

Abstract
Cancers may develop in the background of genomic instability with accumulated mutations. Helicobacter pylori gastritis is characterized by acute foveolitis of the proliferative zone, which is found in any stage of the gastritis as long as the infection persists. Because acute foveolitis targets specifically the proliferative zone of pits, the proliferating epithelial cells are under severe and persistent mutagenic pressure. In H. pylori gastritis, a characteristic morphological change of epithelial cells, the malgun (clear) cell change is frequently present in association with acute foveolitis. Malgun cells have enlarged euchromatic nuclei and abundant cytoplasm. The expression of proliferating cell nuclear antigen and cytokeratin 8 are typically up-regulated in them indicating that they are mitotically and metabolically active. Here, we report evidence for DNA damage and repair in malgun cells. Significant double-strand DNA breaks were shown by the consistent terminal dUTP nick-end labeling in the nuclei of malgun cells. Proteins related to DNA damage and repair, such as Ku, poly(ADP-ribosyl) polymerase, OGG1, and MSH2 were selectively up-regulated in malgun cells. Inducible nitric oxide synthase was also up-regulated. There were occasional bcl2- and p53-expressing cells suggesting that further steps of carcinogenesis took place at the single cell level. Our results suggest that the malgun cell change represents a characteristic morphological sign of cellular genomic damage and repair, and may be implicated in an early stage of carcinogenesis. It is suggested that acute foveolitis of the proliferative zone is a major pathogenetic step of gastric carcinogenesis in H. pylori gastritis.
Malignant tumors develop mostly through multistep carcinogenetic events in the background of genomic instabilities with accumulated mutations. 1,2 Persistent inflammation may provide such a milieu. Inflammation stimulates cells to proliferate, 3 and induces reactive oxygen and nitrogen species, 4,5 which, in turn, induce protein alterations, membrane damage, DNA base oxidation, DNA strand breaks, and chromosomal aberrations, most of which would be involved in carcinogenesis. The effect of oxidative damage on proteins or lipids may be limited temporarily by continual biosynthesis and turnover but, in the case of DNA, it can lead to fixed mutations if polymerases copy damaged templates during DNA replication. Thus, the persistent inflammation puts proliferating cells under intensive mutagenic pressure.
Helicobacter pylori gastritis has been implicated in gastric carcinogenesis. 6,7 It may be a good model of inflammation-associated carcinogenesis, because H. pylori infection induces intense acute and chronic inflammatory reaction that may last for decades. 8,9 In biological systems, it is very unusual to have severe inflammation, especially acute inflammation, persisting for such a long time. The persistent acute inflammation in chronic H. pylori gastritis has been regarded to denote activity of the gastritis. 8,9
The acute inflammation may involve the epithelium as well as the lamina propria. Characteristically, the acute epithelial inflammation, ie, acute foveolitis, is mostly confined to the proliferative zone of the gastric pit, and is present throughout the long course of H. pylori gastritis. 10 The proliferative zone is where epithelial proliferation takes place exclusively in the gastric pit. 11 It is not clear how neutrophils are attracted specifically to the proliferative zone. Whatever the underlying biological mechanism of such a targeting, proliferating epithelial cells are directly damaged by the infiltrating neutrophils; thus, in the long run, atrophic gastritis would ensue because of the suppression of pit repair. At the same time, the targeted acute inflammation would induce intensive mutagenesis in the epithelial cells by introducing abundant reactive oxygen and nitrogen species locally. Thus, it has been proposed that acute foveolitis may be a critical pathogenetic step in the initiation of a vicious cycle in which atrophic gastritis and carcinogenesis would progress simultaneously as long as the H. pylori infection persists. 10
In association with acute foveolitis, nonendocrine epithelial cells with clear, enlarged nuclei and cytoplasm are frequently scattered. 12 They are present as single cells and/or in small clusters. They are mitotically active, and atypical mitotic figures may also be found. Characteristically, the nuclei show a euchromatin pattern with single, prominent nucleoli. For the description of these distinct epithelial cells, a Korean word, “malgun (pronounced as [malg∂n], meaning clear or transparent)” has been adopted to avoid any confusion with the previous descriptions of various clear cell change of the gastrointestinal tract. 13-16 In Korea, in which the prevalence rate of H. pylori infection and gastric cancer incidence remain very high, 17,18 acute foveolitis and the malgun cell change are frequently found in gastric biopsies. 10,12
In this study, we analyzed the pathobiological nature of malgun cells using the terminal dUTP nick-end labeling (TUNEL) method and immunohistochemical staining for DNA repair-related proteins. We found that they were consistently positive for the TUNEL staining and had up-regulated DNA repair proteins. The implications are discussed.
Materials and Methods
Study Population and Histopathological Analysis
For this study, 267 gastric biopsies having pathologically proven H. pylori infection were randomly selected from the surgical pathology files of the Asan Medical Center, Seoul, Korea, between 2000 and 2001. Biopsies were taken for the screening of H. pylori infection and/or the evaluation of gastritis from patients aged from 25 to 72 years who were not treated previously. In the 267 biopsies, H. pylori was confirmed histologically in 137 biopsies but not in 95 biopsies. Neither chemical gastritis nor autoimmune gastritis was included. Typically, biopsies were taken from at least three sites including the antrum and body. Biopsies were fixed in buffered formalin immediately after the gastroscopic procedure. Routine hematoxylin and eosin (H&E) staining was done for conventional examination.
