

Abstract
Patients with serous borderline tumors of the ovary often present with multiple tumors at different sites in the abdominal cavity. Whether different foci of ovarian serous borderline tumors are monoclonal in origin, arising as a consequence of spread from a single ovarian site, or whether such deposits are polyclonal and explained by independent molecular genetic alterations on the background of a field defect, is unknown. So far, only X-chromosome inactivation studies were performed to study this issue. We used a genome-wide allelotyping to assess clonality in 47 metachronous and/or synchronous multifocal tumors from 22 patients, using 59 microsatellite markers. Loss of heterozygosity (LOH) was observed in only 34 of 1969 informative markers in 9 of 22 serous borderline cases studied. Of these cases, 7 showed concordant LOH for at least one polymorphic marker in more than one tumor site. Flanking microsatellite markers enabled identification of identical chromosomal breakpoints in 6 of 7 cases. The LOH results strongly favor a common origin indicated by a likelihood ratio (possibility common origin/possibility independent origin) ranging from 39 to 14,163. Strong additional evidence for monoclonality is provided by the finding of identical microsatellite alterations in all three-tumor sites in one case.
Ovarian serous borderline tumors (SBT) are characterized by either polypoid excrescencies and papillae on the ovarian surface and/or occupying a variable extent of a parenchymal cyst lining. The three features essential for the diagnosis include the formation of cellular buds that appear to drop off the surface of the ovary and float in the intracystic fluid, some degree of nuclear atypia, and lack of destructive stromal invasion. 1 These tumors were originally classified as “borderline” lesions because they behaved in a remarkably indolent manner despite the pathological features suggesting malignancy. Even with widespread tumor deposits at laparotomy and the presence of lymph node involvement, the prognosis of such lesions generally remains excellent. On this basis, tumor nodules, which are found in 20 to 40% of the cases, on the peritoneal surface, omentum or in lymph nodes, are referred to as implants rather than metastases. 2
The pathogenesis of SBT origin at multiple sites remains unclear. Two major hypotheses exist; the first favors a monoclonal origin, arguing that multifocal tumor nodules are formed from neoplastic cells that are shed from a primary ovarian tumor, and seed the pelvic and peritoneal surfaces. The finding that multifocal tumor deposits are associated with exophytic ovarian surface disease 3 supports this view. Furthermore, the characteristic histological appearance of tumor cells, detached from the primary tumor, and floating in cystic fluid is consistent with this hypothesis. The alternative hypothesis favors a polyclonocal origin for multifocal SBT, proposing the existence of a defect in a “field” of susceptible cells of Mullerian origin from which multiple independent tumors arise. 4
So far, only X-chromosome inactivation analysis has been used to study clonality in multifocal SBT. In one study this was combined with loss of heterozygosity (LOH) analysis of the androgen receptor on the X chromosome. 5,6 These studies indicated that a subset of SBT might be of multiclonal origin. However, the reliability of X-chromosome inactivation analysis for clonality analysis in tumors has been challenged. 7 One problem is that tumors may show altered DNA methylation patterns. 8,9 Furthermore, non-random X-chromosome inactivation in germline DNA of healthy and cancer-affected females may complicate the interpretation. 10,11
As an alternative, LOH analysis with multiple microsatellite markers has been used to study clonality in bilateral ovarian carcinomas and bilateral breast cancer. 12,13 This type of approach is directed to stochastic tumor-associated genetic alterations during tumorigenesis and enables calculation of the statistical likelihood of dependent versus independent origin.
In this study, we selected a group of 22 cases of SBT, each comprising a minimum of two separate tumors.Allele-specific LOH patterns in 47 metachronous and/or synchronous multi-focal SBT were determined using 59 microsatellite markers. We found concordant LOH for one to four markers at all tumor sites in seven cases whereas in two cases only one tumor site showed LOH for one marker. No LOH events were found in the remaining 13 tumors. Thus, for the seven cases where the LOH analysis was conclusive, compelling evidence exists for a monoclonal origin.
Materials and Methods
Specimens
Twenty-two cases of ovarian SBT (Leiden University Medical Center and Leyenberg Hospital The Hague, The Netherlands) were included in this study. The tumor tissue was fixed in 10% buffered formalin, embedded in paraffin, and stained with hematoxylin and eosin. Histopathological data are presented in the Results section.
