

Abstract
During protein folding in which few, if any, definable kinetic intermediates are observable, the nature of the transition state is central to understanding the course of the reaction. Current experimental data does not distinguish the relative contributions of side chain immobilization and dehydration phenomena to the major rate-limiting transition state whereas this distinction is central to theoretical models that attempt to simulate the behavior of proteins during folding. Renaturation of the small proteinase inhibitor cystatin under oxidizing versus reducing conditions is the first experimental case in which these processes can be studied independently. Using this example, we show that sidechain immobilization occurs downstream of the major folding transition state. A consequence of this is the existence of states with disordered side chains, which are distinct from kinetic protein folding intermediates and which lie within the folded state free energy well.
Whether a protein folds in a single step or through a rapidly formed, transient intermediate, it is accepted that states observed on the unfolded side of the rate-limiting transition state have disordered side chains and are relatively hydrated. After traversing the barrier, states on the folded side are highly compact and have well defined (frozen) side chain orientations (1–4). Hence, irrespective of whether a folding reaction is two-state or three-state, the transition from non-native to native states involves the removal of water and the exchange of highly favorable packing interactions for side chain conformational entropy. However, we know little about the relative contribution of these processes to the rate-limiting barrier, making this a subject of lively debate (5–7). Here, we present an analysis of the folding behavior of chicken cystatin (a 116-amino acid analog of human cystatin C containing two disulfide bonds; see Fig. 1A) that challenges the idea that the phase transition of side chains from mobile to near crystalline conformations within the protein interior is necessarily an integral component of the major refolding transition state.
Figure 1.

(A) The three-dimensional structure of chicken cystatin (8–10). The single tryptophan residue is highlighted in green and the disulfide bonds in yellow. (B) One-dimensional 1H-NMR spectra of the different states of cystatin were acquired at 298 K on a Bruker DRX-500 spectrometer and were processed by using Felix (Molecular Simulations, Waltham, MA). Fox conditions were 200 μM protein in 50 mM sodium phosphate buffer (pH 7.0) and 10% D2O. Fred was made by refolding a 1.5 mM denatured sample (5.6 M GdnHCl/50 mM Tris⋅HCl, pH 7.0/20 mM DTT/2 mM EDTA) by diluting 5-fold into 50 mM Tris⋅HCl (pH 7.0) and 20 mM DTT in D2O. Ured conditions were 1.5 mM protein in 50 mM Tris⋅HCl (pH 7.0), 5.6 M GdnHCl, 20 mM DTT, 2 mM EDTA, and 10% D2O.
Materials and Methods
Protein Preparation.
Recombinant cystatin (molecular weight 13,100) was expressed in Escherichia coli by using the pIN-III-OmpA system and was purified from a periplasmic extract by papain affinity chromatography (11). Protein concentration was determined by using an extinction coefficient (12) ɛ280 nm = 11,410 M−1⋅cm−1. The protein was stored in 25 mM Tris⋅HCl buffer (pH 7.0) at 4°C in the presence of 1 mM sodium azide. Reduction of both disulfide bonds was obtained by maintaining the ratio of DTT to protein >100 (13).
Equilibrium and Kinetic Data Analysis.
The variation of data with GdnHCl concentration are plotted as a function of denaturant activity rather than concentration to take into account the nonlinear dependence of the free energy of folding with GdnHCl, as described (14–16). GdnHCl unfolding curves could all be described by a single equilibrium transition, which is classically fitted to an expression of the type:
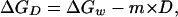 |
1 |
where ΔGD is the change in free energy of folding observed at an activity, D, of denaturant, ΔGw is the change in free energy of folding in water (or the stability of the protein in water), and m is an empirically derived parameter proportional to the change in solvent exposure during the transition (14, 17–18). ΔGD is derived directly from experimental data using:
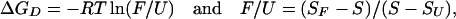 |
2 |
where S is the observed signal at an activity of denaturant D (fluorescence intensity or θ222 nm), SU is the signal of the unfolded form, SF is the signal of the folded form. Sloping baselines for F and U that change independently of the folding transitions under study were corrected by using ref. 19: F/U = [(SF + mF × [D]) − S]/[(S − (SU + mU × [D])].