The histopathological parameters were analyzed according to the Sydney system. 8,9 The gastric mucosa was categorized into the superficial, proliferative, and deep glandular zones according to Harvard and colleagues. 11 The acute gastritis was further divided into acute epithelial and interstitial gastritis. 10 The former, ie, acute foveolitis, was defined to be positive when at least one of the following three criteria were fulfilled in the biopsy: 1) an aggregate of more than five neutrophils within a pit, 2) more than 10 neutrophils infiltrating a pit circumferentially, and/or 3) an inflammatory exudate with more than five neutrophils in the lumen. 10
Malgun cells were readily recognized in the proliferative and surface zones based on the following characteristics: 1) nuclear enlargement, 2) euchromatin pattern, 3) conspicuous nucleoli, and 4) expanded cytoplasm with decreased stainability on H&E staining. 12 They were distinguished from endocrine cells by the characteristic cytological change and the location; endocrine cells were located in the deep glandular zone mostly. If necessary, a triple silver impregnation (modified Warthin-Starry staining) was done according to Genta and colleagues 19 to get a sharper contrast.
TUNEL Staining
To detect cells with DNA damage, the peroxidase in situ TUNEL staining kit (Roche Molecular Biochemicals, Mannheim, Germany) was used according to the manufacturer’s instructions. Briefly, paraffin-embedded sections were rehydrated, treated with 10 μg/ml of proteinase K in phosphate-buffered saline (PBS) for 30 minutes at 37°C, washed with ice-cold PBS four times, incubated in 3% H2O2 in methanol for 5 minutes at room temperature, and washed again. Sections were incubated with 0.1% (v/v) Triton X-100 in PBS for 2 minutes at room temperature. Then, the labeling solution containing the enzyme terminal deoxynucleotidyl transferase was added to the sections for 30 minutes at 37°C. For a negative control, the enzyme was omitted from the labeling solution. To reduce the background, sections were blocked with 3% bovine serum albumin for 20 minutes at room temperature. After washing with PBS, sections were incubated with convert solution, washed again, and incubated in 3,3′-diaminobenzidine solution for 10 minutes at room temperature to visualize the DNA breaks. Two experiments were done on five biopsies separately.
Immunohistochemical Staining
Sources and dilutions of commercially available antibodies used in this study are summarized in Table 1 ▶ . For cytokeratin immunostaining, two monoclonal antibodies were used: monoclonal antibody 35βH11 recognizing low-molecular weight cytokeratin (no. 8) and 35βE12 recognizing high-molecular weight cytokeratins (nos. 1, 5, 10, and 14). Serial sections of 4-μm thickness were prepared from paraffin blocks. Microwave antigen retrieval was applied in 0.01 mol/L of sodium citrate buffer (pH 6.0). Primary antibodies were applied for 2 hours at room temperature. Avidin-biotinylated horseradish peroxidase complex was developed by immersing slides in diaminobenzidine as chromogen and was counterstained with hematoxylin. For every immunostaining, the average percentage of positive cells was obtained by counting 200 malgun cells in three different biopsies.
Table 1.
Immunohistochemical Markers of Malgun Cells
| Antibody | Source | Description/clone | Dilution | Staining | ||
|---|---|---|---|---|---|---|
| Pattern | Intensity* | Frequency (%)† | ||||
| CK8 | Dako‡ | Monoclonal, 35βH11 | 1:50 | C-diffuse | +++ | 100 |
| PARP | Santa Cruz | Monoclonal, Sc-8007 | 1:100 | N-diffuse | +∼++ | 80 ± 15 |
| Ku70 | Santa Cruz | Goat antiserum, Sc-1487 | 1:100 | N-diffuse | +++ | 92 ± 5 |
| Ku80 | Santa Cruz | Goat antiserum, Sc-1484 | 1:200 | N-diffuse | +∼++ | 85 ± 6 |
| OGG1 | Novus | Rabbit antiserum, nb100-106 | 1:80 | N-diffuse | +++ | 75 ± 15 |
| PCNA | Dako | Monoclonal, PC10 | 1:100 | N-diffuse | +∼+++ | 90 ± 5 |
| Ki67 | Dako | Rabbit antiserum, A0047 | 1:200 | N-speckled | +++ | 40 ± 12 |
| iNOS | Santa Cruz | Rabbit antiserum, Sc-651 | 1:200 | N-diffuse | +∼++ | 85 ± 10 |
| MSH2 | Pharmingen | Monoclonal, 6505A | 1:250 | N-diffuse | +∼+++ | 55 ± 8 |
| P53 | Dako | Monoclonal, M887 | 1:1600 | N-diffuse | +++ | Rare |
| Bcl2 | Dako | Monoclonal, M7001 | 1:40 | N-diffuse | +++ | Rare |
| CK: high mol wt | Dako | 35β E12 | 1:50 | − | − | 0 |
| Synaptophysin | Signet | Monoclonal, SY38 | 1:80 | − | − | 0 |
| Chromogranin A | Dako | Monoclonal, DAK-A3 | 1:200 | − | − | 0 |
| Neurofilament | Dako | Monoclonal, 2F11 | 1:100 | − | − | 0 |
| COX-2 | Santa Cruz | Rabbit, Sc-1746 | 1:100 | − | − | 0 |
N, nucleus; C, cytoplasm.
*Graded from negative (−) to very strong (+++).
†The average percentage and range of positive cells were calculated from three different biopsies by counting total 200 malgun cells, respectively.
‡DAKO, Carpinteria, CA; Santa Cruz, Santa Cruz, CA; Novus, Littleton, CO; Pharmingen, San Diego, CA; Signet, North Yorkshire, United Kingdom.
Statistical Analysis
The Fisher’s exact test was used to analyze the correlation between the incidence of H. pylori infection and acute foveolitis or the malgun cell change.
Results
Histopathological Characterization
The histopathological findings are summarized in Table 2 ▶ . In the 267 biopsies, H. pylori was confirmed histologically in 137 biopsies (64.4%) but not in 95 biopsies. The incidence of acute foveolitis was strongly associated with the presence of H. pylori (P < 0.001): 137 and 8 cases had acute foveolitis in the former and latter groups, respectively. As was reported previously, 10 acute foveolitis was primarily confined to the proliferative zone of the gastric mucosa whereas the surface and deep glandular zones were free of significant neutrophilic infiltration (Figure 1) ▶ .
