

Abstract
The human immunodeficiency virus type 1 (HIV-1) Tat protein is a key pathogenic factor in a variety of acquired immune deficiency syndrome (AIDS)-associated disorders. A number of studies have documented the neurotoxic property of Tat protein, and Tat has therefore been proposed to contribute to AIDS-associated neurological diseases. Nevertheless, the bulk of these studies are performed in in vitro neuronal cultures without taking into account the intricate cell-cell interaction in the brain, or by injection of recombinant Tat protein into the brain, which may cause secondary stress or damage to the brain. To gain a better understanding of the roles of Tat protein in HIV-1 neuropathogenesis, we attempted to establish a transgenic mouse model in which Tat expression was regulated by both the astrocyte-specific glial fibrillary acidic protein promoter and a doxycycline (Dox)-inducible promoter. In the present study, we characterized the phenotypic and neuropathogenic features of these mice. Both in vitro and in vivo assays confirmed that Tat expression occurred exclusively in astrocytes and was Dox-dependent. Tat expression in the brain caused failure to thrive, hunched gesture, tremor, ataxia, and slow cognitive and motor movement, seizures, and premature death. Neuropathologies of these mice were characterized by breakdown of cerebellum and cortex, brain edema, astrocytosis, degeneration of neuronal dendrites, neuronal apoptosis, and increased infiltration of activated monocytes and T lymphocytes. These results together demonstrate that Tat expression in the absence of HIV-1 infection is sufficient to cause neuropathologies similar to most of those noted in the brain of AIDS patients, and provide the first evidence in the context of a whole organism to support a critical role of Tat protein in HIV-1 neuropathogenesis. More importantly, our data suggest that the Dox inducible, brain-targeted Tat transgenic mice offer an in vivo model for delineating the molecular mechanisms of Tat neurotoxicity and for developing therapeutic strategies for treating HIV-associated neurological disorders.
Human immunodeficiency virus type 1 (HIV-1) Tat protein plays an important role in the pathogenesis of a number of acquired immune deficiency syndrome (AIDS)-related disorders. 1,2 As one of the early HIV-1 proteins translated from the multiply spliced viral RNA transcripts, 3 Tat transactivates HIV-1 gene expression through interactions with the transactivation responsive element TAR within the HIV-1 long terminal repeat promoter, human cyclin T1, and CDK9. 4 This involves recruitment of an essential multicomponent factor, termed positive transcription elongation factor b (P-TEFb) to the HIV-1 long terminal repeat promoter, and phosphorylation of the C-terminal domain of RNA polymerase II (Pol II). In addition to being a transactivator of HIV-1 gene expression, Tat has also been documented to exert pleiotropic effects on host cells, through direct modulation of gene expression by Tat uptake from extracellular microenvironment, and/or intracellular signaling elicited by interaction of extracellular Tat protein with cell surface receptors. 5-8 Corroborated with these functions, HIV-1 Tat has been demonstrated as secreted from Tat-expressing cells 9-11 and HIV-infected cells, 12,13 and as being capable of entering cells in a biologically active form. 14,15
HIV-1 infects the central nervous system (CNS) of a majority of AIDS patients, 16-18 and often leads to neurological symptoms such as memory loss and impaired cognitive and motor functions. 18,19 More than half of the pediatric AIDS patients, and ∼20 to 25% of HIV-infected adults eventually develop dementia. 17,18,20,21 HIV-associated neuropathologies include reactive astrocytosis and cerebellar atrophy in the early stage of infection, and demyelination, formation of multinucleated giant cells, neuron death, and breakdown of the blood-brain barrier at later stages of the disease. 16,18,19,21-25 The target cells for HIV-1 infection in the brain are microglia/macrophages and astrocytes. 26,27 It has been generally accepted that neurons are most affected, although they are not directly infected. Thus, many indirect mechanisms have been explored and/or proposed for HIV-1 infection-induced neuropathogenesis. Those include HIV-1 viral proteins gp120 and Tat, and cellular factors secreted from HIV-infected macrophages/microglia and astrocytes, such as tumor necrosis factor-α, platelet-activating factor, arachidonic acid metabolites, oxygen-free radicals, nitric oxide, excitatory amino acids, and chemokines. 22,23,28-34 However, the precise in vivo role of these factors in contributing to HIV-associated CNS injury remains to be defined.
A number of studies have implicated Tat protein in HIV-induced neuropathogenesis. Tat is neurotoxic in vitro. 1,2 When injected into the brain, Tat protein causes histological changes similar to those seen in HIV-demented patients. 1,35 A recent study has suggested a positive correlation between the levels of Tat mRNA transcript and HIV- and simian-human immunodeficiency virus-induced encephalitis. 36 The mechanisms proposed for Tat neurotoxicity include direct depolarization of neurons, increased levels of intracellular calcium, increased production/release of proinflammatory cytokines, increased infiltration of macrophages/monocytes, activation of excitatory amino acid receptors, and increased apoptosis. 2 However, the argument has always been whether Tat is present within the CNS or cerebrospinal fluid in HIV-infected individuals at sufficient concentrations, to directly exhibit the acute neurotoxicity observed in most of those studies. Meanwhile, independent studies have shown that at lower concentrations Tat exhibits no acute neurotoxic activity, and instead transactivates gene expression and induces cell proliferation, differentiation, adhesion, and morphological changes. 7,37-39 In agreement with these observations, our recent studies suggest that Tat may affect neuronal function through direct uptake of extracellular Tat protein and subsequent disruption of neuronal metabolic balance of low-density lipoprotein receptor-related protein physiological ligands. 15 In addition, an overwhelming majority of the studies on Tat neurotoxicity has only focused on Tat interaction with neurons, and has not taken into account the interaction among neurons, astrocytes, and other brain cells. Nevertheless, it is certain that Tat interaction with astrocytes and other brain cells including endothelial cells also has adverse effects on neuronal survival and function. 34,40-43
Thus, it has become increasingly evident that the bona fide mechanisms of Tat neurotoxicity should be addressed in the context of a whole organism. There are several Tat transgenic mouse models available, 44-47 but none of them is suitable for studying Tat neurotoxicity. In those models Tat expression occurs constitutively throughout development and in all or most of tissues. As a result, any phenotypic abnormalities observed in the brain or a particular brain region could be caused by, or complicated by, abnormalities that occur in the brain any time during development or that exist in any other tissues or organs of the animal. In the present study we modified the doxycycline (Dox)-regulated gene expression strategy 48 with a brain-specific promoter, ie, the astrocyte-specific glial fibrillary acid protein (GFAP) promoter, 33,49,50 and generated the Dox-inducible and brain-targeted Tat transgenic mouse model. Characterization of phenotypes and neuropathologies of the transgenic mice concludes that Tat protein is a critical pathogenic factor in HIV-associated neuropathogenesis. More importantly, our data suggest that the unique Tat transgenic mouse model is very valuable for understanding the molecular mechanisms of Tat neurotoxicity, and for developing therapeutics for treating HIV-associated neurological diseases.
Materials and Methods
Plasmid Construction
The murine GFAP promoter in C-3123 plasmid (a gift from Dr. Lennart Mucke, University of California, San Francisco, CA 49 ) was released by restriction digestion with EcoRI and BamHI, and cloned into the pTeton (Clontech, Palo Alto, CA) in place of the cytomegalovirus promoter. The recombinant plasmid was designed as pTeton-GFAP. The standard polymerase chain reaction (PCR) cloning technique was used to construct pTRE-Tat86 plasmid. Briefly, HIV-1 Tat gene encoding 86 amino acids from both the first and second exons of HIV-1 isolate HXB2 was obtained by PCR using pBD-Tat86 15 as the template and primers 5′-GGA ATT CAC CAT GGA GCC AGT AGA TCC T-3′ (EcoRI site underlined) and 5′-CGG GAT CCC TAT TCC TTC GGG CCT GT-3′ (BamHI site underlined), and then cloned into EcoRI- and BamHI-digested pTRE vector backbone (Clontech). Similarly, we also constructed a control plasmid pTRE-CAT expressing the chloramphenicol acetyltransferase (CAT) reporter gene. All recombinant plasmids were verified by sequencing.
Cell Cultures, Transfection, and the CAT Assay
HeLa and U87.MG cells were cultured in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum at 37°C with 5% CO2. HeLa (1 × 106 cells/100 mm plate) and U87.MG (0.5 × 106 cells/100 mm plate) cells were transfected with plasmid DNAs as indicated using the standard calcium phosphate method. Transfected cells were cultured in the presence of Dox (1 μg/ml) for 48 hours, and harvested for the CAT reporter gene assay using the phase extraction and direct scintillation counting method as previously described. 51 For U87.MG/iTat stable cells line, we first transfected U87.MG cells with the regulator plasmid pTeton-GFAP, and selected in the presence of 200 μg/ml of G418 for the stable transfectants, called U87.MG/GFAP cells. The stable transfectants were then functionally characterized by transient transfection of the expressor plasmid pTRE-CAT. The cell line with the highest transactivation of the CAT expression in the presence of Dox was selected and transfected with the expressor plasmid pTRE-Tat86. The selection was performed in the presence of 50 μg/ml of hygromycin (Life Technologies, Inc., Gaithersburg, MD) because a plasmid expressing the hygromycin-resistant gene (Clontech) was included in the transfection. The resulting stable cell line was designated U87.MG/iTat cells.