Kinetic data were fully described by a three-state reaction scheme (U ↔ I ↔ F). Rate profiles [observed rate constant for folding/unfolding (kobs) versus denaturant activity] were fitted by using ref. 14:
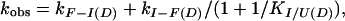 |
3 |
where kF−I(D) and kI−F(D) are rate constants for unfolding and refolding of the intermediate state (Ikin) from or to the folded state (F), and KI/U(D) is the equilibrium constant for the rapid interconversion of the intermediate and unfolded states. Each of these parameters is related to the denaturant activity by:
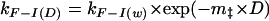 |
4 |
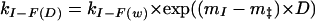 |
5 |
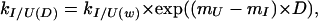 |
6 |
where the m parameters are related to those observed at equilibrium, in that they describe the compactness of each of the states U, I, ‡, and F (where ‡ is the transition state), but, in this case, F is the reference state. These values were then normalized with respect to the unfolded state (where mU = 0) for the purposes of the analysis presented here.
Results and Discussion
Reduction of the two disulfide bonds of cystatin (see Fig. 1A) leads to the formation of a stable, highly compact molten globule. The near-UV CD spectrum of the folded oxidized protein (Fox) shows a negative ellipticity in contrast to the folded reduced form (Fred), which has lost all ellipticity in this region, indicating that the environment of side chains contributing to this region within the Fred state is rotationally averaged (Fig. 2A). The far-UV CD spectrum of the two forms of the protein (Fig. 2B), which reports the conformation of the amide backbone, demonstrates that the secondary structure is largely maintained after both disulfide bonds are broken. Analysis of the relative compactness of Fred compared with Fox by size exclusion chromatography shows that the reduced protein remains as compact as the oxidized protein (Fig. 2C). Thus, reduction of the disulfide bonds is sufficient to cause “melting” of the side chains whereas compactness and secondary structure are retained.
Figure 2.

(A) Near-UV CD spectra were recorded for 100 μM Fox and Fred on a Jobin Yvon CD6 spectropolarimeter (pathlength 1 cm). For Fox, conditions were 25 mM Tris⋅HCl (pH 7.0). Fred was refolded from a denaturing solution of 5.6 M GdnHCl and 100 mM DTT in 25 mM Tris⋅HCl (pH 7.0) by a five-fold dilution into 25 mM Tris⋅HCl (pH 7.0). (B) Far-UV CD spectra after a 10-fold dilution of the solutions above with water. The ratio of reducing agent to protein is maintained at 200:1, and all buffers were degassed to prevent any reoxidation. (C) Molecular volumes were evaluated using an Biosep-sec 3000 HPLC column (Phenomenex, Torrance, CA). Proteins were injected at a concentration of 100 μM. Fox and Fred both elute with an apparent molecular weight of 15,000 [50 mM phosphate buffer (pH 7.0) ± 20 mM DTT], and Uox and Ured elute with apparent molecular weights of 50,000 and 59,000, respectively (as above with 6.5 M GdnHCl). In D and E are represented the fluorescence spectra of oxidized and reduced cystatin, respectively, in both their folded and unfolded states. The presence of DTT in the reduced samples prevents the direct comparison of oxidized and reduced spectra. However, peak wavelengths (λmax) of 345 and 350 nm can be reported for folded and unfolded species, respectively, in both oxidizing and reducing conditions. The fluorescence intensity of the tryptophan residue in Fox and Fred is quenched with respect to unfolded states suggesting exposure to a specific polar residue. Conditions are as detailed in the legend to Fig. 3.