Table 2.
Acute Foveolitis and Malgun Cell Change in 267 Randomly Selected Biopsies
| Histopathologic parameters | Number of positive cases | P value* | |
|---|---|---|---|
| H. pylori-positive gastritis (n = 172) | H. pylori-negative gastritis (n = 95) | ||
| Acute foveolitis | 137 | 8 | <0.001 |
| Malgun cell change | 152 | 41 | <0.001 |
*Statistical analysis was done by Fisher’s exact test.
Figure 1.
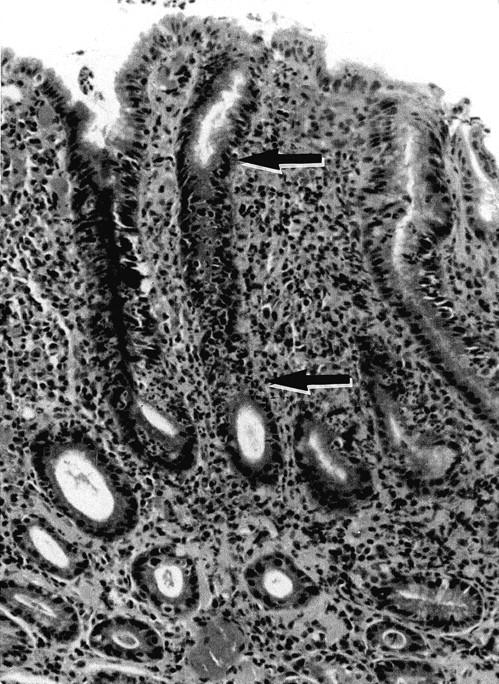
Acute foveolitis of the proliferative zone. Note intense neutrophilic infiltration targeted to the proliferative zone that is typically elongated in H. pylori gastritis (the segment between the arrows). The surface epithelium and deep glands are free of significant neutrophilic infiltration. The interstitial infiltration primarily consists of lymphocytes, plasma cells, and histiocytes. H&E staining; original magnification, ×60.
Malgun cells were present in 193 biopsies (72.3%). The incidence of the malgun cell change was also strongly associated with the presence of H. pylori (P < 0.001): 152 and 41 biopsies had malgun cell change in the H. pylori-positive, and -negative biopsies, respectively (Table 2) ▶ . In a positive biopsy, malgun cells were usually numerous, comprising up to 10% of total epithelial cells of the proliferative and/or surface zone. Malgun cells appeared to be generated at the proliferative zone where they were frequently in close association with the infiltrating neutrophils (Figure 2) ▶ . Topographically, they distributed much more frequently in the antrum than in the body. In contrast to H. pylori gastritis in Americans, Korean and Japanese H. pylori gastritis was characterized by intense acute foveolitis in the antrum rather than the body (manuscript in preparation).
Figure 2.
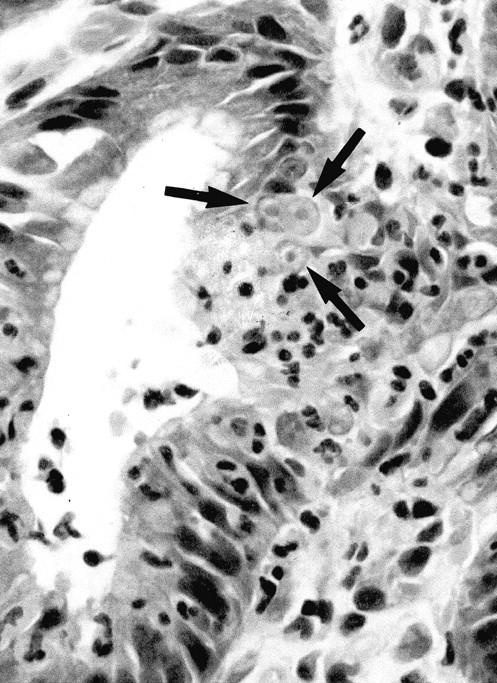
The malgun cell change. Epithelial cells in contact with infiltrating neutrophils undergo the malgun cell change (arrows). Malgun cells have enlarged, round nuclei with prominent nucleoli. The nucleus consists of euchromatin, seem is in contrast to the hyperchromatic nuclei of adjacent epithelial cells. Malgun cells are noted as single cells or clusters in the proliferative and surface zones. H&E staining; original magnification, ×400.
The malgun cell change was readily recognized on freshly fixed tissues, but was less pronounced when the fixation was delayed or inadequate. It could be most effectively demonstrated by triple silver staining (modified Warthin-Starry stain). All other epithelial cells were stained with the silver impregnation in the nuclei and cytoplasm whereas malgun cells were not stained at all (Figure 3) ▶ . Thus, only the nuclei of malgun cells were stained lightly with hematoxylin, rendering a sharp contrast from adjacent cells.
Figure 3.
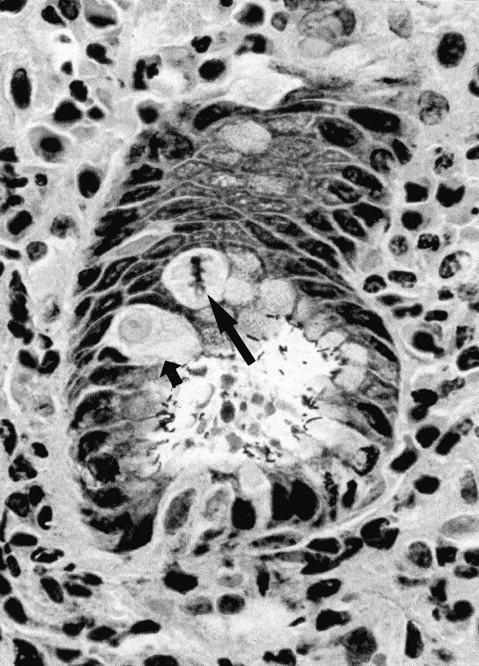
Malgun cells at the proliferative zone. Note two malgun cells in the interphase (small arrow) and metaphase (large arrow), respectively. By triple silver staining, malgun cells are not stained by silver impregnation but faintly counterstained by hematoxylin. Adjacent epithelial cells are densely stained by silver impregnation. Note numerous H. pylori attached to the epithelium at the glandular lumen. Triple silver impregnation staining; original magnification, ×400.