Creation of Inducible Tat Transgenic Mice
Inducible Tat transgenic mouse colonies (GT-tg) were obtained by generation of two separate transgenic lines Teton-GFAP mice (G-tg) and TRE-Tat86 mice (T-tg), and then cross-breeding of these two lines of transgenic mice. Briefly, a DNA fragment (2238 bp) containing the Teton-GFAP gene, along with downstream simian virus 40 splicing and polyadenylation sequences, was released by XhoI and PvuII digestion of the pTeton-GFAP plasmid and purified by agarose gel electrophoresis, and microinjected into fertilized eggs of F1 females obtained from mating between C3HeB and FeJ mice (Jackson Laboratories, Bar Harbor, ME). Founder transgenic animals were crossed with C57BL/6 mice to generate stable G-tg transgenic lines. Similarly, T-tg transgenic lines were obtained using a DNA fragment (1189 bp) released by XhoI and PvuII digestion of the pTRE-Tat86 plasmid. Founder animals and progeny carrying the transgenes were identified by PCR analysis of genomic DNA, which was extracted from mouse tail clippings (∼0.5 to 1 cm long) using the Wizard genomic DNA isolation kit (Promega, Madison, WI). For amplification of the Teton-GFAP transgene, the primers 5′-GCT CCA CCC CCT CAG GCT ATT CAA-3′ and 5′-TAA AGG GCA AAA GTG AGT ATG GTG-3′ were used, whereas the TRE-Tat86 transgene was amplified with the primers 5′-CGG TGG GAG GCC TAT ATA AGC-3′ and 5′-AAC TGC AGT TAT TCC TTC GGG CCT GTC GG-3′. Fifty ng of genomic DNA was used in the PCR reactions, with a program of one cycle of 94°C for 3 minutes, 35 cycles of 94°C for 1 minute, 60°C for 1 minute, and 72°C for 1 minute, and one cycle of 72°C for 7 minutes. The expected sizes of the amplified fragments for Teton-GFAP and TRE-Tat86 transgenes were 420 bp and 250 bp, respectively. Southern blot analysis was also performed to determine the presence and the copy number of the transgenes in mice using the 32P-labeled transgene probes.
Dox Treatment of Transgenic Mice
The animals were housed in the Laboratory Animal Care Center of Indiana University School of Medicine with a 12-hour light and 12-hour dark photoperiod. Water and food are provided ad libitum. All animal procedures were approved by the Institutional Biosafety Committee. In all experiments unless stated otherwise, comparisons were made between wild-type mice treated with Dox and Tat transgenic mice treated with and without Dox. Mice at postnatal day 21 (P21) were given Dox (Sigma, Louis, MO) in drinking water (in aluminum foil-wrapped bottles) at a concentration of 6 mg/ml for 7 days. Empty vehicle treatment was included as a control. Animals were sacrificed after the last day of Dox treatment, ie, P28, and the brains were removed and divided saggitally. One hemibrain was processed for RNA or protein isolation. The other hemibrain was fixed overnight in phosphate-buffered saline (PBS)-buffered 4% paraformaldehyde and processed for paraffin embedding.
RNA Isolation and Reverse Transcriptase (RT)-PCR Analysis of Tat Expression
Total RNA was isolated using the Trizol Reagents (Life Technologies, Inc.) according to the manufacturer’s instructions. Tat expression was analyzed using the Titan One Tube RT-PCR System kit (Boehringer Mannheim, Indianapolis, IN), with Tat-specific primers 5′-GGA ATT CAC CAT GGA GCC AGT AGA TCC T-3′ and 5′-CGG GAT CCC TAT TCC TTC GGG CCT GT-3′. RT-PCR was performed on a PE Thermocycler 9600 (PE Applied Biosystem, Foster City, CA) with a program of 50°C for 30 minutes, 94°C for 2 minutes, followed by 30 cycles of 94°C for 30 seconds, 60°C for 30 seconds, and 72°C for 30 seconds, and one cycle of 72°C for 7 minutes. Control RT-PCR reactions were performed in the absence of RT and genomic DNA templates to rule out the possibilities of genomic DNA contamination in RNA preparations and nonspecific amplification. In addition, the mouse glyceraldehyde-3-phosphate dehydrogenase gene (GAPDH) was included in the RT-PCR as an internal control, with GAPDH-specific primers 5′-CTC AGT GTA GCC CAG GAT GC-3′ and 5′-ACC ACC ATG GAG AAG GCT GG-3′.
Immunohistochemistry
To ensure objective assessments and reliability of results, brain sections from mice to be compared in any given experiment were blind-coded and processed in parallels. Codes were revealed only after the analysis was complete.
Five-μm paraffin sections were cut on a microtome and mounted directly on glass slides for histochemical staining. The sections were deparaffinized in xylene and rehydrated, then stained using the Immunocruz Staining System (Santa Cruz Biotechnologies, Santa Cruz, CA) according to the manufacturer’s instruction. Anti-Tat antibody was a gift from Dr. Avindra Nath (University of Kentucky, Lexington, KY). All other antibodies used in our studies were from Santa Cruz Biotechnologies, including rabbit antibodies directed against GFAP (a marker for astrocytes) and CD14 (a marker for monocytes), and goat antibodies directed against MAP-2 (a marker for neurons) and CD68 (a marker for macrophages/microglia). Briefly, deparaffinized sections were unmasked by heating the sections at 95°C for 5 minutes in 10 mmol/L of sodium citrate buffer (pH 6.0) in a microwave with buffer exchanges for three times. After cooling to room temperature, the sections were pretreated in methanol containing 0.3% hydrogen peroxide (H2O2) for 30 minutes, blocked with 2.5% bovine serum albumin and 2% normal serum for 2 hours at room temperature, and then incubated with the appropriate primary antibodies at room temperature for 3.5 hours or at 4°C overnight, followed by an appropriate secondary biotinylated antibody for an additional 2 hours at room temperature. Bound antibodies were visualized with 3,3′-diaminobenzidine as the chromogen. All incubations were performed under humidified conditions, and slides were washed three times between steps with PBS. Finally, sections were counterstained in 1% hematoxylin. Omission of the primary antibodies in parallel stainings was included as a control. Stained sections were examined and bright-field microscopic images were captured with a Zeiss color camera mounted on a Zeiss Axiovert M200 fluorescence microscope using a ×20 plan apochromat objective, unless stated otherwise. A yellow background was generated by the microscope when the transmission light was selected to capture the immunohistochemical staining images. Image analysis was performed using an Axiovision 3.0 software package (Zeiss).
Cell Counting
The number of immunolabeled cells was counted manually on bright-field microscopic images in 1 of every 10 saggital sections (coded) by three independent individuals. All cell countings were location-matched between each group of mice. One frame for the visual field of 100 × 100 μm was used for cell counting. The analysis was performed on three animals of each group. Three different visual fields were chosen randomly within the same microanatomical brain regions of each animal. Average values of cell counts were calculated from the pooled data.
Enzyme-Linked Immunosorbent Assay (ELISA) Analysis for Tat and Interferon (IFN)-γ
Tat secretion in the supernatants of cell cultures and IFN-γ production in the brain homogenate were determined by Due Set ELISA Development System (R&D Systems, Minneapolis, MN) according to the manufacturer’s instruction. U87.MG/iTat stable cells were treated with or without 1 μg/ml of Dox, and the cell-free supernatants (0.2 ml) were collected for Tat determination at time points as indicated. For IFN-γ, the hemibrain was homogenized in 25 mmol/L of HEPES, pH 7.4, containing a cocktail of protease inhibitor called Complete TM (Boehringer Mannheim) on ice for 10 minutes. The homogenates were obtained by centrifugation at 4°C and 2000 × g for 5 minutes, followed by 13,000 × g for 30 minutes. The protein concentration was determined using a Bio-Rad DC Protein Assay kit (Bio-Rad, Hercules, CA). Serial dilutions of the supernatants or brain homogenates were performed to ensure the readings in the linear range. Recombinant Tat protein 15 and IFN-γ standards (R&D Systems) were used to calibrate the assay and determine the levels of Tat or IFN-γ. Antibodies for Tat and IFN-γ ELISA assay were from the National Institutes of Health AIDS Reagents Program and R&D Systems, respectively.
In Situ Apoptosis Detection
Five-μm paraffinized brain sections were deparaffinized and rehydrated as described above. Cell apoptosis was determined with the terminal deoxynucleotidyl-mediated dUTP nick end-labeling (TUNEL)-based TdT-FragEL DNA Fragmentation Detection kit (Oncogene, Boston, MA). Briefly, deparaffinized sections were treated in 10 mmol/L of Tris-HCl, pH 8.0, containing 20 μg/ml of proteinase K at room temperature for 20 minutes, and in 3% H2O2 at room temperature for 5 minutes. The sections were then equilibrated with 1× TdT buffer, and incubated with TdT-labeling reaction mixture at 37°C for 2 hours. The labeling reaction was terminated by addition of the stop solution supplied in the kit, and incubation at room temperature for 5 minutes. The sections were rinsed with TBS (20 mmol/L Tris-HCl, pH 7.6, and 140 mmol/L NaCl) between steps. Finally, cell apoptosis was visualized with incubation of the sections in 3,3′-diaminobenzidine at room temperature for 15 minutes. Counterstain was performed in 0.3% methyl green solution after a thorough rinse with distilled water.
Statistical Analysis
Values were expressed as means ± SEM. Comparisons among groups were made using the Student’s t-test. A P value of <0.05 was considered statistically significant (*), and P < 0.001 highly significant (**).