Microdissection, DNA Isolation, and Quantification
Ovarian SBT tissue from 10-μm serial hematoxylin-stained paraffin sections was microdissected with a 10-G needle and incubated for 12 hours in 186 μl PK1 buffer [10 mmol/L Tris (pH 8.3), 50 mmol/L KCl, 2.5 mmol/L MgCl2, 0.45% NP40, 0.45% Tween 20, 0.01% gelatin], 5% Chelex (Chelex 100; Bio-Rad Laboratories, Hercules, CA), and 10 μl proteinase K (10 mg/ml) at 56°C followed by a 10-minute incubation at 100°C to inactivate proteinase K. Subsequently, the resin was removed from the supernatant following a 10-minute centrifugation at 30,000 rpm. Non-tumoral DNA was extracted from archival paraffin blocks from the same patient. DNA content was quantitated using Picogreen double-stranded DNA (dsDNA) quantitation reagent (Molecular Probes Europe BV, Leiden, The Netherlands), an ultra-sensitive fluorescent nucleic acid stain for quantitating dsDNA in solution, according to the manufacturer’s instructions.
Microsatellite Marker PCR
Detection of LOH was performed by polymerase chain reaction (PCR) as described by Weber and May 14 with 59 microsatellite markers covering all chromosomes. A list of markers is available upon request (G.J.Fleuren@LUMC.nl). In brief, working solutions of 10 ng/μl DNA were prepared to circumvent PCR artifacts. 15 PCR was performed in a total reaction volume of 12 μl, containing 5 pmol forward and 5 pmol reverse primer, 0.1 mg/ml BSA, 10 mmol/L Tris-HCl (pH 8.3), 50 mmol/L KCl, 1.5 mmol/L MgCl2, 0.01% (w/v) gelatin, 200 μmol/L dATP, 200 μmol/L dGTP, 2.5 μmol/L dCTP, 200 μmol/L dTTP, 0.1 μl [α-32P]dCTP (3000 Ci/mmol, 10 mCi/ml, Amersham Pharmacia Biotech, s’-Hertogenbosch, The Netherlands.) and 1.0 units AmpliTaq Gold polymerase (Applied Biosystems Inc., Foster City, CA) for 10 minutes at 96°C, 33 cycles of 1 minute at 94°C, 2 minutes at 55°C, 1 minute at 72°C, with a delay extension step of 6 minutes at 72°C in a thermal cycler (MJ Research, Watertown, MA). The PCR samples were denatured with 2 volumes of 0.3% xylene-cyanol, 0.3% bromphenol blue, 10 mmol/L EDTA (pH 8.0), and 90% formamide. Electrophoresis was performed on a 0.4 mm 6.5% polyacrylamide gel containing 7 mol/L urea. After drying, a x-ray film was exposed to the gel. Allele intensities were quantitated using a phosphor imager (Phosphor Imager 445 SI, Molecular Dynamics, Sunnyvale, CA). LOH was assessed by calculation the imbalance factor (IF) ie, the ratio of the normal allele intensities divided by the ratio of the tumor allele intensities. An IF, in at least two independent experiments, higher than 1.7 was interpreted as LOH. 16 Whenever LOH was found, additional flanking microsatellite markers were tested to identify chromosomal breakpoints.
Statistics
For each patient the probability of the observed pattern of LOH in the pair of tumors was calculated assuming an independent origin (PIO) and assuming a common origin (PCO). These probabilities depend on the number of informative (heterozygous) markers (n), and the probability of LOH at each individual marker (p). Subsequently the likelihood ratio (LR = PCO/PIO) was calculated, which indicates how likely the observed LOH pattern is under the assumption of common origin compared to independent origin.
For a case where m2 is the number of markers for which LOH was observed in both tumors for the same allele and m1 the number of markers wherein only one of the two tumors LOH was observed, the PIO is
 |
To calculate the PCO, it should be taken into account that LOH may occur both before and following genetic divergence of the tumor. If we note with p1, the probability that LOH occurred before divergence and with p2 the probability of LOH occurred after the genetic divergence, then
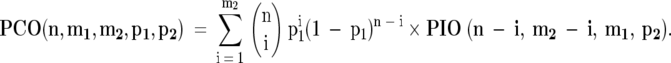 |
In the calculations it was assumed that p1 = p2 and that p1 + p2 = p. In the above formulae, the possibility of LOH for different alleles in the respective tumors was ignored, as no such cases were observed in this data set. Only the 59 microsatellite markers selected for the allelotyping were included in the calculations; additional flanking markers to identify chromosomal breakpoints (indicated by the asterisks in Figure 1 ▶ ) were excluded since LOH on the same chromosome arm is not an independent event.