Malgun cells had smooth nuclear envelopes with slight folding occasionally (Figures 2 and 3) ▶ . The chromatin pattern was unremarkable without demonstrable clumping. Nucleoli were typically single and conspicuous. Malgun cells in mitosis were distinguished from other epithelial cells by the characteristic cytoplasm (Figure 3) ▶ . The mitotic activity indicated that they were not just degenerating but metabolically active cells; occasionally atypical mitotic figures were noted. 12
TUNEL Staining
Based on their typical morphology and distribution pattern, malgun cells were readily distinguished on TUNEL staining. Typically, they had abundant, clear cytoplasm and a large nucleus that was more than twice the size of that of adjacent cells. The majority (more than 90%) of malgun cells were positively stained by the TUNEL method, while no adjacent cell was convincingly stained (Figure 4) ▶ . The staining was limited to the nucleus; the cytoplasm remained clear. The positively stained malgun cells were distributed in the proliferative zone as well as in the surface zone. No staining was present in the negative controls omitting the terminal deoxynucleotidyl transferase (data not shown).
Figure 4.
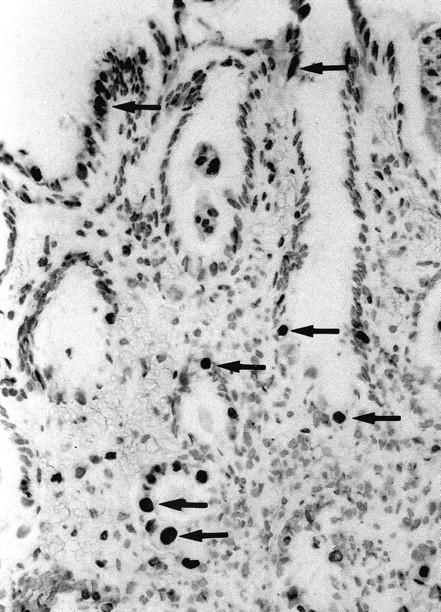
TUNEL staining of malgun cells. Note numerous malgun cells stained positively in the nuclei (arrows). They have enlarged, round nuclei and abundant cytoplasm, which are typical of malgun cells. They are scattered in the proliferative and surface zone, showing a typical distribution of the malgun cell change. TUNEL staining; original magnification, ×120.
Immunohistochemical Study
The results of immunohistochemical staining are summarized in Table 1 ▶ . The typical cytological features of malgun cells were preserved well after the immunohistochemical staining. As previously reported, cytokeratin 8 was consistently up-regulated in all malgun cells in every biopsy, suggesting that cytokeratin 8 was the most sensitive marker for the malgun cell change (Figure 5A) ▶ . Characteristically, cytokeratin filaments were in concentric arrangement along the cytoplasmic membrane leaving a perinuclear halo of clear cytoplasm. Neither malgun cells nor adjacent epithelial cells showed any immunostaining for high-molecular weight cytokeratin (data not shown).
Figure 5.
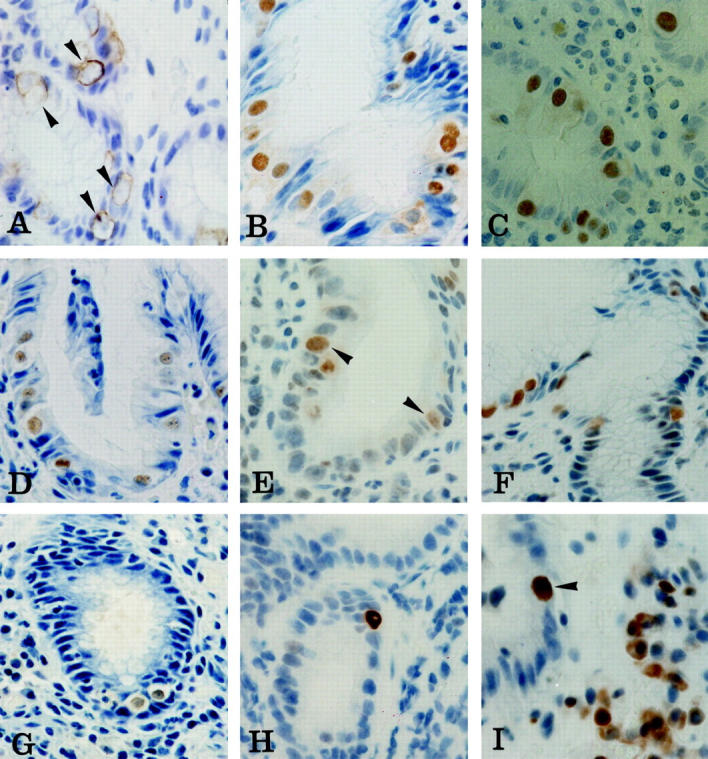
Immunohistochemical staining of malgun cells. A: Cytokeratin 8. Cytokeratin filaments are in concentric arrangement along the cytoplasmic membrane leaving a perinuclear halo of clear cytoplasm (arrowheads). Note enlarged nuclei that are only faintly stained by hematoxylin counterstaining. B–G: Ku70, PARP, proliferating cell nuclear antigen, OGG1, MSH2, and iNOS, respectively. Most malgun cells are immunostained in the nuclei. Note enlarged, round nuclei and abundant, clear cytoplasm of positively stained malgun cells. Nucleoli are seen in some malgun cells. H: p53. Note an epithelial cell strongly immunostained in the nuclei. The enlarged nuclei and prominent, unstained nucleoli are compatible with those of a malgun cell. I: bcl2. Note a malgun cell strongly immunostained in the nuclei (arrowhead). The nucleus is enlarged and the cytoplasm is abundant. Some mononuclear inflammatory cells are positively immunostained in the cytoplasm. Original magnifications: ×400 (A–C, E, I); ×240 (D, F, G, H).