Results
Astrocyte-Specific and Dox-Dependent Gene Expression in Vitro
Our goal was to determine the roles of Tat in HIV-1 neuropathogenesis. Because of possible acute neurotoxicity of Tat, we decided to use the tetracycline-regulated gene expression strategy, in which gene expression can be regulated by simple administration or withdrawal of tetracycline or its derivatives. This system consists of two components: 48 a tetracycline-responsive regulator (tTA) that is a fusion of Escherichia coli tet repressor to the transactivation domain of VP16 protein of herpes simplex virus under the cytomegalovirus (CMV) promoter, and a tTA-dependent, minimal RNA polymerase II promoter-driven expressor. Based on the relationship between tetracycline and gene expression, this system has evolved into two versions, ie, the Tet-off system in which addition of tetracycline prevents transcription by binding to tTA, and genes of interest are expressed only after tetracycline is removed, and the Tet-on system that uses a reverse tTA element (rtTA) and only becomes transcriptionally active when tetracycline or its derivatives such as Dox are present. A recent study has shown that a long-term administration of tetracycline has potential adverse effects on normal memory. 52 Thus, we chose the Tet-on system in our proposed studies. Moreover, to achieve brain-specific Tat expression, we decided to modify the Tet-on system by integrating into the regulator the astrocyte-specific GFAP promoter. Therefore, we made two transgenic constructs expressing each of these components. One was the regulator pTeton-GFAP, in which the CMV promoter was replaced by a 2.1-kb GFAP promoter 49 (Figure 1a) ▶ . The other was the expressor pTRE-Tat86, in which the HIV-1 HXB2 Tat cDNA (86 amino acids in length) was cloned under the control of the tetO promoter (Figure 1a) ▶ . Although Tat proteins from primary HIV-1 isolates have more than 86 amino acids, the 86-amino acid Tat gene that we used contains the most conserved domains of the Tat sequence of many HIV isolates (KT Jeang: HIV-1 Tat: structure and function. “The Human Retroviruses and AIDS Compendium on Line”: http://hiv-web.lanl.gov, 1994). First, we investigated the specificity of the GFAP promoter in the context of the pTeton backbone, ie, pTeton-GFAP. We transfected astroglial U87.MG cells and nonastroglial HeLa cells with pTRE-CAT expressing the CAT reporter gene, in combination with pTeton-GFAP, or pTeton (Figure 1a) ▶ . pCMVβGal was included to normalize variations of transfection efficiency among transfections. As expected, in the absence of Dox treatment, transfection of pTRE-CAT with pTeton or pTeton-GFAP gave rise to little CAT activity in HeLa and U87.MG cells (Figure 1b) ▶ . In the presence of Dox (1 μg/ml), co-transfection of pTRE-CAT with pTeton exhibited comparable levels of CAT expression between HeLa cells and U87.MG cells, which was 44.6 ± 3.8-fold and 40.4 ± 2.5-fold induction, respectively (Figure 1b) ▶ . In contrast, in the presence of Dox treatment, co-transfection of pTRE-CAT with pTeton-GFAP transactivated CAT expression by 7.8 ± 0.7-fold in U87.MG cells, but only 1.4 ± 0.3-fold in HeLa cells (Figure 1b) ▶ . These results confirmed that the GFAP promoter activity was astrocyte-specific. The inducibility of the CAT gene expression by the GFAP promoter, ie, sixfold to sevenfold was similar to the activity of the constitutive GFAP promoter. 53,54
Figure 1.

Transgenic constructs and Dox-dependent GFAP promoter activity. a: Transgenic constructs. pTRE-CAT was constructed as an expressor control for in vitro analysis. The GFAP promoter was cloned in place of the CMV promoter of pTeton regulator, while the Tat cDNA encoding HIV-1 HXB2 Tat (86 amino acids) was cloned into the pTRE. Both pTeton and pTRE were purchased from Clontech. b: The astrocyte-specific activity of the GFAP promoter. Astroglial U87.MG cells were transfected with pTeton and pTRE-CAT (open bars), or pTeton-GFAP and pTRE-CAT (filled bars). Epithelial-derived HeLa cells were included as a control. Transfected cells were cultured in the media supplemented with or without Dox (1 μg/ml) for 48 hours, and harvested for analysis of the CAT activity. Data represent means ± SEM of triplicate samples. c: Tat secretion from U87.MG/iTat cells. U87.MG/iTat cells were generated by sequential stable transfection of pTeton-GFAP and pTRE-Tat86, as described in the Materials and Methods section. ELISA was performed to determine Tat protein in the supernatants of U87.MG/iTat cells in the absence (open bars) or presence (filled bars) of 1 μg/ml Dox, at time points as indicated. Recombinant Tat protein was used as standards. Data represent means ± SEM of triplicate samples.
To further ensure the validity of the GFAP promoter-based Tet-on strategy for studying the effects of Tat in the CNS, we determined whether Tat was secreted out of astrocytes when expressed in astrocytes. Tat secretion as an extracellular protein is required for subsequent interaction of Tat with neurons, which mimics the brain of HIV-infected individuals. Thus, we stably transfected pTeton-GFAP and pTRE-Tat86 DNAs sequentially into U87.MG cells, and generated an inducible Tat astroglial cell line called U87.MG/iTat (see Material and Methods for details). We treated U87.MG/iTat cells with or without Dox, and determined levels of Tat protein in the culture supernatants. In the presence of Dox (1 μg/ml), Tat protein was detected in the supernatants at 12 hours after Dox treatment, and increased to a maximal level at 24 hours after Dox treatment (Figure 1c) ▶ . In contrast, little Tat protein was detected in the supernatants of U87.MG/iTat cells in the absence of Dox treatment (Figure 1c) ▶ . The concentrations of Tat protein in the supernatants were determined to be in the range of 100 to 850 pg/ml. These results confirmed that Tat was secreted once expressed in astrocytes. In addition, we did not detect cell death in Dox-treated U87.MG/iTat cells by the propidium iodide-staining method (data not shown), suggesting that Tat protein has no apparent adverse effects on U87.MG cells. These data together provided the initial evidence to support use of the modified Tet-on system to study Tat interaction with neurons in the transgenic mouse model.
Generation of the Dox-Regulated, Brain-Targeted Tat Transgenic Mouse Colonies
Next, we began to generate the Dox-regulated, brain-targeted Tat transgenic mice. Our strategy was to generate separate transgenic lines of mice containing the Teton-GFAP transgene (designated G-tg) or TRE-Tat86 transgene (designated T-tg), and then to crossbreed these two lines of transgenic mice to obtain the Dox-regulated, brain-targeted Tat bigenic mice (designated GT-tg; Figure 2a ▶ ). Both Teton-GFAP and TRE-Tat86 transgenes were released by restriction digestion (XhoI and PvuII) from their respective plasmid DNAs pTeton-GFAP and pTRE-Tat86 (Figure 1a) ▶ , purified, and microinjected into fertilized oocytes (C3HeB × FeJ). A total of five G-tg transgenic founders and seven T-tg transgenic founders were obtained. Crossing with the wild-type C57BL/6 mice was performed to stabilize each transgene. Then, G-tg transgenic mice were paired and crossed with T-tg transgenic mice to obtain GT-tg bigenic mice carrying both Teton-GFAP and TRE-Tat86 transgenes. The presence of transgenes was confirmed using genomic DNA PCR of the mouse tail clippings with transgene-specific primers (see Materials and Methods). Homozygous genotypes were confirmed by crossbreeding transgenic or bigenic mice with wild-type C57BL/6 mice. Representative genotyping results of transgenic and bigenic mice were shown in Figure 2b ▶ . The copy numbers of transgenes were determined to be in the range of 3 to 10 (data not shown). A total of four GT-tg bigenic colonies was obtained. Although only GT-tg bigenic mice were needed for most of the studies (see below), we had also maintained the parental G-tg and T-tg transgenic lines. This would allow us to regenerate GT-tg bigenic mice in case the unexpected happens.
Figure 2.

Creation of Tat transgenic colonies. a: The creation scheme of the Dox-inducible and brain-targeted Tat transgenic mice. Teton-GFAP (G-tg) and TRE-Tat86 (T-tg) transgenic mice were generated independently, and then crossbred to obtain the Teton-GFAP and TRE-Tat86 bigenic mice (GT-tg). b: Representative genotyping of transgenic mice. Genomic genotyping of transgenic mice was performed by PCR of transgenes in the genomic DNAs isolated from the mouse tail. Fifty ng of genomic DNA was used as templates in the PCR reaction. PCR products were analyzed on 1% agarose gel. The expected sizes of PCR products were 250 bp for TRE-Tat86 and 420 bp for Teton-GFAP, respectively. For each transgene, genomic PCR reactions using primers specific for GAPDH were also performed as a control. Stnd, pTeton-GFAP and pTRE-Tat86 plasmids as templates; Wt, wild-type mice; t, PCR with primers specific for TRE-Tat transgene; g, PCR with primers specific for Teton-GFAP transgene.
Tat Expression in the Brain of Inducible Tat Bigenic Mice
Because there are no reliable anti-Tat antibodies available for Western blot analysis of native Tat protein, we decided to use RT-PCR, as well as in situ hybridization, immunohistochemical staining (see below), and ELISA analysis, to determine Tat expression in the brain of Tat transgenic mice. We first performed RT-PCR to determine whether Dox would induce Tat expression in the brain. We fed the GT-tg bigenic mice immediately after weaning, ie, 21 days of age, with Dox-containing (6 mg/ml) drinking water for 7 consecutive days, 55,56 and then harvested the brain for RNA isolation. The representative results showed that Dox treatment in the drinking water induced Tat mRNA expression in both heterozygous and homozygous GT-tg bigenic mice, and no Tat mRNA expression in the wild-type control mice (Figure 3a) ▶ . To ensure no genomic DNA contamination in the RNAs isolated, RT-PCR reactions without addition of the RT were always performed as controls (data not shown). However, as reported elsewhere, 57 there was leaky expression of Tat mRNA transcripts in the GT-tg bigenic mice in the absence of Dox treatment (Figure 3a) ▶ . We then determined whether Tat expression was regulated by Dox. We fed the GT-tg bigenic mice with the drinking water containing different concentrations of Dox. Consistent with previous studies, 55,56 the results showed that Tat expression began at 1.5 mg/ml of Dox, reached a plateau at 6 mg/ml of Dox, with no further increases when Dox concentration exceeded 6 mg/ml (Figure 3b) ▶ . These results together showed that Tat expression in the brain of GT-tg bigenic mice was induced and regulated by Dox. Tat expression in the brain was also detected in supernatants of the primary astrocyte cultures prepared from the brain tissue of the GT-tg bigenic mice, as well as in the brain homogenates of the GT-tg bigenic mice using ELISA analysis (data not shown). In addition, there was a positive correlation between the copy numbers of the expressor TRE-Tat86 transgene and levels of Tat mRNA transcripts in GT-tg bigenic lines (data not shown). Nonetheless, the differences of Tat expression among the bigenic lines appeared well within the range of Tat expression at the concentrations of Dox examined (Figure 3b) ▶ . Based on the maximal inducibility of Tat mRNA expression in the presence of Dox, these four GT-tg bigenic lines were designated as low-expressor line GT-tg181, intermediate-expressor lines GT-tg182 and GT-tg272, and high-expressor line GT-tg271. The high-expressor line GT-tg271 was used throughout our studies unless stated otherwise.