Figure 1.

Concordance of LOH in bilateral SBT and one implant (OV112). Patient identification numbers are underlined. Lt, left ovary; Rt, right ovary; Im, implant. Open symbols: retention of heterozygosity (ROH); red: LOH; gray: non-informative; blue: allelic gain. Asterisk, Microsatellite markers added to the allelotyping to identify breakpoints. Boxed microsatellite markers delineate identical chromosomal breakpoints. Chromosomes for which no LOH was observed were not included in the figure.
Results
Gross Pathology and Histopathology
All but 2 (OV11, 99) of the 22 bilateral ovarian SBT had exophytic ovarian surface involvement. Non-invasive SBT implants were present on the abdominal peritoneal surface in eight patients (OV1, 9, 12, 23, 25, 28, 105, 112) although only three of these (OV12, 23, 112) contained sufficient DNA following microdissection to permit inclusion in the LOH analysis. There was close to a 50% difference in tumor size between the ovaries in cases OV2, 3, 8, 11, 15, 24, 28, 99, 101, and 105. In a further seven cases (OV1, 6, 9, 23, 25, 112, 114) there was no size difference between the ovaries and the size was not adequately documented to permit comparison in the remaining five cases (OV10, 12, 16, 26, 103).
Histologically, nearly all tumors showed the typical pathological features of SBT. However, one case displayed pronounced tubal metaplasia (OV15) and a second showed extensive fibroblast proliferation (OV25). Interestingly, these changes were apparent in both ovarian tumors in these two patients (Figure 2, A and B) ▶ .
Figure 2.

A and B: Hematoxylin and eosin-stained sections of SBT showing pronounced tubal metaplasia (OV15, ×40) (A), and a striking stromal reaction (OV25, ×10) (B), in bilateral ovarian neoplasm.
Loss of Heterozygosity
LOH was identified 38 times in 1969 informative PCR reactions in 9 of the 22 cases studied (Figure 1) ▶ . In two cases, OV24 (D9S53, D9S103), and OV105 (D7S2847, D7S2202)), LOH was represented by two markers on the same chromosome arm, hence, these two events must be considered as non-independent. Therefore the total number of LOH events was considered to be 34. No LOH was detected in 13 cases (OV 3, 6, 8, 9, 11, 12, 23, 25, 28, 99, 101, 103, 114). LOH was observed in 13 of the 23 investigated chromosomes; the most frequently involved include 6q, 8p, 13q, 22q, and Xq, showing LOH for two cases. Typical examples of autoradiographs demonstrating LOH are shown in Figure 3 ▶ .
Figure 3.
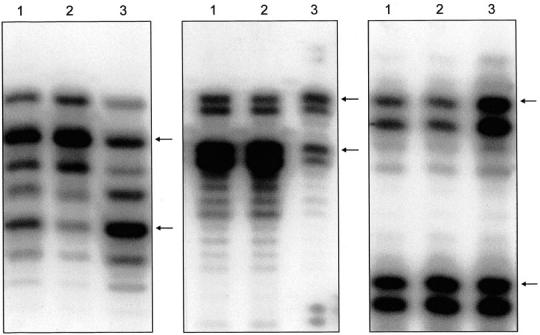
An autoradiograph showing loss of the lower allele for D9S103 in OV24 (Figure 3A) ▶ , loss of the upper allele for DXS6801 in OV105 (Figure 3B) ▶ , and loss of the upper allele for D12S2070 in OV2 (Figure 3C) ▶ ; left ovarian SBT (lane 1), right ovarian SBT (lane 2), and control tissue (lane 3). Arrows indicate the two (heterozygous) alleles in the control tissue.
