlin SR: A dual thrombin receptor system for platelet activation. Nature 1998, 394:690-694 [DOI] [PubMed] [Google Scholar]
- 41.Nakanishi-Matsui M, Zheng YW, Sulciner DJ, Weiss EJ, Ludeman MJ, Coughlin SR: PAR3 is a cofactor for PAR4 activation by thrombin. Nature 2000, 404:609-613 [DOI] [PubMed] [Google Scholar]
- 42.Mendelsohn J: The epidermal growth factor receptor as a target for cancer therapy. Endocr Relat Cancer 2001, 8:3-9 [DOI] [PubMed] [Google Scholar]
- 43.Giovannucci E: Insulin, insulin-like growth factors and colon cancer: a review of the evidence. J Nutr 2001, 131:3109S-3120S [DOI] [PubMed] [Google Scholar]
- 44.Dignass AU, Lynch-Devaney K, Podolsky DK: Hepatocyte growth factor/scatter factor modulates intestinal epithelial cell proliferation and migration. Biochem Biophys Res Commun 1994, 202:701-709 [DOI] [PubMed] [Google Scholar]
- 45.Maoret JJ, Anini Y, Rouyer-Fessard C, Gully D, Laburthe M: Neurotensin and a non-peptide neurotensin receptor antagonist control human colon cancer cell growth in cell culture and in cells xenografted into nude mice. Int J Cancer 1999, 80:448-454 [DOI] [PubMed] [Google Scholar]
- 46.Stepan VM, Sawada M, Todisco A, Dickinson CJ: Glycine-extended gastrin exerts growth-promoting effects on human colon cancer cells. Mol Med 1999, 5:147-159 [PMC free article] [PubMed] [Google Scholar]
- 47.Walz DA, Fenton JW: The role of thrombin in tumor cell metastasis. Invasion Metastasis 1994, 14:303-308 [PubMed] [Google Scholar]
- 48.Nierodzik ML, Kajumo F, Karpatkin S: Effect of thrombin treatment of tumor cells on adhesion of tumor cells to platelets in vitro and tumor metastasis in vivo. Cancer Res 1992, 52:3267-3272 [PubMed] [Google Scholar]
- 49.Wojtukiewicz MZ, Tang DG, Ciarelli JJ, Nelson KK, Walz DA, Diglio CA, Mammen EF, Honn KV: Thrombin increases the metastatic potential of tumor cells. Int J Cancer 1993, 54:793-806 [DOI] [PubMed] [Google Scholar]
- 50.Chiang HS, Yang RS, Huang TF: Thrombin enhances the adhesion and migration of human colon adenocarcinoma cells via increased beta 3-integrin expression on the tumour cell surface and their inhibition by the snake venom peptide, rhodostomin. Br J Cancer 1996, 73:902-908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rickles FR, Falanga A: Molecular basis for the relationship between thrombosis and cancer. Thromb Res 2001, 102:V215-V224 [DOI] [PubMed] [Google Scholar]
- 52.Yuan F, Chen Y, Dellian M, Safabakhsh N, Ferrara N, Jain RK: Time-dependent vascular regression and permeability changes in established human tumor xenografts induced by an anti-vascular endothelial growth factor/vascular permeability factor antibody. Proc Natl Acad Sci USA 1996, 93:14765-14770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saclarides TJ: Angiogenesis in colorectal cancer. Surg Clin North Am 1997, 77:253-260 [DOI] [PubMed] [Google Scholar]
- 54.Zacharski LR, Memoli VA, Morain WD, Schlaeppi JM, Rousseau SM: Cellular localization of enzymatically active thrombin in intact human tissues by hirudin binding. Thromb Haemost 1995, 73:793-797 [PubMed] [Google Scholar]