Figure 3.

Tat expression in the brain of the GT-tg bigenic mice. a: Dox-dependent Tat expression. GT-tg bigenic mice, 21 days old, were fed with Dox (6 mg/ml)-containing water for 7 consecutive days, and the brain was harvested. Age-matched wild-type mice were also included as a control. Total RNA was then isolated from the brain using the Trizol Reagent (Life Technologies, Inc.). Tat expression was analyzed by RT-PCR of the total RNA using Tat-specific primers. RT-PCR using primers specific for GAPDH, and in the absence of RT were performed as controls. *, Heterozygous GT-tg bigenic mice. b: Tat expression is Dox dose-dependent. GT-tg bigenic mice were fed with drinking water containing Dox at the concentrations as indicated, and Tat expression was analyzed as described above. c: Exclusive Tat expression in the brain. GT-tg bigenic mice were treated with Dox (6 mg/ml) as above, and sacrificed for tissue harvest. Tat expression was then determined by RT-PCR as above. The amplification products in the tissues except brain were determined to be nonspecific, because no hybridization signals were detected on the Southern blot using a Tat-specific probe (data not shown).
We also determined whether the modified Tet-on strategy would specifically target Tat expression within the brain. We fed the GT-tg bigenic mice with the Dox-containing water for 7 days, and harvested various organs of the GT-tg bigenic mice, such as eye, heart, kidney, liver, lung, spleen, and thymus. We performed RT-PCR for Tat expression in those tissues. The results showed that Tat expression occurred in the brain, but not in other tissues or organs, after the GT-tg bigenic mice were systematically exposed to Dox (Figure 3c) ▶ . The amplification products of genomic DNA in these nonbrain tissues or organs was confirmed to be nonspecific by Southern blot analysis using a TRE-Tat transgene-specific DNA probe (data not shown). Moreover, consistent with the astroglial GFAP expression pattern, 58,59 in situ hybridization detected higher levels of Tat mRNA in the cerebellum, cerebellar cortex, hippocampus, brain stem, tectum, and white matter tracts in the brains of GT-tg bigenic mice treated with Dox, much fewer Tat mRNA transcripts in the brains of GT-tg bigenic mice treated without Dox, and no Tat mRNA transcripts in the brains of wild-type mice (data not shown).
Developmental and Behavioral Abnormalities of Tat-Expressing Mice
To assess the effects of Tat expression on mouse development, we fed the mice at 21 days of age with Dox (6 mg/ml)-containing water and recorded the changes of the body weight. Before initiation of Dox treatment, GT-tg bigenic mice appeared to be normal, and indistinguishable from the age-matched wild-type mice, G-tg transgenic, or T-tg transgenic mice. When treated with Dox, the GT-tg bigenic mice exhibited significant growth failure (Figure 4a) ▶ , which has also been reported in the mice expressing the NL4-3 Δgag/pol transgene. 60,61 By the end of the 7-day Dox treatment (6 mg/ml), the wild-type mice gained 4.51 ± 0.26 g (n = 8, P < 0.001), the G-tg transgenic mice for 4.32 ± 0.21 g (n = 6, P < 0.001), and the T-tg transgenic mice for 4.47 ± 0.25 g (n = 8, P < 0.001), whereas the GT-tg bigenic mice showed little growth (0.66 ± 0.21 g, n = 20, the bigenic line GT-tg271, P > 0.05) (Figure 4b) ▶ . To ascertain that the growth failure was because of Tat expression, we treated the GT-tg bigenic mice with different concentrations of Dox (Figure 4c) ▶ . In agreement with induction of Tat expression (Figure 3b) ▶ , the least growth was found in mice fed with 6 mg/ml of Dox (0.76 ± 0.27 g, n = 5, P > 0.05), followed by 3 mg/ml of Dox (1.81 ± 0.28 g, n = 6; P < 0.001), 1.5 mg/ml Dox (3.45 ± 0.24 grams, n = 5; P < 0.001), and no Dox treatment (3.27 ± 0.19 g, n = 6; P < 0.001), whereas the wild-type mice gained 4.21 ± 0.23 g (n = 4, P < 0.001) when treated with Dox at the highest concentration, ie, 6 mg/ml. Similar patterns of growth failure were obtained in other three bigenic lines GT-tg181, GT-tg182, and GT-tg272 (data not shown). These results together demonstrated that growth retardation was because of induction of Tat gene expression in the brain, other than an insertional mutation with neurodevelopmental consequences, or other unknown nonspecific effects.
Figure 4.

Tat expression-induced growth failure. a: Appearance of the GT-tg bigenic mice treated with or without Dox. b: Effects of Tat expression on body growth of mice. GT-tg bigenic mice, 21 days of age (filled bars), were fed with Dox-containing water (6 mg/ml) for 7 consecutive days and monitored for body growth at days 1, 3, and 7. Age-matched wild-type (open bars), G-tg transgenic (dotted bars), and T-tg transgenic (hatched bars) mice were included as controls. Tat expression in the brain of the mice were confirmed by RT-PCR. c: Dox dose dependence of Tat-induced growth failure. GT-tg bigenic mice were fed with drinking water containing Dox at 1.5 mg/ml (open triangle), 3 mg/ml (open circle), and 6 mg/ml (filled square). The drinking water containing no Dox was included in the GT-tg bigenic mice (open diamond), while 6 mg/ml of Dox was added in the drinking water for the age-matched wild-type mice (open square). Body weight represents means ± SEM of mice in each group.
In addition to growth failure, we also noted a number of behavioral changes and/or neurological signs inTat-expressing bigenic mice. Those changes included hunched posture, tremor, ataxia, hindlimb weakness, slow motor and cognitive movement, and seizures (Table 1) ▶ . By week 1 to week 2 after Dox treatment, affected mice were 16 of 16 in the bigenic line GT-tg181, 9 of 10 in the bigenic line GT-tg182, 17 of 22 in the bigenic line GT-tg271, and 12 of 15 in the bigenic line GT-tg272. In the absence of Dox, bigenic lines exhibited similar phenotypic features, but to a much lesser extent, ie, 6 of 14 in the bigenic line GT-tg181, 2 of 8 in the bigenic line GT-tg182, 6 of 15 in the bigenic line GT-tg271, and 4 of 12 in bigenic line GT-tg272. In addition, some of the affected mice died at an early age (3 to weeks), typically during and perhaps as a result of seizures. The death rate was 5 of 17 of affected mice in the bigenic line GT-tg271, 3 of 12 of affected mice in the bigenic line GT-tg272, and 1 of 6 of affected mice in the bigenic line GT-tg271. The severity of the abnormal changes and death rate appeared to be correlated with the levels of Tat expression in the brain. Interestingly, the GT-tg bigenic mice that survived the first 2 weeks after Dox treatment showed some improvement in body growth for several weeks, but progressively developed tremor, ataxia, and seizures, and eventually died by 3 months of age.
Table 1.
Tat Expression and Abnormalities in Mice
| Dox | Wt | GT-tg181 | GT-tg182 | GT-tg271 | GT-tg272 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| − | + | − | + | − | + | − | + | − | + | |
| Litters | 7 | 5 | 3 | 3 | 2 | 2 | 4 | 5 | 3 | 4 |
| Animals | 35 | 24 | 14 | 16 | 8 | 10 | 15 | 22 | 12 | 15 |
| Affected | 0 | 0 | 6 | 16 | 2 | 9 | 6 | 17 | 4 | 12 |
| Death | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 5 | 0 | 3 |
Developmental and behavioral abnormalities in Tat-expressing mice. Crossbreedings within each GT-tg bigenic line were performed. Animals from each breeding were fed with Dox (6 mg/ml)-containing water at age of 21 days, as described above, and monitored for body weight and other abnormalities including hunched poster, tremor, ataxia, slow motor and cognitive movement, seizure, and/or premature death. +, treated with Dox; −, treated without Dox (empty vehicle only).
Tat Expression-Induced Neuropathologies
We then characterized the neuropathologies of Tat-expressing mice. We first performed hematoxylin and eosin staining of the brain sections. Histological examination revealed complete collapse of the cerebellum, progressive loss of the cortex, and significant vacuolation in the brain of the GT-tg bigenic mice treated with Dox, and the dead GT-tg bigenic mice from Dox treatment (Figure 5) ▶ . The cerebellum was severely afflicted in the brain of the GT-tg bigenic mice treated with Dox, or the dead GT-tg bigenic mice from Dox treatment and as result, became disintegrated during the tissue processing (Figure 5, C and D) ▶ . In addition, Tat expression was associated with increased hematoxylin and eosin staining in the nuclei, suggesting that Tat expression resulted in cell death, mostly neurons determined by cell morphology. Although inflammation (encephalitis or meningitis) was only occasionally observed, hydroencephalitis or edema was often present in the GT-tg bigenic mice when treated with Dox.