This conclusion is supported by 1H-NMR spectroscopy (Fig. 1B). The oxidized protein shows a well dispersed spectrum characteristic of a native state whereas reduction leads to a loss of dispersion. Resonances belonging to residues throughout the molecule are shifted away from their original positions. The resulting spectrum shows that side chains have become mobile across the whole volume of the molecule, a feature that is characteristic of a molten globule state (20). Interestingly, despite the loss of fixed contacts between residues, the reduced protein retains high solubility and unfolds through a distinct, sharp, and fully reversible equilibrium transition (Fig. 3). A comparison of the free energy of folding of the reduced and oxidized proteins gives values of −7.8 and −15.6 kcal⋅mol−1, respectively. Moreover, the m values for these transitions, which represent the extent of solvent exclusion during folding, are very similar (Table 1), implying that the reduced, folded state is almost as compact (m(F/U)red/m(F/U)ox = 0.8) as the native oxidized state. The changes in tryptophan fluorescence on guanidinium denaturation are also indicative of conformational similarity between Fox and Fred. In both cases, there is an increase in indole fluorescence intensity on unfolding, showing that in Fox and Fred the indole group has a similar quenching environment (Fig. 2 D and E).
Figure 3.

Equilibrium unfolding in GdnHCl. The unfolding curves of Fox (○) and Fred (□) are shown as probed by tryptophan fluorescence (A) and far-UV CD (B). (A) Open symbols, unfolding titration; filled symbols, refolding titration. Fluorescence emission was measured with a Hitachi F-4,500 spectrofluorimeter between 300 and 410 nm by using an excitation wavelength of 290 nm (slit widths were 5 nm on both excitation and emission, 25°C). Final protein concentration was 5 μM in 50 mM Tris⋅HCl (pH 7.0). Buffers for the reduced proteins all contained 20 mM DTT. (B) Far-UV CD spectra were measured as described for Fig. 2. The data were fitted to Eq. 1, and values determined for ΔGeq(F/U) and meq(F/U) are listed in Table 1.
Table 1.
Thermodynamic parameters for the different states of cystatin observed both at equilibrium and kinetically*
| Oxidized cystatin | Reduced cystatin | |
|---|---|---|
| ΔGeq(F/U) | −15.6/−13.2† ± 1.0 | −7.8 ± 0.9 |
| meq(F/U) | −8.6 ± 0.3 | −6.8 ± 0.5 |
| c1/2 | 3.1 ± 0.3 | 1.9 ± 0.2 |
| kfold(I →F) | 192 ± 30 | 100 ± 7 |
| kunf(F → I) | 1.4 × 10−5 ± 4 × 10−6 | 0.12 ± 0.04 |
| ΔGkin(F/I) | −9.5 ± 0.2 | −4.0 ± 0.2 |
| Kkin(I/U) | 8,755 ± 8,200 | 264 ± 180 |
| ΔGkin(I/U) | −5.3/−3.0† ± 0.3 | −3.3 ± 0.3 |
| mkin(F/U) | −8.4 ± 0.5 | −7.4 ± 0.7 |
| mkin(‡/U) | −5.2 ± 0.5 | −4.7 ± 0.1 |
| mkin(I/U) | −4.8 ± 0.5 | −4.4 ± 0.2 |
The free energies of folding, ΔGeq(F/U), of both oxidized and reduced cystatin in the absence of denaturant are shown together with the meq(F/U) values and the activity of GdnHCl at the midpoint for unfolding, c1/2. These values represent best fits to the data with the lowest deviation and always fell within the uncertainty of parameters calculated from less precise data. Also shown are folding and unfolding rates for the rate-limiting step Ikin ↔ F observed for both oxidized and reduced cystatin, the deduced values of ΔGkin(F/I), and the extrapolated m values for the folded state, the transition state ‡, and the kinetic intermediate Ikin with respect to the unfolded state. The values determined from the fit to the kinetic data are represented graphically in Fig. 4B.
ΔG in kcal⋅mol−1, m in kcal⋅mol−2⋅liter, c1/2 in M (GdnHCl activity), rate constants in s−1.
†Free energy values with respect to Ured.