In addition, malgun cells were frequently immunostained for DNA repair proteins including Ku (Figure 5B) ▶ , poly(ADP-ribosyl) polymerase (PARP) (Figure 5C) ▶ , proliferating cell nuclear antigen (Figure 5D) ▶ , OGG1 (Figure 5E) ▶ , and MSH2 (Figure 5F) ▶ . Malgun cells had distinct nuclear staining in every biopsy immunostained for the markers; however, the intensity and percentage of positive cells varied mildly among the cases depending on the antibodies (Table 1) ▶ . No other epithelial cells reacted for the aforementioned markers. Malgun cells were also specifically immunostained for inducible nitric oxide synthase (iNOS) in the nuclei and, faintly, in the cytoplasm as well (Figure 5G) ▶ .
To analyze the expression of proteins implicated in carcinogenesis, we screened the samples for p53-, and bcl2-positive cells by immunohistochemistry. Rarely, epithelial cells with strong p53 immunostaining were noted. They were present in biopsies with severe gastritis, and were consistent with malgun cells morphologically (Figure 5H) ▶ . Some bcl2-positive malgun cells were also present (Figure 5I) ▶ ; they were also infrequent, but still more frequent than p53-positive cells. The staining was mostly in the nuclei. Inflammatory mononuclear cells were also immunostained for bcl2, but they showed cytoplasmic immunostaining as well, and were distinguishable obviously from malgun cells by their shape, size, and location.
As reported previously, Ki67 immunostaining was localized to the proliferative zone. 11,12 Among the Ki67-positive cells, both malgun and nonmalgun cells were included; 12 in most biopsies, the number of nonmalgun cells was more than double that of malgun cells. The immunostaining was typically speckled in both cell types. 12 Synaptophysin, chromogranin A, and neurofilament were consistently negative among malgun cells, indicating the nonendocrine nature of malgun cells. Malgun cells were not immunostained for Cox-2 either. Frequently, deep glandular cells were immunostained for Cox-2 in the cytoplasm (data not shown).
Discussion
Malgun cells were distinguished from adjacent epithelial cells by the specific TUNEL staining. There were reports of TUNEL staining in H. pylori gastritis previously, showing scattered epithelial cells positively stained for TUNEL. 20-25 They were invariably interpreted as apoptotic cells; however, in our view, the cytological appearance and distribution of TUNEL-positive cells in most reports are quite reminiscent of the malgun cells described herein. Their frequency also supports that notion, for it was noted that the number of TUNEL-positive cells correlated positively with the severity of gastritis, 24,25 while the number was low in intestinal metaplasia. 25 The malgun cell change occurs in acute foveolitis, but is rare, if detectable at all, in the intestinal metaplasia. 12
In the TUNEL staining, terminal deoxynucleotidyl transferase incorporates polynucleotide tails onto free DNA ends, either blunt-ends or overhanging 3′-OH ends; thus, TUNEL staining mostly detects DNA double-strand breaks. Because DNA double-strand breaks are most abundant in apoptotic cells, the TUNEL staining has generally been regarded to indicate apoptosis. 26 However, our observations strongly suggest that malgun cells are not apoptotic but viable. First, it is difficult to find karyorrhectic debris in the gastric epithelium even in severe gastritis. Second, nuclei of malgun cell are typically large and euchromatic with prominent nucleoli, being compatible with those of metabolically active cells. Third, they frequently show mitotic activity including atypical mitoses. Fourth, they express proliferating cell nuclear antigen and/or Ki67. 12 Lastly, cytokeratin 8 is characteristically up-regulated in malgun cells. 12 Cytokeratin 8 has been implicated in protection of cellular apoptosis induced by tumor necrosis factor family receptors 27 and/or Fas. 28 Cytokeratin 8 competes with tumor necrosis factor receptor-1 for the binding with TRADD (tumor necrosis factor receptor1-associated death domain protein), which is a key factor for the activation of caspase 8. 29 Cytokeratin 8 and 18 are expressed commonly and persistently in carcinomas, whereas they collapse to punctate inclusions of variable size in apoptotic cells. 30 Taken together, our data suggest that malgun cells are not apoptotic but rather protected against apoptosis. Thus, it is suggested that the positive TUNEL staining does not invariably represent apoptosis but extensive DNA damage, mostly of double-strand breaks. It appears that malgun cell change is a morphologically identifiable reaction pattern to significant DNA damage of cells that remain viable.
Such a notion was further supported by the up-regulation of various DNA repair proteins in malgun cells. The biological functions of the selected DNA repair proteins are summarized in Table 3 ▶ . In mammalian cells, five major DNA repair pathways have been described: homologous recombinational repair, nonhomologous end joining, base excision repair, nucleotide excision repair, and mismatch repair. 31 Every pathway has its own major target of DNA damage to repair.
Table 3.