Figure 5.

Disruption of cerebella and cortex and neuronal death in the brain of Tat-expressing mice. H&E staining was performed in the sections of brain from wild-type mice treated with Dox (6 mg/ml) for 7 days, GT-tg bigenic mice treated with or without Dox (6 mg/ml) for 7 days, and the GT-tg bigenic mice that died 5 days after initiation of Dox treatment (shown as GT-tg + Dox*). The representative images of A–D were taken from the whole brain section, while the representative images of E–L were taken in the cortex region of the brain.
Because astrocytosis is a hallmark of HIV-induced neuropathologies, 62-64 we next determined whether Tat expression in the astrocytes would induce astrocytosis. We performed immunohistochemical staining of brain sections using anti-GFAP antibody, and anti-Tat antibody (a gift from Dr. A. Nath, University of Kentucky, Lexington, KY). We also included a normal rabbit IgG as a staining control (data not shown). Consistent with levels of Tat expression, the astrocytes in the brain of the Dox-treated GT-tg bigenic mice had a morphology typical of reactive astrocytosis, 65 showing enlarged cell bodies and an increased GFAP immunoreactivity (Figure 6) ▶ . The GT-tg bigenic mice also showed some morphological changes of astrocytes in the absence of Dox treatment and perhaps the GFAP immunoreactivity, but to a much lesser extent. Meanwhile, the age-matched, wild-type control mice showed a normal distribution and morphology of GFAP-positive astrocytes.
Figure 6.

Astrocytosis, loss of neuron dendrite, and neuronal apoptosis in the brain of Tat-expressing mice. The serial sections of the brain of the wild-type mice treated with Dox and GT-tg bigenic mice treated with or without Dox were immunolabeled with anti-Tat antibody (A–C), anti-GFAP antibody (D–F), and anti-MAP2 antibody (G–I). TUNEL staining was also performed in the brain sections (J–L). The representative images were taken in the cortex region of the brain. Original magnifications: ×20 (A–F and J–L); ×64 (G–I).
We then evaluated the integrity of neurons in the brain of Tat-expressing mice. We performed immunohistochemical staining of brain sections using an antibody against a dendrite marker, microtubule-associated protein 2 (MAP2). Surprisingly, digital-scanning microscopic imaging showed significant neuronal and dendritic changes in cerebellar, cortical, and hippocampal neurons. Compared to the age-matched, wild-type normal mice, the GT-tg bigenic mice exhibited considerable loss of dendritic processes, and atrophic and tortuous neuronal body (Figure 6) ▶ . The terminal-deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) staining further confirmed a significant increase in positively stained neurons (apoptotic neurons) in the brain of the Dox-treated GT-tg bigenic mice (Figure 6) ▶ . The apoptotic neurons appeared to be in a close proximity to Tat-positive regions including cerebellum, cortex, and hippocampus.
Tat Expression and the CNS Infiltration
Macrophage/monocyte infiltration into the CNS plays an important role in AIDS-associated dementia. HIV-1 Tat protein is chemotactic by itself, or through up-regulation of chemokine and chemokine receptor expression. Thus, we next examined the relationship between Tat expression and macrophage/monocyte and lymphocyte infiltration. We performed immunohistochemical staining of the brain sections with antibodies against CD14 (monocyte marker) and CD68 (macrophage/microglia marker), as well as antibodies against CD4 (CD4 T lymphocytes) and CD8 (CD8 T lymphocytes). Examination of stained brain sections revealed that there were considerable increases in CD14-, CD4-, and CD8-positive cells, but fewer changes in CD68-positive cells in the brain of the GT-tg bigenic mice treated with Dox (Figure 7a) ▶ . There also appeared even more CD14-, CD4-, and CD8-positive cells in the brain of the dead GT-tg bigenic mice from Dox treatment. Moreover, the CD4- and CD8-positive cells in the brain of the dead GT-tg bigenic mice exhibited a morphology typical of T lymphocytes in an activated stage (Figure 7a) ▶ . Quantitative analysis of these brain sections showed that the number of CD4-positive cells was 165.2 ± 24.1 in the wild-type mice and 135.8 ± 15.2 in the GT-tg bigenic mice in the absence of Dox treatment, which was increased to 250.6 ± 34.4 in the GT-tg bigenic mice when treated with Dox, and 481.5 ± 40.2 in the dead Dox-treated GT-tg bigenic mice. Meanwhile, the number of CD8-positive cells was 15.3 ± 3.2 in the wild-type mice and 35.4 ± 4.6 in the GT-tg bigenic mice in the absence of Dox treatment, which was increased to 141.2 ± 12.7 in the GT-tg bigenic mice when treated with Dox, and 253.3 ± 17.8 in the dead Dox-treated GT-tg bigenic mice. Similarly, the number of CD14-positive cells also showed considerable increases, ie, 35.6 ± 5.2 in the wild-type mice, 60.1 ± 4.8 in the GT-tg bigenic mice treated without Dox, 85.6 ± 5.9 in the GT-tg bigenic mice treated with Dox, and 161.4 ± 14.3 in the dead Dox-treated GT-tg bigenic mice. Nevertheless, no marked changes were noted in the number of CD68-positive cells, ie, 65.3 ± 6.3 in the wild-type mice, 67. 3 ± 4.8 in the GT-tg bigenic mice treated without Dox, 70.6 ± 3.4 in the Dox-treated GT-tg bigenic mice, and 68.4 ± 6.2 in the dead Dox-treated GT-tg bigenic mice (Figure 7b) ▶ .
Figure 7.
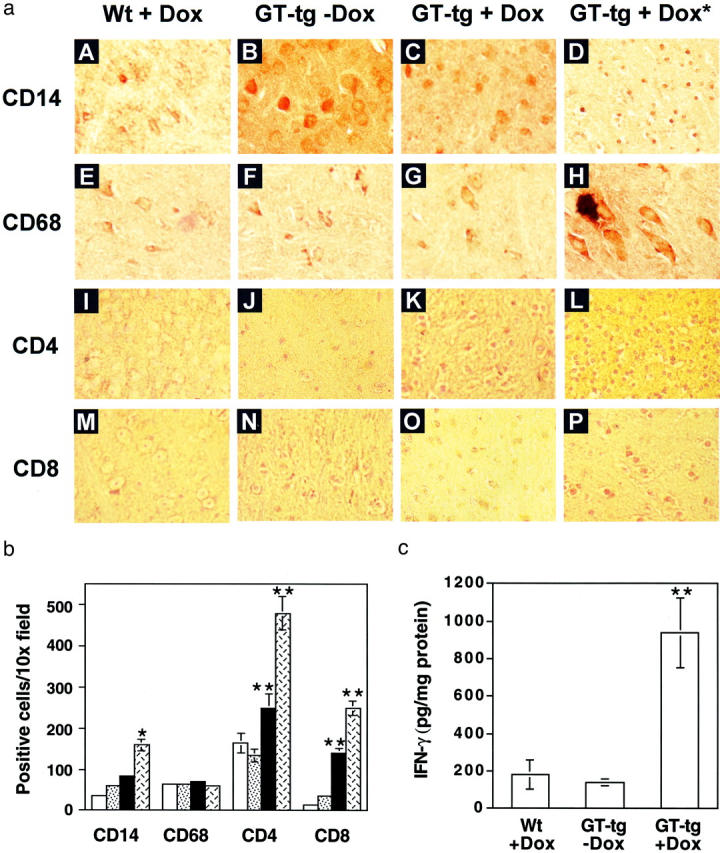
The CNS infiltration induced by Tat expression. a: Increased staining of CD14-, CD4-, and CD8-positive, but not CD68-positive cells in the brain of Tat-expressing mice. The serial sections of the brain of the wild-type mice treated with Dox, the GT-tg bigenic mice treated with or without Dox, and the dead GT-tg bigenic mice resulting from Dox treatment (shown as GT-tg + Dox*) were immunolabeled with anti-CD14 antibody (A–D), anti-CD68 antibody (E–H), anti-CD4 antibody (I–L), and anti-CD8 antibody (M–P). The representative images were taken in the cortex region of the brain. b: Quantitation of CD14-, CD68-, CD4-, and CD8-positive cells in the cortex region of immunolabeled sections from A. Cell countings of positively stained cells were performed as described in the Materials and Methods section. Open bars, wild-type mice treated with Dox; dotted bars, GT-tg bigenic mice treated without Dox; filled bars, GT-tg bigenic mice treated with Dox; hatched bars, dead GT-tg bigenic mice 5 days after initiation of Dox treatment. c: IFN-γ production in the brain of Tat-expressing mice. Brain homogenates were prepared from the other hemibrain of the mice as described in the Materials and Methods section. The levels of IFN-γ production in the brain homogenates were determined by ELISA. Data represent means ± SEM of mice in each group (b and c).
T lymphocytes normally enter the CNS only after they are activated by contact with helper cells and other antigens. 66,67 The CNS infiltration of activated CD4 and CD8 cells has been demonstrated in viral infection-induced neurological disorders. 68,69 Thus, we next determined whether T lymphocytes infiltrated into the CNS of the Tat-expressing mice were activated. We prepared brain homogenates and measured production of IFN-γ, a molecular marker for CD8 cell activation. Consistent with the morphological changes (Figure 7a) ▶ , the results showed that IFN-γ production was markedly increased from 184.6 ± 78.8 pg/mg protein in the Dox-treated wild-type mice and 140.9 ± 18.2 pg/mg protein in the GT-tg bigenic mice treated without Dox to 939.8 ± 185.9 pg/mg protein in the Dox-treated GT-tg bigenic mice (Figure 7C) ▶ , confirming that those infiltrated T lymphocytes were activated.