Of the 9 cases with LOH, 2 cases (OV16, 26) showed LOH for a single microsatellite marker in only one of the tumor deposits. The remaining 7 cases showed concordant LOH (ie, same allele lost for a microsatellite marker) for at least one polymorphic marker in all tumor sites (ie, 26 of the total 34 observed losses (77%)). Flanking microsatellite markers enabled identification of chromosomal breakpoints in 6 of these 7 cases (Figure 1) ▶ . Of note, in two cases (OV10, 105) identical breakpoints were even present on two different chromosomes. Six additional LOH events were restricted to one tumor site in 5 (OV1, 2, 10, 15, 24) of the 7 cases; two cases (OV105, 112) were completely concordant (see Figure 1 ▶ ). In three of these cases (OV2, 15, 24) there was close to a 50% difference in tumor size and LOH was restricted in these cases to the larger of the two tumors.
Attention is drawn to case OV112, which comprises a bilateral ovarian tumor with non-invasive abdominal peritoneal implants. In addition to LOH at chromosome 10q in all three-tumor sites, the tumor deposits showed an identical novel band for D15S1232 that was not present in the paired normal DNA. Sequencing of the PCR products confirmed the presence of an extra GAAA-repeat in all three-tumor samples. These results were duplicated using additional paraffin blocks to exclude tumor heterogeneity (Figure 4) ▶ . The presence of a hereditary non-polyposis colorectal cancer (HNPCC)-related mutator phenotype in this patient was excluded because it did not meet the Amsterdam Criteria (data not shown). 17
Figure 4.

Gain of allele for D15S1232 in OV112; right ovarian SBT (lane 1), left ovarian SBT (lanes 2 and 3), abdominal implant (lane 4), and control tissue (lane 5). Arrow indicates the one (homozygous) allele in the control tissue. The extra allele in the tumor samples is indicated by an asterisk.
Statistical Analysis
There were nine cases with LOH. For some markers LOH was observed in more than one individual. The distribution of LOH was in agreement with expectations assuming that LOH for a given marker occurs independently of the presence of LOH at other chromosome arms. The probability to observe LOH, based on the prevalence of LOH and the actual number of informative markers, was calculated to be 0.07 per patient per heterozygous marker. The LR for all seven representative cases varied from 39 to 14,163, strongly favoring a common origin (Table 1) ▶ .
Table 1.
Probability Assuming a Common Origin (PCO), Probability Assuming an Independent Origin (PIO) and Likelihood Ratio (LR=PCO/PIO)
| Case | M2* | PCO | PIO | LR |
|---|---|---|---|---|
| OV1 | 2 | 5.4 10−2 | 2.2 10−4 | 245 |
| OV2 | 1 | 3.8 10−2 | 9.1 10−4 | 42 |
| OV10 | 2 | 3.3 10−2 | 5.4 10−5 | 614 |
| OV15 | 1 | 4.3 10−2 | 1.1 10−3 | 39 |
| OV24 | 2 | 6.3 10−2 | 3.1 10−4 | 202 |
| OV105 | 3 | 7.7 10−3 | 5.5 10−7 | 14,163 |
| OV112 | 1 | 8.0 10−4 | 8.5 10−7 | 940 |
*M2 is the number of markers for which LOH was observed in both tumors.
Discussion
We have allelotyped 22 cases of multifocal SBT with 59 microsatellite markers covering all chromosomes to asses the multiclonal versus monoclonal origin of these tumors. Thirteen (59%) of the 22 cases showed no LOH for any of the 59 microsatellite markers in this study. Of the nine cases (41%) with LOH, seven cases showed concordant LOH for one to four markers representing different chromosome arms. In 26 of 38 LOH events the upper allele was lost. Preferential loss of the upper allele may be due to degradation of the DNA. This may result in artifactual LOH of the upper allele, especially when neoplastic paraffin-embedded tissue samples are compared with fresh (frozen) normal control tissues. In the present study, the control DNA was from the same tissue source (ie, formalin-fixed, paraffin-embedded) as the SBT samples. Moreover, quantitative evaluation of allele intensities rather than visual assessment of the alleles enables the calculation of the IF, thus normalizing allele intensity values. 18
No discordant LOH (ie, different allele lost for a microsatellite marker showing bilateral LOH) was observed in any of these seven cases. In six of these cases, both ovaries were involved, whereas in a seventh case (OV112) additional tumor deposits were identified in the abdominal peritoneum. No difference was found with respect to age at diagnosis and clinical data for the cases with and without LOH. However, since clinical follow-up is limited, we formally cannot exclude the possibility that both groups are biologically different subsets of SBT. For this reason the cases without LOH were excluded in calculating the chance to observe LOH for a marker.