Biological Functions of DNA Repair Proteins
| Proteins | DNA repair pathway | Functions | References |
|---|---|---|---|
| Ku | NHEJ | Binds to double-strand DNA breaks and activates DNA-PK complex | 33-35 |
| OGG1 | BER | DNA glycosylase/AP lyase involved in the first step of BER | 37-39 |
| PARP | BER/NHEJ | Binds to single- and double-strand DNA breaks and poly(ADP)ribosylates nuclear proteins | 40-41 |
| MSH2 | MMR | Corrects DNA replication errors | 42-44 |
| PCNA | MMR/NER? | Binds to MSH and XPG | 45-47 |
HRR, homologous recombinational repair; NHEJ, nonhomologous end joining; BER, base excision repair; NER, nucleotide excision repair; and MMR, mismatch repair; PCNA, proliferating cell nuclear antigen.
Homologous recombinational repair and nonhomologous end joining are responsible for the repair of double-strand breaks, which are directly connected to the genomic instability. 32 Mammalian cells repair the majority of double-strand breaks by nonhomologous end joining, which is not based on the homologous recombination, and, thus, may not repair the lost sequence information accurately. Ku, a heterodimer of Ku70 and Ku80, is a key player in nonhomologous end joining. It binds the DNA double-strand break, 33 and, then, recruits and activates a protein kinase, DNA-PK. 34 Thus formed, a DNA-PK complex aligns the ends of the double-strand breaks. It has been shown that Ku is up-regulated by the DNA damage induced by ionizing radiation in a p53- and ATM-dependent manner. 35
Base excision repair is a pathway protecting cells against single-base DNA damage caused by oxidizing and methylating agents. H. pylori gastritis poses severe, persistent oxidative stress on the gastric mucosa. 36 Infiltrating neutrophils produce abundant superoxide radicals (O2−), hydroxyl radicals (.OH), and potent hydrochlorous acid (HOCl). Oxidative stress is a major cause of DNA damage, including base modifications and single- and double-strand breaks. 37 Among the base damages, 8-hydroxyguanine is a particularly abundant adduct in the damaged DNA by oxidative stress as well as chemical carcinogens. 38 8-Hydroxyguanine is highly mutagenic, yielding G:C to T:A transversions on the replication by DNA polymerases. Ogg1 is a DNA glycosylase/AP lyase that is required in the first step of base excision repair, which eliminates 8-hydroxyguanine. 38 It has been implicated in carcinogenesis. 39
PARP is another key enzyme essential for base excision repair. 40 It is immediately activated by genomic DNA breaks and catalyzes the synthesis of poly(ADP-ribose) from the respiratory coenzyme NAD+. 41 The synthesis of poly(ADP-ribose) is directly proportional to the number of single- and double-strand breaks in the genomic DNA. 41 PARP poly(ADP) ribosylates itself, p53, histones, and many other nuclear proteins. In mammalian cells, poly(ADP-ribosyl)ation of histones causes major changes in nucleosomal architecture, rendering chromosomal decondensation, ie, euchromatin. 41 The PARP-dependent chromosomal decondensation is of particular interest, because it is compatible with the characteristic euchromatin pattern of malgun cell nuclei. The characteristic nuclear change in malgun cells may be induced at least partly by the activation of PARP. Such chromosomal change activates various nuclear functions, facilitates the DNA repair, and may be related to the characteristic exclusion of malgun cells by the triple silver impregnation.
Nucleotide excision repair is to repair large helix-distorting damage induced by UV light or chemotherapeutics. 31 Mismatch repair is primarily responsible for the postreplication correction, 42 and is also involved in the removal of 8-hydroxyguanine. 43 Mismatch repair mutations are implicated in the etiology of hereditary nonpolyposis colorectal cancer syndrome. 44 MSH2 is one of the major enzymes involved in mismatch repair; its inactivation in human tumors greatly increases spontaneous mutation rates. 42 Proliferating cell nuclear antigen is an auxiliary protein of DNA polymerase δ 45 ; it binds to MSH 46 and ERCC5/XPG, 47 suggesting that it also is involved in mismatch repair and/or nucleotide excision repair.
The up-regulation of iNOS may have a significant implication in the pathogenesis of the malgun cell change. Excessive toxic nitric oxide metabolites, ie, reactive nitrogen species, have been implicated in carcinogenesis. 5,48,49 Reactive nitrogen species may cause DNA damage directly, and may inhibit the repair of DNA damage posed by reactive oxygen species simultaneously. 49,50 Thus, excessive nitrogen oxide induced by iNOS may damage DNA along with oxidative stress synergistically. Further investigations are required to elucidate the mechanism of iNOS up-regulation in the malgun cell change.
Taken together, our data indicate that the malgun cell change is a morphological phenomenon of gastric epithelial cells reflecting considerable DNA damage and active repair in a persistent mutagenic environment. The reactive oxygen and nitrogen species would induce extensive mutations and deregulation of key proteins implicated in carcinogenesis. The presence of p53-positive malgun cells supports that notion. Because most malgun cells were clearly negative for p53, the strong immunoreaction for p53 in occasional cells suggests that the gene has been mutated in those particular cells rather than reflects a mild up-regulation in reaction to DNA damage. Alteration of p53 gene is common in gastric carcinoma. 51 The presence of the p53- and bcl2-positive cells strongly suggests that early steps of carcinogenesis may begin at the single cell level.
Most reactive oxygen and nitrogen species are in a gaseous state. They may thus penetrate cells easily, but their spatial range of activity is limited because their half-life is generally very short. The deleterious effect of reactive oxygen and nitrogen species would increase exponentially as the distance to the target cell decreases. In acute foveolitis, the proliferating epithelial cells are in direct contact with the activated neutrophils. Thus, it seems that acute foveolitis provides one of the worst possible combinations in terms of the safety of the gastric mucosa. 10 Obviously, acute inflammation is designed to deal with new challenges for only a short period. If acute inflammation were to linger for decades as in the case of acute foveolitis in H. pylori gastritis, the development of serious sequelae should not be surprising. To prevent pathological consequences of H. pylori gastritis including gastric carcinogenesis, the molecular pathobiological mechanisms underlying the specific neutrophilic infiltration to the proliferative zone require investigation.