Discussion
In this study, we demonstrated that brain-targeted expression of HIV-1 Tat protein was sufficient to cause a number of developmental and behavioral abnormalities including failure to thrive, hunched posture, tremor, ataxia, slow motor and cognitive movement, seizures, and premature death, and extensive neuropathological changes such as loss of cerebellum and cortex, neuronal death (apoptosis), astrocytosis, degeneration of neuronal dendrites, and the CNS infiltration of monocytes and activated T lymphocytes. The phenotypic and neuropathological features noted in the GT-tg bigenic mice correlated with the level and spatial distribution of Tat mRNA and/or protein expression and occurred in four different GT-tg bigenic lines when treated with Dox but not in these bigenic mice treated without Dox, or in the Dox-treated wild-type normal mice. Therefore, all of these findings did not result from an insertional mutation, or other unknown nonspecific effects, and support the notion that HIV-1 Tat protein has direct and wide-ranging pathogenic actions in the CNS and has an important role in HIV-associated neuropathogenesis.
Unlike most of the published studies, we attempted to address Tat neurotoxicity in the context of a whole organism. To this end, we integrated the astrocyte-specific GFAP promoter into the inducible Tet-on gene expression system. We chose the GFAP promoter to target astrocytes as Tat-expressing cells in the CNS for the following reasons: 1) Tat has direct and acute neurotoxic activity, which may not allow Tat interaction with neurons as an extracellular (soluble) factor if Tat is expressed in neurons; 2) there are no microglia/macrophage-specific promoters available, although HIV-1 primarily infects microglia/macrophages in the CNS; 3) Tat has been found to have no apparent cytotoxicity on astrocytes; 4) increasing evidence suggests that astrocytes are involved in the pathogenesis of HIV-associated neurological diseases; 23,70-73 5) the GFAP promoter-driven gene expression in astrocytes has been successfully used to characterize the CNS functions of a number of genes including HIV-1 gp120, lipoprotein E4 (ApoE4), and cytokines such as interleukin (IL)-3, IL-6, IL-12, tumor necrosis factor-α, and IFN-α. 53 . Our results confirmed that the astrocyte-specific GFAP promoter effectively targeted Tat expression in astrocytes, and in a Dox-dependent manner both in vivo and in vitro (Figures 1 and 3) ▶ . Nevertheless, consistent with an earlier report, 57 our results indicated that this modified system exhibited a minimal level (leaky) of Tat expression in the absence of Dox (Figure 3) ▶ . However, our results showed that the inducibility of Tat expression by Dox treatment fell within the range of Tat expression among the GT-tg bigenic lines containing different copies of the transgene TRE-Tat86 (data not shown), which may be because of the inherited lower activity of the GFAP promoter. 33,50,53,54 Interestingly, it has been recently demonstrated that Dox has significant protective effects on neurons against apoptosis induction and global ischemia. 74,75 Therefore, the actual adverse effects of Tat on the development, behavior, and neuropathology of the GT-tg mice would likely be much greater.
Our results showed that Tat expression induced growth failure, and other apparent abnormalities such as hunched posture, tremor, ataxia, and slow motor and cognitive movement, seizures, and subsequent premature death (Figure 4 ▶ and Table 1 ▶ ). Similar results have been reported in several HIV-1 transgenic studies, 60,61,76 as well as HIV-infected children and adults. 77-80 Although the mechanisms are currently unknown, a multifactorial etiology seems likely. At the cellular level, Tat expression in the brain of the GT-tg bigenic mice resulted in degeneration of cerebellum and cortex, and neuronal death (Figure 5) ▶ . Compared to mild cerebellar atrophy often identified in the brain of HIV-infected individuals, 81-84 Tat-expressing mice exhibited complete collapse of the cerebellum, which may likely result from rapid induction and localized accumulation of Tat protein in the cerebellum within such a short period of time, ie, 7 days, because less severe cerebellar damage was evident in the brain of GT-tg bigenic mice treated with Dox at the concentrations of 1.5 and 3 mg/ml (data not shown). Moreover, induction of astrocytosis, loss of neuron dendrites, and neuron apoptosis were also noted in the brains of Tat-expressing mice (Figure 6) ▶ . Similar neuropathologies have been described in the brain of HIV-infected individuals. 35, 62-64, 85 Consistent with the results obtained from a rat model, 35 our data demonstrated that Tat increased the CNS infiltration of monocytes and T lymphocytes (Figure 7, a and b) ▶ , which we further confirmed was accompanied by increased production of potential neurotoxic molecules such as IFN-γ (Figure 7c) ▶ . This activity is apparently because of Tat expression and secretion, and/or Tat-mediated modulation of chemokines/chemokine receptors and cytokines in the CNS.
Although the exact molecular basis for the neurodegenerative changes seen in the brain of Tat-expressing GT-tg bigenic mice, including the unusual diffuse nuclear and perinuclear TUNEL-staining pattern in the brain of Tat-expressing mice, is under investigation in the laboratory, it is undoubtedly clear that the changes are a combined result of Tat expression in astrocytes, Tat interactions with neurons, other brain cells, including microglia and brain capillary endothelial cells, and infiltrated monocytes and T lymphocytes, and the interactions among those cells. More importantly, the unique transgenic mouse model will help to define the molecular mechanisms of Tat neurotoxicity, and develop and evaluate novel therapeutics for treating HIV-associated neuropathologies.
Acknowledgments
We thank Dr. Lennart Mucke for the GFAP promoter plasmid, Dr. Avindra Nath for anti-Tat antibodies, and Dr. Loren Field for technical assistance on the transgenic work.
Footnotes
Address reprint requests to Johnny J. He, Department of Microbiology and Immunology, Indiana University School of Medicine, R4 302, 1044 W. Walnut St., Indianapolis, IN 46202. E-mail: jjhe@iupui.edu.
Supported by the National Institutes of Health (grants R01NS39804 and R01MH65158 to J. J. H.) and the Ralph W. and Grace M. Showalter Trust Foundation (an award to J. J. H.).
References
- 1.Rappaport J, Joseph J, Croul S, Alexander G, Del Valle L, Amini S, Khalili K: Molecular pathway involved in HIV-1-induced CNS pathology: role of viral regulatory protein, Tat. J Leukoc Biol 1999, 65:458-465 [DOI] [PubMed] [Google Scholar]
- 2.Nath A, Mattson MP, Magnuson DSK, Jones M, Berger JR: Role of viral proteins in HIV-1 neuropathogenesis with emphasis on Tat. NeuroAIDS 1998, :1 [Google Scholar]
- 3.Sodroski J, Patarca R, Rosen C, Wong-Staal F, Haseltine W: Location of the trans-activating region on the genome of human T-cell lymphotropic virus type III. Science 1985, 229:74-77 [DOI] [PubMed] [Google Scholar]
- 4.Wei P, Garber ME, Fang SM, Fischer WH, Jones KA: A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 1998, 92:451-462 [DOI] [PubMed] [Google Scholar]
- 5.Cupp C, Taylor JP, Khalili K, Amini S: Evidence for stimulation of the transforming growth factor beta 1 promoter by HIV-1 Tat in cells derived from CNS. Oncogene 1993, 8:2231. [PubMed] [Google Scholar]
- 6.Ganju RK, Munshi N, Nair BC, Liu ZY, Gill P, Groopman JE: Human immunodeficiency virus Tat modulates the Flk-1/KDR receptor, mitogen-activated protein kinases, and components of focal adhesion in Kaposi’s sarcoma cells. J Virol 1998, 72:6131-6137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Milani D, Zauli G, Neri LM, Marchisio M, Previati M, Capitani S: Influence of the human immunodeficiency virus type 1 Tat protein on the proliferation and differentiation of PC12 rat pheochromocytoma cells. J Gen Virol 1993, 74:2587-2594 [DOI] [PubMed] [Google Scholar]
- 8.Zauli G, Gibellini D, Milani D, Mazzoni M, Borgatti P, La Placa M, Capitani S: Human immunodeficiency virus type 1 Tat protein protects lymphoid, epithelial, and neuronal cell lines from death by apoptosis. Cancer Res 1993, 53:4481-4485 [PubMed] [Google Scholar]
- 9.Ensoli B, Barillari G, Salahuddin SZ, Gallo RC, Wong-Staal F: Tat protein of HIV-1 stimulates growth of cells derived from Kaposi’s sarcoma lesions of AIDS patients. Nature 1990, 345:84-86 [DOI] [PubMed] [Google Scholar]
- 10.Chang HC, Samaniego F, Nair BC, Buonaguro L, Ensoli B: HIV-1 Tat protein exits from cells via a leaderless secretory pathway and binds to extracellular matrix-associated heparan sulfate proteoglycans through its basic region. AIDS 1997, 11:1421-1431 [DOI] [PubMed] [Google Scholar]