The calculated likelihood ratios, although showing a wide variation (range, 39 to 14,163), strongly support the hypothesis of a common origin of bilateral ovarian SBT. The LR rapidly increases with the number of markers showing LOH illustrating the power of this statistical approach compared to the non-quantitative X-chromosome inactivation analysis. Evidence for a common origin is further provided by the fact that additional flanking markers enabled the identification of identical chromosomal breakpoints in all tumor sites for six of seven cases (OV1, 2, 10, 15, 105, 112).
LOH was restricted to one SBT in seven cases (OV1, 2, 10, 15, 24, 16, 26); (concordant LOH was present in two of these seven cases (OV2 and OV15)). Unilateral LOH does not exclude the possibility of a monoclonal origin since this can be attributed to on-going genetic alterations. 19 This is supported by the observation that LOH was confined to the dominant (primary) tumor site for one microsatellite marker in two cases (OV2, 15) and for two microsatellite markers in one case (OV24). Unilateral LOH can also be the result of heterogeneity within the tumor cell population, but this was largely excluded by the consistency of the results from different paraffin blocks of the same SBT in our study.
Additional evidence for monoclonality is presented by case OV112, in which we identified an identical microsatellite alteration (repeat extension) for D15S1232 in all three-tumor sites. Although the mechanism producing expansion or reduction of tri- or tetra nucleotide repeats in non-HNPCC tumors is not known, identical microsatellite alterations constitute a powerful marker of clonality. 20
Apart from the LOH results, evidence for a common origin is presented by two cases (OV15, 25) showing similar atypical histological features in both tumor sites. OV15 was one of the seven cases in which LOH was identified for one polymorphic marker at both tumor sites. Unfortunately, OV25 was non-contributory as LOH was not identified in this case.
Evidence for independent tumorigenesis of multifocal SBT is presented in two studies based on X-chromosome inactivation analysis. 5,6 Lu et al 5 examined eight cases of which five showed the same pattern of X-chromosome inactivation in different tumor sites. Since there is a 50% chance of inactivating the same X chromosome in genetically non-related tumors, no conclusions could be drawn from these five cases. The remaining three cases revealed alternate patterns of X inactivation. Likewise, Gu et al 6 reported on seven patients with multifocal SBT of which six had different patterns of X inactivation.
The use of X-chromosome inactivation for analysis of clonality is based on the occurrence of random X-chromosome inactivation in early embryogenesis. Since, for a specific progenitor, all daughter cells will inherit the same inactivated X chromosome, this can be used as a marker of clonality. 21 However, recent insights in tumor-related and technical aspects explain why X-chromosome inactivation studies are far from ideal for assessing clonality.
Tumor-related changes interfering with X-chromosome inactivation are best illustrated by the finding of random X-chromosome inactivation in up to 50% of invasive cancers. 22 This may be due to X-chromosome aneuploidy and abnormalities in methylation pattern as these changes are frequently observed in malignancy. 22 For SBT, these findings were underscored by the study of Buller et al 7 who found 72% of random X-chromosome inactivation in a series of 44 cases with an unilateral SBT.
Furthermore, technical problems like incomplete digestion of DNA samples and contamination of normal tissues, may flaw the interpretation of X-chromosome inactivation patterns. We previously showed that the amount of input DNA is a critical factor for reliable microsatellite PCR since insufficient DNA input frequently results in artifactual loss or gain of alleles. 15 Our results indicate a minimum amount of 10 ng/μl for DNA extracted from paraffin blocks whereas we calculated that the amount of DNA extracted by Gu et al, 6 from 400 to 600 microdissected cells was less than 0.25 ng/μl. At this concentration, PCR artifacts occur in over 30%, 15 which may explain to a certain extent the discrepancy with our results.