Another question remains to be solved: how malgun cells having such extensive DNA damage can escape apoptosis and/or cell-cycle arrest? We might adduce that there is up-regulation of bcl2, but the number of positive cells are too small to explain the survival of so many malgun cells for a long time. The overexpression of cytokeratin 8 may indeed protect malgun cells against apoptosis. Whatever the eventual answer(s) might turn out to be, we might speculate that it constitutes a significant property acquired by those cells on their way to overt malignancy.
Acknowledgments
We thank Sangho Ahn, Sol Baik, and Jenam Koh for the immunohistochemical staining.
Footnotes
Address reprint requests to Inchul Lee, M.D., Ph.D., Department of Pathology, University of Ulsan College of Medicine, 388-1 Poongnap-Dong, Songpa-Gu, Seoul 138-736, Korea. E-mail: iclee@amc.seoul.kr.
Supported by a grant from the Functional Analysis of Human Genome Project in the 21C Frontier Research and Development Program of the Ministry of Science and Technology of Korea.
References
- 1.Weinberg RA: Oncogenes, antioncogenes, and the molecular basis of multistep carcinogenesis. Cancer Res 1989, 49:3713-3721 [PubMed] [Google Scholar]
- 2.Lengauer C, Kinzler KW, Vogelstein B: Genetic instabilities in human cancers. Nature 1998, 396:643-649 [DOI] [PubMed] [Google Scholar]
- 3.Cohen SM, Ellwein LB: Cell proliferation in carcinogenesis. Science 1990, 249:1007-1011 [DOI] [PubMed] [Google Scholar]
- 4.Cerutti PA: Prooxidant states and tumor promotion. Science 1985, 227:375-381 [DOI] [PubMed] [Google Scholar]
- 5.Burney S, Caulfield JL, Niles JC, Wishnok JS, Tannenbaum SR: The chemistry of DNA damage from nitric oxide and peroxynitrite. Mutat Res 1999, 424:37-49 [DOI] [PubMed] [Google Scholar]
- 6.Correa P: Helicobacter pylori as a pathogen and carcinogen. J Physiol Pharmacol 1997, 48(Suppl 4):19-24 [PubMed] [Google Scholar]
- 7.Sipponen P, Marshall BJ: Gastritis and gastric cancer. Western countries. Gastroenterol Clin North Am 2000, 29:579-592 [DOI] [PubMed] [Google Scholar]
- 8.Price AB: The Sydney system: histological division. J Gastroenterol Hepatol 1991, 6:209-222 [DOI] [PubMed] [Google Scholar]
- 9.Dixon MF, Genta RM, Yardley JH, Correa P: Classification and grading of gastritis: the updated Sydney system. Am J Surg Pathol 1996, 20:1161-1181 [DOI] [PubMed] [Google Scholar]
- 10.Yu E, Lee HK, Kim HR, Lee MS, Lee I: Acute inflammation of proliferative zone in Helicobacter pylori gastritis. Pathol Res Pract 1999, 195:689-697 [DOI] [PubMed] [Google Scholar]
- 11.Harvard TJ, Sarsfield P, Wotherspoon AC, Steer HW: Increased gastric epithelial cell proliferation in Helicobacter pylori associated follicular gastritis. J Clin Pathol 1996, 49:68-71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee H, Jang J, Ahn S, Gong M, Choi E, Lee I: “Malgun” (clear) cell change of gastric epithelium in chronic Helicobacter pylori gastritis. Pathol Res Pract 2000, 196:541-551 [DOI] [PubMed] [Google Scholar]
- 13.Pfeiffer CJ, Weibel J: The antral clear cell—a new cell type discovered in the pyloric-antral mucosa of the ferret. J Ultrastruct Res 1969, 29:550-562 [DOI] [PubMed] [Google Scholar]
- 14.Ordonez NG, Mackay B, el-Naggar A, Bannayan GA, Duncan J: Clear cell carcinoid tumour of the stomach. Histopathology 1993, 22:190-193 [DOI] [PubMed] [Google Scholar]
- 15.Jewell LD, Barr JR, McCaughey WT, Nguyen GK, Owen DA: Clear-cell epithelial neoplasms of the large intestine. Arch Pathol Lab Med 1988, 112:197-199 [PubMed] [Google Scholar]
- 16.Domoto H, Terahata S, Senoh A, Sato K, Aida S, Tamai S: Clear cell change in colorectal adenomas: its incidence and histological characteristics. Histopathology 1999, 34:250-256 [DOI] [PubMed] [Google Scholar]
- 17.Youn HS, Ko GH, Chung MH, Lee WK, Cho MJ, Rhee KH: Pathogenesis and prevention of stomach cancer. J Korean Med Sci 1996, 11:373-385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahn YO: Cancer in Korea: present features. Jpn J Clin Oncol 2002, 32:S32-S36 [DOI] [PubMed] [Google Scholar]
- 19.Genta RM, Robason GO, Graham DY: Simultaneous visualization of Helicobacter pylori and gastric morphology: a new stain. Hum Pathol 1994, 525:221-226 [DOI] [PubMed] [Google Scholar]
- 20.Ishida M, Gomyo Y, Tatebe S, Ohfuji S, Ito H: Apoptosis in human gastric mucosa, chronic gastritis, dysplasia and carcinoma: analysis by terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labelling. Virchows Arch 1996, 428:229-235 [DOI] [PubMed] [Google Scholar]
- 21.Steininger H, Faller G, Dewald E, Brabletz T, Jung A, Kirchner T: Apoptosis in chronic gastritis and its correlation with antigastric autoantibodies. Virchows Arch 1998, 433:13-18 [DOI] [PubMed] [Google Scholar]