Having established that the two folded states differ mainly in the mobility of side chains and reversibly interconvert with the unfolded form, we can then compare the rates of formation and decay of these different end-states. To this end, the kinetics of folding and unfolding were measured as a function of the final activity of denaturant (as shown in Fig. 4A), allowing us to extract all kinetic parameters (listed in Table 1). We fully expected reduced cystatin to fold and unfold on a much more rapid time-scale than the oxidized form because of the lack of a requirement to dock the side chains within a crystalline core. Kinetic measurements of one of the best defined folding reactions, that of apomyoglobin, have illustrated this point by reporting rate constants on the microsecond scale for U→Ikin (21–23). In contrast, the folding rates of the two forms of cystatin in the absence of denaturant occur on similar time-scales; in fact, the reduced protein folds to the molten globule state slightly more slowly than its oxidized counterpart folds to the native state. [Slow folding to molten globule states has to our knowledge only been reported thus far for one other species, the A-state of cytochrome c (24).] Moreover, the diagram in Fig. 4B reflects the remarkable similarity in the thermodynamic properties of both the kinetic intermediate (Ikin), which gives rise to the roll-over in the kinetic plot, and the transition state through which the two forms of the protein pass on refolding. It should be noted that the energy profile for the oxidized state has been modified to account for the entropic cost of the two disulfide bonds. This requires raising the free energy of the unfolded reference state according to the calculation of chain entropy described in the legend.
Figure 4.

(A) Chevron plots for the folding reactions of Fox and Fred indicating the population of a kinetic intermediate. Open circles represent oxidized cystatin: folding was initiated by 1 + 10 dilution of 11 μM Uox [in 50 mM Tris⋅HCl (pH7.0) and 6.3 M GdnHCl], against 10 volumes of buffer or buffered GdnHCl in an Applied Photophysics (Surrey, U.K.) stopped-flow apparatus (λexcitation = 290 nm and slit width 4 nm). Fluorescence emission above 320 nm was recorded. Unfolding was initiated by 1 + 10 dilution of 11 μM Fox [in 50 mM Tris⋅HCl (pH 7.0)] as above. Open squares represent Fred: Refolding was carried out in two successive steps: a 50 μM stock solution of Ured [in 50 mM Tris⋅HCl (pH7.0) buffer, 6.3 M GdnHCl] was diluted to 3.3 M GdnHCl, where Fred (10 μM) is still not populated significantly (A). This solution was then refolded as for Fox. Unfolding was also carried out in two steps. A 100 μM stock protein solution in 5.6 M GdnHCl was diluted 20-fold, with 50 mM Tris⋅HCl (pH 7.0), before unfolding as for Fox. Fresh Fred was made every 20 min. All solutions contained 20 mM DTT. All experiments were carried out at 20°C. The data were fitted to Eq. 3, and the parameters determined are listed in Table 1. The inset to A shows typical unfolding and refolding transients. (B) A free energy plot for the folding reactions of both Fox (○) and Fred (□) is shown in which the reaction coordinate is the m values of the different states relative to the unfolded state. The plot representing folding in oxidizing conditions is offset vertically with respect to that in a reducing environment to account for the destabilizing effect of the disulfide bonds on Uox. Using the free energies of folding of cystatin in the reduced and oxidized states, and the redox potential measured in the folded state (−5.5 kcal⋅mol−1; results not shown), the resulting thermodynamic cycle provides a value of + 2.3 kcal⋅mol−1 for the free energy of Uox with respect to Ured. In agreement with this, a destabilization of +2.1 kcal⋅mol−1 was obtained from a theoretical calculation of the entropic effect of introducing a cross-link in a random coil polymer (32). (C) A schematic representation of the nature of the states identifiable in the refolding profile is shown. Hydrophilic side chains are shaded in purple, and hydrophobic side chains in green. When conditions favor folding, the protein initially buries many of its hydrophobic residues and forms secondary structure. Such species appear to be significantly native-like in fold topology (28). To bury further hydrophobic side chains, the protein has to expel many of the remaining water molecules, and can only do this at a high enthalpic cost through a transition state that contains structurally constrained water molecules as described (36). Moreover, once past the transition state, rigid packing interactions are only formed when the entropic cost for this immobilization is adequately compensated.