In contrast to analysis of clonality by X inactivation, LOH is an irreversible genetic event acquired during tumorigenesis rather than an epigenetic phenomenon like methylation. The weakness of this approach is that in the absence of informative markers and the failure to detect LOH it is likely to underestimate the frequency of clonality. Also, due to stringent demands on DNA input concentration, only three of the eight non-invasive peritoneal implants contained sufficient lesional tissue to be included in the analysis. Of these three cases, only one (OV112) showed LOH and allelic gain, which was entirely concordant for the three different tumor sites. Although this is indicative for a monoclonal origin of the peritoneal implants, on the basis of only one case, it is not possible to generalize this result. However, assuming a monoclonal origin of bilateral ovarian SBT, it is reasonable to speculate that the different peritoneal deposits arise through seeding from a dominant ovarian tumor mass. This is supported by the frequent occurrence of implants in the presence of an ovarian SBT with predominantly exophytic growth patterns. Furthermore, there is a tendency for concentration of implants close to the primary tumor. 3 On the other hand, one must also try to explain why, on occasion, SBT deposits occupy sinusoids in abdominal or even extra-abdominal lymph nodes. These findings support the earlier suggestion that SBT must have at least limited capacity to metastasize. 23 Why, in general, these lymph node metastases behave in such an indolent manner is intriguing but is not unique to SBT. A comparable situation exists for benign metastasizing leiomyoma of the uterus. 24 In conclusion, our results strongly support a monoclonal origin of at least a subset of bilateral ovarian SBT.
Acknowledgments
We thank Leyenburg Hospital, The Hague, The Netherlands for their support in providing pathology material.
Footnotes
Address reprint requests to Prof. Dr. G.J. Fleuren, Department of Pathology, Leiden University Hospital, Albinusdreef 2, Building 1, P1–41, P.O. Box 9600, 2300 RC Leiden, The Netherlands. E-mail: G.J.Fleuren@LUMC.nl.
Supported by Grant ZON MW, The Netherlands.
References
- 1.Russel P: Surface epithelial-stromal tumors of the ovary. Kurman RJ eds. Blaustein’s Pathology of the Female Genital Tract 1994:pp 705-782 Springer-Verlag New York, Berlin, Heidelberg
- 2.Russel P: Surface epithelial-stromal tumors of the ovary. Fox H eds. Obstetrical and Gynaecological Pathology 1995:pp 743-822 Churchill-Livingstone, Inc. New York
- 3.Segal GH, Hart WR: Ovarian serous tumors of low malignant potential (serous borderline tumors): the relationship of exophytic surface tumor to peritoneal implants. Am J Surg Pathol 1992, 16:577-581 [DOI] [PubMed] [Google Scholar]
- 4.Saretzki G, Hoffmann U, Rohlke P, Psille R, Gaigal T, Keller G, Hofler H, Loning T, Petersen I, Dietel M: Identification of allelic losses in benign, borderline, and invasive epithelial ovarian tumors and correlation with clinical outcome. Cancer 1997, 80:1241-1249 [DOI] [PubMed] [Google Scholar]
- 5.Lu KH, Bell DA, Welch WR, Berkowitz RS, Mok SC: Evidence for the multi-focal origin of bilateral and advanced human serous borderline ovarian tumors. Cancer Res 1998, 58:2328-2330 [PubMed] [Google Scholar]
- 6.Gu J, Roth LM, Younger C, Michael H, Abdul-Karim FW, Zhang S, Ulbright TM, Eble JN, Cheng L: Molecular evidence for the independent origin of extra-ovarian papillary serous tumors of low malignant potential. J Natl Cancer Inst 2001, 93:1147-1152 [DOI] [PubMed] [Google Scholar]
- 7.Buller RE, Sood AK, Lallas T, Buekers, Skilling JS: Association between nonrandom X-chromosome inactivation and BRCA1 mutation in germline DNA of patients with ovarian cancer. J Natl Cancer Inst 1999, 91:339-346 [DOI] [PubMed] [Google Scholar]
- 8.Yang DH, Smith ER, Cohen C, Wu H, Patriotis C, Godwin AK, Hamilton TC, Xu XX: Molecular events associated with dysplastic morphologic transformation and initiation of ovarian tumorigenicity. Cancer 2002, 94:2380-2392 [DOI] [PubMed] [Google Scholar]