- 11.Zauli G, La Placa M, Vignoli M, Re MC, Gibellini D, Furlini G, Milani D, Marchisio M, Mazzoni M, Capitani S: An autocrine loop of HIV type-1 Tat protein responsible for the improved survival/proliferation capacity of permanently Tat-transfected cells and required for optimal HIV-1 LTR transactivating activity. J Acquir Immune Defic Syndr Hum Retrovirol 1995, 10:306-316 [PubMed] [Google Scholar]
- 12.Westendorp MO, Frank R, Ochsenbauer C, Stricker K, Dhein J, Walczak H, Debatin KM, Krammer PH: Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature 1995, 375:497-500 [DOI] [PubMed] [Google Scholar]
- 13.Xiao H, Neuveut C, Tiffany HL, Benkirane M, Rich EA, Murphy PM, Jeang KT: Selective CXCR4 antagonism by Tat: implications for in vivo expansion of coreceptor use by HIV-1. Proc Natl Acad Sci USA 2000, 97:11466-11471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frankel AD, Pabo CO: Cellular uptake of the Tat protein from human immunodeficiency virus. Cell 1988, 55:1189-1193 [DOI] [PubMed] [Google Scholar]
- 15.Liu Y, Jones M, Hingtgen CM, Bu G, Laribee N, Tanzi RE, Moir RD, Nath A, He JJ: Uptake of HIV-1 Tat protein mediated by low-density lipoprotein receptor-related protein disrupts the neuronal metabolic balance of the receptor ligands. Nat Med 2000, 6:1380-1387 [DOI] [PubMed] [Google Scholar]
- 16.Gabuzda DH, Hirsch MS: Neurologic manifestations of infection with human immunodeficiency virus. Clinical features and pathogenesis. Ann Intern Med 1987, 107:383-391 [DOI] [PubMed] [Google Scholar]
- 17.Glass JD, Wesselingh SL, Selnes OA, McArthur JC: Clinical-neuropathologic correlation in HIV-associated dementia. Neurology 1993, 43:2230-2237 [DOI] [PubMed] [Google Scholar]
- 18.Price RW, Brew B, Sidtis J, Rosenblum M, Scheck AC, Cleary P: The brain in AIDS: central nervous system HIV-1 infection and AIDS dementia complex. Science 1988, 239:586-592 [DOI] [PubMed] [Google Scholar]
- 19.Navia B, Cho ES, Petito CK, Price RW: The AIDS dementia complex. II Neuropathology. Ann Neurol 1986, 19:525-535 [DOI] [PubMed] [Google Scholar]
- 20.Lipton SA, Gendelman HE: Dementia associated with the acquired immunodeficiency syndrome. N Engl J Med 1995, 332:934-940 [DOI] [PubMed] [Google Scholar]
- 21.Wiley CA, Achim C: Human immunodeficiency virus encephalitis is the pathological correlate of dementia in acquired immunodeficiency syndrome. Ann Neurol 1994, 36:673-676 [DOI] [PubMed] [Google Scholar]
- 22.Epstein LG, Gendelman HE: Human immunodeficiency virus type 1 infection of the nervous system: pathogenetic mechanisms [see comments]. Ann Neurol 1993, 33:429-436 [DOI] [PubMed] [Google Scholar]
- 23.Genis P, Jett M, Bernton EW, Boyle T, Gelbard HA, Dzenko K, Keane RW, Resnick L, Mizrachi Y, Volsky DJ: Cytokines and arachidonic metabolites produced during human immunodeficiency virus (HIV)-infected macrophage-astroglia interactions: implications for the neuropathogenesis of HIV disease. J Exp Med 1992, 176:1703-1718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petito CK, Cash KS: Blood-brain barrier abnormalities in the acquired immunodeficiency syndrome: immunohistochemical localization of serum proteins in postmortem brain. Ann Neurol 1992, 32:658-666 [DOI] [PubMed] [Google Scholar]
- 25.Power C, Kong PA, Crawford TO, Wesselingh S, Glass JD, McArthur JC, Trapp BD: Cerebral white matter changes in acquired immunodeficiency syndrome dementia: alterations of the blood-brain barrier. Ann Neurol 1993, 34:339-350 [DOI] [PubMed] [Google Scholar]
- 26.Lee SC, Liu W, Brosnan CF, Dickson DW: Characterization of primary human fetal dissociated central nervous system cultures with an emphasis on microglia. Lab Invest 1992, 67:465-476 [PubMed] [Google Scholar]
- 27.Tornatore C, Chandra R, Berger JR, Major EO: HIV-1 infection of subcortical astrocytes in the pediatric central nervous system. Neurology 1994, 44:481-487 [DOI] [PubMed] [Google Scholar]
- 28.Bukrinsky MI, Nottet HS, Schmidtmayerova H, Dubrovsky L, Flanagan CR, Mullins ME, Lipton SA, Gendelman HE: Regulation of nitric oxide synthase activity in human immunodeficiency virus type 1 (HIV-1)-infected monocytes: implications for HIV-associated neurological disease. J Exp Med 1995, 181:735-745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gelbard HA, Nottet HS, Swindells S, Jett M, Dzenko KA, Genis P, White R, Wang L, Choi YB, Zhang D: Platelet-activating factor: a candidate human immunodeficiency virus type 1-induced neurotoxin. J Virol 1994, 68:4628-4635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giulian D, Lachman LB: Interleukin-1 stimulation of astroglial proliferation after brain injury. Science 1985, 228:497-499 [DOI] [PubMed] [Google Scholar]
- 31.Lipton SA, Sucher NJ, Kaiser PK, Dreyer EB: Synergistic effects of HIV coat protein and NMDA receptor-mediated neurotoxicity. Neuron 1991, 7:111-118 [DOI] [PubMed] [Google Scholar]
- 32.Pulliam L, Herndier BG, Tang NM, McGrath MS: Human immunodeficiency virus-infected macrophages produce soluble factors that cause histological and neurochemical alterations in cultured human brains. J Clin Invest 1991, 87:503-512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Toggas SM, Masliah E, Rockenstein EM, Rall GF, Abraham CR, Mucke L: Central nervous system damage produced by expression of the HIV-1 coat protein gp120 in transgenic mice [see comments]. Nature 1994, 367:188-193 [DOI] [PubMed] [Google Scholar]
- 34.Conant K, Garzino-Demo A, Nath A, McArthur JC, Halliday W, Power C, Gallo RC, Major EO: Induction of monocyte chemoattractant protein-1 in HIV-1 Tat-stimulated astrocytes and elevation in AIDS dementia. Proc Natl Acad Sci USA 1998, 95:3117-3121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones M, Olafson K, Del Bigio MR, Peeling J, Nath A: Intraventricular injection of human immunodeficiency virus type 1 (HIV-1) Tat protein causes inflammation, gliosis, apoptosis, and ventricular enlargement. J Neuropathol Exp Neurol 1998, 57:563-570 [DOI] [PubMed] [Google Scholar]
- 36.Hudson L, Liu J, Nath A, Jones M, Raghavan R, Narayan O, Male D, Everall I: Detection of the human immunodeficiency virus regulatory protein Tat in CNS tissues. J Neurovirol 2000, 6:145-155 [DOI] [PubMed] [Google Scholar]
- 37.Helland DE, Welles JL, Caputo A, Haseltine WA: Transcellular transactivation by the human immunodeficiency virus type 1 Tat protein. J Virol 1991, 65:4547-4549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ensoli B, Buonaguro L, Barillari G, Fiorelli V, Gendelman R, Morgan RA, Wingfield P, Gallo RC: Release, uptake, and effects of extracellular human immunodeficiency virus type 1 Tat protein on cell growth and viral transactivation. J Virol 1993, 67:277-287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kolson DL, Buchhalter J, Collman R, Hellmig B, Farrell CF, Debouck C, Gonzalez-Scarano F: HIV-1 Tat alters normal organization of neurons and astrocytes in primary rodent brain cell cultures: rGD sequence dependence. AIDS Res Hum Retroviruses 1993, 9:677-685 [DOI] [PubMed] [Google Scholar]
- 40.Taylor JP, Cupp C, Diaz A, Taylor JP, Cupp C, Diaz A, Chowdhury M, Khalili K, Jimenez SA, Amini S: Activation of expression of genes coding for extracellular matrix proteins in Tat-producing glioblastoma cells. Proc Natl Acad Sci USA 1992, 89:9617-9621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hofman FM, Chen P, Incardona F, Zidovetzki R, Hinton DR: HIV-1 Tat protein induces the production of interleukin-8 by human brain-derived endothelial cells. J Neuroimmunol 1999, 94:28-39 [DOI] [PubMed] [Google Scholar]
- 42.Zidovetzki R, Wang JL, Chen P, Jeyaseelan R, Hofman F: Human immunodeficiency virus Tat protein induces interleukin 6 mRNA expression in human brain endothelial cells via protein kinase C- and cAMP-dependent protein kinase pathways. AIDS Res Hum Retroviruses 1998, 14:825-833 [DOI] [PubMed] [Google Scholar]
- 43.Kutsch O, Oh J, Nath A, Benveniste EN: Induction of the chemokines interleukin-8 and IP-10 by human immunodeficiency virus type 1 Tat in astrocytes. J Virol 2000, 74:9214-9221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Choi J, Liu RM, Kundu RK, Sangiorgi F, Wu W, Maxson R, Forman HJ: Molecular mechanism of decreased glutathione content in human immunodeficiency virus type 1 Tat-transgenic mice. J Biol Chem 2000, 275:3693-3698 [DOI] [PubMed] [Google Scholar]
- 45.Garza HH, Jr, Prakash O, Carr DJ: Aberrant regulation of cytokines in HIV-1 TAT72-transgenic mice. J Immunol 1996, 156:3631-3637 [PubMed] [Google Scholar]
- 46.Vellutini C, Horschowski N, Philippon V, Gambarelli D, Nave KA, Filippi P: Development of lymphoid hyperplasia in transgenic mice expressing the HIV Tat gene. AIDS Res Hum Retroviruses 1995, 11:21-29 [DOI] [PubMed] [Google Scholar]
- 47.Vogel J, Hinrichs SH, Reynolds RK, Luciw PA, Jay G: The HIV Tat gene induces dermal lesions resembling Kaposi’s sarcoma in transgenic mice. Nature 1988, 335:606-611 [DOI] [PubMed] [Google Scholar]