The formation of this intermediate must occur within the dead time of the stopped-flow apparatus because, at all times, only a single refolding transient is measured (an example is shown in the inset to Fig. 4A). No significant burst phase amplitude is observed on folding, suggesting that Ikin is spectroscopically silent with respect to U under these conditions. According to the m value analysis, the transient intermediate, whether disulfide-bridged or not, has excluded approximately half of the solvent from the protein interior, showing that the chain becomes relatively compact. Kinetics alone cannot be used to establish whether this kinetic intermediate is on-pathway. The two schemes shown below are equally applicable to the rate plot in Fig. 4A:
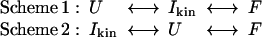 |
In the first scheme, Ikin is a productive intermediate that serves as the ground state from which the rate-limiting transition state is acquired. By contrast, Scheme 2 defines Ikin as a misfolded and off-pathway species. In this case, the rate-limiting step in folding results from a combination of breaking non-native interactions to create a more unfolded ground state, followed by a slow conversion to the folded state (25–27). In the case of cystatin, the former scheme is favored. As discussed previously (28), if Ikin had to be unfolded before the chain could fold (Scheme 2), the rate of conversion from U to F would be untenably large: k(U → F) = kfold(obs) × K(I/U) = 192 × 8,755 = 1.7 × 106 s−1. This rate is commensurate with the fastest measured propagation of an α–helix and is unlikely to be applicable to the formation of a complex α/β protein such as cystatin. Similar arguments have been used to demonstrate that Ikin states of IL-1β (29), N-PGK, (30) and pseudoazurin (31) are also on-pathway. In addition, the Ikin state is a nonrandom distribution of conformations. If the intermediate state constituted an ensemble of randomly collapsed chains, then the entropic penalty for introducing disulfide bridges into the molecule would be approximately the same as on the fully unfolded, random population (Uox is destabilized by ≈2.3 kcal⋅mol−1 compared with Ured, as illustrated in Fig. 4B). Hence, the effect of disulfides bonds on the relative stability would be small. In reality, the increase in the stability of Ikin is similar in both the oxidized and reduced forms and thus is close to that expected for a state in which the backbone of the polymer is completely ordered in Ikin but random in U (32).
More intriguing is the close similarity in the size of the major transition state barrier in the reduced and oxidized species. In the case of oxidized cystatin, two processes occur as Ikin is converted to the folded state; water must be removed and side-chains must acquire rigid and precise packing. In the case of folding the reduced protein, side-chain immobilization is not achieved, yet the barrier height is the same. One possibility is that the folding barrier for both oxidized and reduced protein results from their progression during refolding to similar thermodynamic traps that require some unfolding to escape. In such circumstances, however, the m values of the transition should be markedly less than those of the kinetic intermediates. This is not observed experimentally (28). The inescapable conclusion is that the major contributor to the folding barrier is the removal of water. To some extent, the conclusion is consistent with φ value analysis that reveals that φ‡ even for core residues averages ≈0.4 and rarely exceeds 0.7 (33). If precise and fixed interactions between side chains were a feature of the transition state, then one would expect values to be higher if only for a nucleus of core residues. However, φ‡ values are entirely derived from the relative effect of mutations on the rates of folding and unfolding. Hence, this type of analysis defines the degree of native contact energy acquired by a specified side chain in the transition state. It does not distinguish whether, for instance, high φ‡ values are attributable to a high degree of local desolvation or the fixing of local side chain contacts in a native-like conformation.
Moreover, thermodynamic analysis shows that the transition from kinetic intermediate to the rate-limiting transition state is enthalpically unfavorable but entropically favorable (34, 35). The entropic stability of the transition state is consistent with the necessity to desolvate the core of the protein before the interior side chains can be docked. In passing from the transition state to the folded state, the situation is reversed; an entropic penalty paid by side-chain immobilization is outweighed by the enthalpic contribution. Although the characteristics of this last phase of folding can be partially explained by the exchange of favorable van der Waals interactions for the unfavorable fixing of side chain conformation, the high enthalpy of the transition state barrier is more mysterious. It has previously been attributed to the protein chain reaching a critical stage of dehydration in which water molecules are trapped in environments in which they cannot bond either to groups on the protein or to other water molecules in bulk solvent (34). This interpretation of the role of water in providing a high enthalpic barrier to the folding process is fully consistent with the theoretical dehydration model of Rank and Baker (36) and with the physical phenomenon we describe here. The results above indicate that complications introduced into such interpretations by a requirement to consider side chain immobilization are unnecessary. Side chain immobilization occurs downstream of the major transition state.