- 9.Jacobs IJ, Kohler MF, Wiseman RW, Marks JR, Whitaker R, Kerns BAJ, Humphrey P, Berchuck A, Ponder BAJ, Bast RC: Clonal origin of epithelial ovarian carcinoma: analysis by loss of heterozygosity, p53 mutation, and X-chromosome inactivation. J Natl Cancer Inst 1992, 84:1793-1798 [DOI] [PubMed] [Google Scholar]
- 10.Gale RE, Wheadon H, Linch DC: X-chromosome inactivation patterns using HPRT and PGK polymorphisms in haematologically normal and post-chemotherapy females. Br J Haematol 1991, 79:193-197 [DOI] [PubMed] [Google Scholar]
- 11.Vogelstein B, Fearon ER, Hamilton SR, Preisinger AC, Willard HF, Michelson AM: Clonal analysis using recombinant DNA probes from the X chromosome. Cancer Res 1987, 47:4806-4813 [PubMed] [Google Scholar]
- 12.Abeln ECA, Kuipers-Dijkshoorn NJ, Berns EMJJ, Henzen-Logmans SC, Fleuren GJ, Cornelisse CJ: Molecular genetic evidence for unifocal origin of advanced epithelial ovarian cancer and for minor clonal divergency. Br J Cancer 1995, 72:1330-1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Imyanitov EN, Suspitsin EN, Grigoriev MY, Togo AV, Kuligina ES, Belogubova EV, Pozharisski KM, Turkevich EA, Rodriques C, Cornelisse CJ, Hanson KPTC: Concordance of allelic imbalance profiles in synchronous and metachronous bilateral breast carcinomas. Int J Cancer 2002, 100:557-564 [DOI] [PubMed] [Google Scholar]
- 14.Weber JL, May PE: Abundant class of human DNA polymorphisms which can be typed using the polymerase chain reaction. Am J Hum Genet 1989, 44:388-396 [PMC free article] [PubMed] [Google Scholar]
- 15.Sieben NLG, ter Haar NT, Cornelisse CJ, Fleuren GJ, Cleton-Jansen AM: PCR artifacts in LOH and MSI analysis of microdissected tumor cells. Hum Pathol 2000, 31:1414-1419 [PubMed] [Google Scholar]
- 16.Nash MA, Ferrandina G, Gordinier M, Loercher A, Freedman RS: The role of cytokines in both the normal and malignant ovary. Endocr Relat Cancer 1999, 6:93-107 [DOI] [PubMed] [Google Scholar]
- 17.Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Fodde R, Vasen H, Bigas-Rodriquez M, Srivastava SA: A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 1998, 58:5248-5257 [PubMed] [Google Scholar]
- 18.Zauber NP, Sabbath-Solitare M, Marotta SP, McMahon L, Bishop DT: Comparison of allelic ratios from paired blood and paraffin-embedded normal tissue for use in a polymerase chain reaction to assess loss of heterozygosity. Mol Diagn 1999, 4:29-35 [DOI] [PubMed] [Google Scholar]
- 19.Bonsing BA, Corver WE, Fleuren GJ, Cleton-Jansen AM, Devilee P, Cornelisse CJ: Allelotype analysis of flow-sorted breast cancer cells demonstrates genetically related diploid and aneuploid subpopulations in primary tumours and lymph node metastases. Genes Chromosomes Cancer 2000, 28:173-183 [DOI] [PubMed] [Google Scholar]
- 20.Mao L, Lee DJ, Tockman MS, Erozan YS, Askin F, Sidransky D: Microsatellite alterations as clonal markers for the detection of human cancer. Proc Natl Acad Sci USA 1994, 91:9871-9875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lyon MF: X-chromosome inactivation and developmental patterns in mammals. Biol Rev 1972, 47:1-35 [DOI] [PubMed] [Google Scholar]
- 22.Cheng L, Gu J, Eble JN, Bostwick DG, Yonger C, MacLennan GT, Abdul-Karim FW, Geary WA, Koch MO, Zang S, Ulbright TM: Molecular genetic evidence for different clonal origin of components of human renal angiomyolipomas. Am J Surg Pathol 2001, 25:1231-1236 [DOI] [PubMed] [Google Scholar]
- 23.Scully RE, Young RH, Clement PB: Surface epithelial-stromal tumors; serous tumors. Rosai J Sobin LH eds. Atlas of Tumor Pathology. Tumors of the Ovary, Maldeveloped Gonads, Fallopian Tube, and Broad Ligament 1998:pp 51-79 Armed Forces Institute of Pathology Bethesda, MD
- 24.Kayser K, Zink S, Schneider T, Dienemann H, Andre S, Kaltner H, Schuring MP, Zick Y, Gabius HJ: Benign metastasizing leiomyoma of the uterus: documentation of clinical, immunohistochemical, and lectin-histochemical data of ten cases. Virchows Arch 2000, 437:284-292 [DOI] [PubMed] [Google Scholar]