- 48.Gossen M, Bujard H: Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci USA 1992, 89:5547-5551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johnson WB, Ruppe MD, Rockenstein EM, Price J, Sarthy VP, Verderber LC, Mucke L: Indicator expression directed by regulatory sequences of the glial fibrillary acidic protein (GFAP) gene: in vivo comparison of distinct GFAP-lacZ transgenes. Glia 1995, 13:174-184 [DOI] [PubMed] [Google Scholar]
- 50.Mucke L, Abraham CR, Ruppe MD, Rockenstein EM, Toggas SM, Mallory M, Alford M, Masliah E: Protection against HIV-1 gp120-induced brain damage by neuronal expression of human amyloid precursor protein. J Exp Med 1995, 181:1551-1556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.He J, Furmanski P: Sequence specificity and transcriptional activation in the binding of lactoferrin to DNA. Nature 1995, 373:721-724 [DOI] [PubMed] [Google Scholar]
- 52.Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER: Control of memory formation through regulated expression of a CaMKII transgene. Science 1996, 274:1678-1683 [DOI] [PubMed] [Google Scholar]
- 53.Campbell IL, Stalder AK, Akwa Y, Pagenstecher A, Asensio VC: Transgenic models to study the actions of cytokines in the central nervous system. Neuroimmunomodulation 1998, 5:126-135 [DOI] [PubMed] [Google Scholar]
- 54.Campbell IL: Transgenic mice and cytokine actions in the brain: bridging the gap between structural and functional neuropathology. Brain Res Brain Res Rev 1998, 26:327-336 [DOI] [PubMed] [Google Scholar]
- 55.Chen J, Kelz MB, Zeng G, Sakai N, Steffen C, Shockett PE, Picciotto MR, Duman RS, Nestler EJ: Transgenic animals with inducible, targeted gene expression in brain. Mol Pharmacol 1998, 54:495-503 [DOI] [PubMed] [Google Scholar]
- 56.Mansuy IM, Winder DG, Moallem TM, Osman M, Mayford M, Hawkins RD, Kandel ER: Inducible and reversible gene expression with the rtTA system for the study of memory. Neuron 1998, 21:257-265 [DOI] [PubMed] [Google Scholar]
- 57.Kistner A, Gossen M, Zimmermann F, Jerecic J, Ullmer C, Lubbert H, Bujard H: Doxycycline-mediated quantitative and tissue-specific control of gene expression in transgenic mice. Proc Natl Acad Sci USA 1996, 93:10933-10938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brenner M, Kisseberth WC, Su Y, Besnard F, Messing A: GFAP promoter directs astrocyte-specific expression in transgenic mice. J Neurosci 1994, 14:1030-1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Galou M, Pournin S, Ensergueix D, Ridet JL, Tchelingerian JL, Lossouarn L, Privat A, Babinet C, Dupouey P: Normal and pathological expression of GFAP promoter elements in transgenic mice. Glia 1994, 12:281-293 [DOI] [PubMed] [Google Scholar]
- 60.Santoro TJ, Bryant JL, Pellicoro J, Klotman ME, Kopp JB, Bruggeman LA, Franks RR, Notkins AL, Klotman PE: Growth failure and AIDS-like cachexia syndrome in HIV-1 transgenic mice. Virology 1994, 201:147-151 [DOI] [PubMed] [Google Scholar]
- 61.Leonard JM, Abramczuk JW, Pezen DS, Rutledge R, Belcher JH, Hakim F, Shearer G, Lamperth L, Travis W, Fredrickson T: Development of disease and virus recovery in transgenic mice containing HIV proviral DNA. Science 1988, 242:1665-1670 [DOI] [PubMed] [Google Scholar]
- 62.Persidsky Y, Limoges J, McComb R, Bock P, Baldwin T, Tyor W, Patil A, Nottet HS, Epstein L, Gelbard H, Flanagan E, Reinhard J, Pirruccello SJ, Gendelman HE: Human immunodeficiency virus encephalitis in SCID mice [see comments]. Am J Pathol 1996, 149:1027-1053 [PMC free article] [PubMed] [Google Scholar]
- 63.Eddleston M, Mucke L: Molecular profile of reactive astrocytes—implications for their role in neurologic disease. Neuroscience 1993, 54:15-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.da Cunha A, Jefferson JJ, Tyor WR, Glass JD, Jannotta FS, Vitkovic L: Control of astrocytosis by interleukin-1 and transforming growth factor-beta 1 in human brain. Brain Res 1993, 631:39-45 [DOI] [PubMed] [Google Scholar]
- 65.Ridet JL, Malhotra SK, Privat A, Gage FH: Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci 1997, 20:570-577 [DOI] [PubMed] [Google Scholar]
- 66.Hickey WF, Hsu BL, Kimura H: T-lymphocyte entry into the central nervous system. J Neurosci Res 1991, 28:254-260 [DOI] [PubMed] [Google Scholar]
- 67.Wekerle H: T-cell autoimmunity in the central nervous system. Intervirology 1993, 35:95-100 [DOI] [PubMed] [Google Scholar]
- 68.Lane TE, Liu MT, Chen BP, Asensio VC, Samawi RM, Paoletti AD, Campbell IL, Kunkel SL, Fox HS, Buchmeier MJ: A central role for CD4(+) T cells and RANTES in virus-induced central nervous system inflammation and demyelination. J Virol 2000, 74:1415-1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Marcondes MC, Burudi EM, Huitron-Resendiz S, Sanchez-Alavez M, Watry D, Zandonatti M, Henriksen SJ, Fox HS: Highly activated CD8(+) T cells in the brain correlate with early central nervous system dysfunction in simian immunodeficiency virus infection. J Immunol 2001, 167:5429-5438 [DOI] [PubMed] [Google Scholar]
- 70.Levi G, Patrizio M, Bernardo A, Petrucci TC, Agresti C: Human immunodeficiency virus coat protein gp120 inhibits the beta-adrenergic regulation of astroglial and microglial functions. Proc Natl Acad Sci USA 1993, 90:1541-1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tornatore C, Nath A, Amemiya K, Major EO: Persistent human immunodeficiency virus type 1 infection in human fetal glial cells reactivated by T-cell factor(s) or by the cytokines tumor necrosis factor alpha and interleukin-1 beta. J Virol 1991, 65:6094-6100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Saito Y, Sharer LR, Epstein LG, Michaels J, Mintz M, Louder M, Golding K, Cvetkovich TA, Blumberg BM: Overexpression of nef as a marker for restricted HIV-1 infection of astrocytes in postmortem pediatric central nervous tissues. Neurology 1994, 44:474-481 [DOI] [PubMed] [Google Scholar]
- 73.Benos DJ, Hahn BH, Shaw GM, Bubien JK, Benveniste EN: gp120-mediated alterations in astrocyte ion transport. Adv Neuroimmunol 1994, 4:175-179 [DOI] [PubMed] [Google Scholar]
- 74.Tikka T, Usenius T, Tenhunen M, Keinanen R, Koistinaho J: Tetracycline derivatives and ceftriaxone, a cephalosporin antibiotic, protect neurons against apoptosis induced by ionizing radiation. J Neurochem 2001, 78:1409-1414 [DOI] [PubMed] [Google Scholar]
- 75.Yrjanheikki J, Keinanen R, Pellikka M, Hokfelt T, Koistinaho J: Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci USA 1998, 95:15769-15774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Franks RR, Ray PE, Babbott CC, Bryant JL, Notkins AL, Santoro TJ, Klotman PE: Maternal-fetal interactions affect growth of human immunodeficiency virus type 1 transgenic mice. Pediatr Res 1995, 37:56-63 [DOI] [PubMed] [Google Scholar]
- 77.Laue L, Pizzo PA, Butler K, Cutler GB, Jr: Growth and neuroendocrine dysfunction in children with acquired immunodeficiency syndrome. J Pediatr 1990, 117:541-545 [DOI] [PubMed] [Google Scholar]
- 78.Grunfeld C, Feingold KR: Metabolic disturbances and wasting in the acquired immunodeficiency syndrome. N Engl J Med 1992, 327:329-337 [DOI] [PubMed] [Google Scholar]
- 79.Guarino A, Tarallo L, Guandalini S, Troncone R, Albano F, Rubino A: Impaired intestinal function in symptomatic HIV infection. J Pediatr Gastroenterol Nutr 1991, 12:453-458 [DOI] [PubMed] [Google Scholar]
- 80.Wanke CA, Falutz JM, Shevitz A, Phair JP, Kotler DP: Clinical evaluation and management of metabolic and morphologic abnormalities associated with human immunodeficiency virus. Clin Infect Dis 2002, 34:248-259 [DOI] [PubMed] [Google Scholar]
- 81.Tagliati M, Simpson D, Morgello S, Clifford D, Schwartz RL, Berger JR: Cerebellar degeneration associated with human immunodeficiency virus infection. Neurology 1998, 50:244-251 [DOI] [PubMed] [Google Scholar]
- 82.DeCarli C, Civitello LA, Brouwers P, Pizzo PA: The prevalence of computed tomographic abnormalities of the cerebrum in 100 consecutive children symptomatic with the human immune deficiency virus. Ann Neurol 1993, 34:198-205 [DOI] [PubMed] [Google Scholar]
- 83.Elovaara I, Poutiainen E, Raininko R, Valanne L, Virta A, Valle SL, Lahdevirta J, Iivanainen M: Mild brain atrophy in early HIV infection: the lack of association with cognitive deficits and HIV-specific intrathecal immune response. J Neurol Sci 1990, 99:121-136 [DOI] [PubMed] [Google Scholar]
- 84.Aboulafia DM, Saxton EH, Koga H, Diagne A, Rosenblatt JD: A patient with progressive myelopathy and antibodies to human T-cell leukemia virus type I and human immunodeficiency virus type 1 in serum and cerebrospinal fluid. Arch Neurol 1990, 47:477-479 [DOI] [PubMed] [Google Scholar]
- 85.Persidsky Y, Gendelman HE: Development of laboratory and animal model systems for HIV-1 encephalitis and its associated dementia. J Leukoc Biol 1997, 62:100-106 [DOI] [PubMed] [Google Scholar]