Acknowledgments
We thank Ian McGilloway for help with some of the preliminary experiments. We are also indebted to Janko Kos and Vito Turk (J. S. Institute) for the generous gift of cystatin isolated from chicken egg-white. This work was supported by a Wellcome Trust project grant, TEMPUS fellowship (to E.Z.), and Wellcome Trust and Biotechnology and Biological Sciences Research Council for spectrometers. R.A.S. is the holder of a Wellcome Trust Research Career Development Fellowship. J.P.W. and A.R.C. are fellows of the Lister Institute.
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Shakhnovich E I, Finkelstein A V. Biopolymers. 1989;28:1667–1680. doi: 10.1002/bip.360281003. [DOI] [PubMed] [Google Scholar]
- 2.Matoushek A, Kellis J T, Jr, Serrano L, Fersht A R. Nature (London) 1989;340:122–126. doi: 10.1038/340122a0. [DOI] [PubMed] [Google Scholar]
- 3.Matoushek A, Kellis J T, Jr, Serrano L, Bycroft M, Fersht A R. Nature (London) 1990;346:440–445. doi: 10.1038/346440a0. [DOI] [PubMed] [Google Scholar]
- 4.Ha J-H, Loh S N. Nat Struct Biol. 1998;5:730–737. doi: 10.1038/1436. [DOI] [PubMed] [Google Scholar]
- 5.Kay M S, Baldwin R L. Nat Struct Biol. 1996;3:439–445. doi: 10.1038/nsb0596-439. [DOI] [PubMed] [Google Scholar]
- 6.Ptitsyn O B. Nat Struct Biol. 1996;3:488–490. doi: 10.1038/nsb0696-488. [DOI] [PubMed] [Google Scholar]
- 7.Segawa S I, Sugihara M. Biopolymers. 1984;23:2473–2488. doi: 10.1002/bip.360231122. [DOI] [PubMed] [Google Scholar]
- 8.Bode W, Engh R A, Musil D, Thiele U, Huber R, Karshikiv A, Brzin J, Kos J, Turk V. EMBO J. 1988;7:2593–2599. doi: 10.1002/j.1460-2075.1988.tb03109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dieckmann T, Mitschang L, Hofman M, Kos J, Turk V, Auerswald E A, Jaenicke R, Oschkinat H. J Mol Biol. 1993;234:1048–1059. doi: 10.1006/jmbi.1993.1658. [DOI] [PubMed] [Google Scholar]
- 10.Engh R A, Dieckmann T, Bode W, Auerswald E A, Turk V, Huber R, Oschkinat H. J Mol Biol. 1993;234:1060–1069. doi: 10.1006/jmbi.1993.1659. [DOI] [PubMed] [Google Scholar]
- 11.Anastasi A, Brown M A, Kembhavi A A, Nicklin M J H, Sayers C A, Sunter D C, Barett A J. Biochem J. 1983;211:129–138. doi: 10.1042/bj2110129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Auerswald E A, Genenger G, Mentele R, Lenzen S, Assfalg-Machleidt I, Mitschang L, Oschkinat H, Fritz H. Eur J Biochem. 1991;200:131–138. doi: 10.1111/j.1432-1033.1991.tb21059.x. [DOI] [PubMed] [Google Scholar]
- 13.Björk I, Ylinenjärvi K. Biochemistry. 1992;31:8597–8602. doi: 10.1021/bi00151a029. [DOI] [PubMed] [Google Scholar]
- 14.Parker M J, Spencer J, Clarke A R. J Mol Biol. 1995;253:771–786. doi: 10.1006/jmbi.1995.0590. [DOI] [PubMed] [Google Scholar]
- 15.Staniforth R A, Burston S G, Smith C J, Jackson G S, Badcoe I G, Atkinson T, Holbrook J J, Clarke A R. Biochemistry. 1993;32:3842–3851. doi: 10.1021/bi00066a003. [DOI] [PubMed] [Google Scholar]
- 16.Johnson C M, Fersht A R. Biochemistry. 1995;34:6975–6804. doi: 10.1021/bi00020a026. [DOI] [PubMed] [Google Scholar]
- 17.Myers J K, Pace C N, Scholtz J M. Protein Sci. 1995;4:2138–2148. doi: 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alonso D O V, Dill K A. Biochemistry. 1991;30:5974–5985. doi: 10.1021/bi00238a023. [DOI] [PubMed] [Google Scholar]
- 19.Santoro M M, Bolen D W. Biochemistry. 1988;27:8063–8068. doi: 10.1021/bi00421a014. [DOI] [PubMed] [Google Scholar]
- 20.Baum J, Dobson C M, Evans P A, Hanly C. Biochemistry. 1989;24:4570–4577. [Google Scholar]
- 21.Jamin M, Baldwin R L. Nat Struct Biol. 1996;3:613–618. doi: 10.1038/nsb0796-613. [DOI] [PubMed] [Google Scholar]
- 22.Gilmanshin R, Williams S, Callender R H, Woodruff W H, Dyer R B. Proc Natl Acad Sci USA. 1997a;94:3709–3713. doi: 10.1073/pnas.94.8.3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ballew R M, Sabelko J, Gruebele M. Proc Natl Acad Sci USA. 1996;93:5759–5764. doi: 10.1073/pnas.93.12.5759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Colón W, Roder H. Nat Struct Biol. 1996;3:1019–1025. doi: 10.1038/nsb1296-1019. [DOI] [PubMed] [Google Scholar]
- 25.Kiefhaber T. Proc Natl Acad Sci USA. 1995;92:9029–9033. doi: 10.1073/pnas.92.20.9029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sosnick T R, Mayne L, Hiller R, Englander S W. Nat Struct Biol. 1994;1:149–156. doi: 10.1038/nsb0394-149. [DOI] [PubMed] [Google Scholar]
- 27.Bryngelson J D, Onuchic J N, Socci N D, Wolynes P G. Proteins. 1995;21:167–195. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 28.Clarke A R, Waltho J P. Curr Opin Biotechnol. 1997;8:400–410. doi: 10.1016/s0958-1669(97)80060-2. [DOI] [PubMed] [Google Scholar]
- 29.Heidary D K, Gross L A, Roy M, Jennings P A. Nat Struct Biol. 1997;4:725–731. doi: 10.1038/nsb0997-725. [DOI] [PubMed] [Google Scholar]
- 30.Hosszu L L P, Craven C J, Parker M J, Lorch M, Spencer J, Clarke A R, Waltho J P. Nat Struct Biol. 1997;4:801–804. doi: 10.1038/nsb1097-801. [DOI] [PubMed] [Google Scholar]
- 31.Capaldi A P, Ferguson S J, Radford S E. J Mol Biol. 1999;286:1621–1632. doi: 10.1006/jmbi.1998.2588. [DOI] [PubMed] [Google Scholar]
- 32.Betz S F. Protein Sci. 1993;2:1551–1558. doi: 10.1002/pro.5560021002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fersht A R. Structure and Mechanism in Protein Science. New York: Freeman; 1998. [Google Scholar]
- 34.Parker M J, Lorch M, Sessions R B, Clarke A R. Biochemistry. 1998;37:2538–2545. doi: 10.1021/bi972459k. [DOI] [PubMed] [Google Scholar]
- 35.Boczko E M, Brooks C L. Science. 1995;269:393–396. doi: 10.1126/science.7618103. [DOI] [PubMed] [Google Scholar]
- 36.Rank J A, Baker D. Protein Sci. 1997;6:347–354. doi: 10.1002/pro.5560060210. [DOI] [PMC free article] [PubMed] [Google Scholar]