- 22.Hoshi T, Sasano H, Kato K, Ohara S, Shimosegawa T, Toyota T, Nagura H: Cell damage and proliferation in human gastric mucosa infected by Helicobacter pylori—a comparison before and after H pylori eradication in non-atrophic gastritis. Hum Pathol 1999, 30:1412-1417 [DOI] [PubMed] [Google Scholar]
- 23.Moss SF, Valle J, Abdalla AM, Wang S, Siurala M, Sipponen P: Gastric cellular turnover and the development of atrophy after 31 years of follow-up: a case-control study. Am J Gastroenterol 1999, 94:2109-2014 [DOI] [PubMed] [Google Scholar]
- 24.Mizuki I, Shimoyama T, Fukuda S, Liu Q, Nakaji S, Munakata A: Association of gastric epithelial apoptosis with the ability of Helicobacter pylori to induce a neutrophil oxidative burst. J Med Microbiol 2000, 49:521-524 [DOI] [PubMed] [Google Scholar]
- 25.Scotiniotis IA, Rokkas T, Furth EE, Rigas B, Shiff SJ: Altered gastric epithelial cell kinetics in Helicobacter pylori-associated intestinal metaplasia: implications for gastric carcinogenesis. Int J Cancer 2000, 85:192-200 [PubMed] [Google Scholar]
- 26.Ben-Sasson SA, Sherman Y, Gavrieli Y: Identification of dying cells—in situ staining. Methods Cell Biol 1995, 46:29-39 [PubMed] [Google Scholar]
- 27.Caulin C, Ware CF, Magin TM, Oshima RG: Keratin-dependent, epithelial resistance to tumor necrosis factor-induced apoptosis. J Cell Biol 2000, 149:17-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gilbert S, Loranger A, Daigle N, Marceau N: Simple epithelium keratins 8 and 18 provide resistance to fas-mediated apoptosis. The protection occurs through a receptor-targeting modulation. J Cell Biol 2001, 154:763-774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Inada H, Izawa I, Nishizawa M, Fujita E, Kiyono T, Takahashi T, Momoi T, Inagaki M: Keratin attenuates tumor necrosis factor-induced cytotoxicity through association with TRADD. J Cell Biol 2001, 155:415-425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oshima RG: Apoptosis and keratin intermediate filaments. Cell Death Differ 2002, 9:486-492 [DOI] [PubMed] [Google Scholar]
- 31.Bernstein C, Bernstein H, Payne CM, Garewal H: DNA repair/pro-apoptotic dual-role proteins in five major DNA repair pathways: fail-safe protection against carcinogenesis. Mutat Res 2002, 511:145-178 [DOI] [PubMed] [Google Scholar]
- 32.van Gent DC, Hoeijmakers JH, Kanaar R: Chromosomal stability and the DNA double-stranded break connection. Nat Rev Genet 2001, 2:196-206 [DOI] [PubMed] [Google Scholar]
- 33.Blier PR, Griffith AJ, Craft J, Hardin JA: Binding of Ku protein to DNA. Measurement of affinity for ends and demonstration of binding to nicks. J Biol Chem 1993, 268:7594-7601 [PubMed] [Google Scholar]
- 34.Barnes DE: DNA damage: air-breaks? Curr Biol 2002, 12:R262-R264 [DOI] [PubMed] [Google Scholar]
- 35.Brown KD, Lataxes TA, Shangary S, Mannino JL, Giardina JF, Chen J, Baskaran R: Ionizing radiation exposure results in up-regulation of Ku70 via a p53/ataxia-telangiectasia-mutated protein-dependent mechanism. J Biol Chem 2000, 275:6651-6656 [DOI] [PubMed] [Google Scholar]
- 36.Naito Y, Yoshikawa T: Molecular and cellular mechanisms involved in Helicobacter pylori-induced inflammation and oxidative stress. Free Radic Biol Med 2002, 33:323-336 [DOI] [PubMed] [Google Scholar]
- 37.Karanjawala ZE, Murphy N, Hinton DR, Hsieh CL, Lieber MR: Oxygen metabolism causes chromosome breaks and is associated with the neuronal apoptosis observed in DNA double-strand break repair mutants. Curr Biol 2002, 12:397-402 [DOI] [PubMed] [Google Scholar]
- 38.Boiteux S, Radicella JP: The human OGG1 gene: structure, functions, and its implication in the process of carcinogenesis. Arch Biochem Biophys 2000, 377:1-8 [DOI] [PubMed] [Google Scholar]
- 39.Shinmura K, Yokota J: The OGG1 gene encodes a repair enzyme for oxidatively damaged DNA and is involved in human carcinogenesis. Antioxid Redox Signal 2001, 3:597-609 [DOI] [PubMed] [Google Scholar]
- 40.Satoh MS, Lindahl T: Enzymatic repair of oxidative DNA damage. Cancer Res 1994, 54(Suppl 7):1899s-1901s [PubMed] [Google Scholar]
- 41.D’Amours D, Desnoyers S, D’Silva I, Poirier GG: Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J 1999, 342:249-268 [PMC free article] [PubMed] [Google Scholar]
- 42.Marti TM, Kunz C, Fleck O: DNA mismatch repair and mutation avoidance pathways. J Cell Physiol 2002, 191:28-41 [DOI] [PubMed] [Google Scholar]
- 43.Colussi C, Parlanti E, Degan P, Aquilina G, Barnes D, Macpherson P, Karran P, Crescenzi M, Dogliotti E, Bignami M: The mammalian mismatch repair pathway removes DNA 8-oxodGMP incorporated from the oxidized dNTP pool. Curr Biol 2002, 12:912-918 [DOI] [PubMed] [Google Scholar]
- 44.Lynch HT, Smyrk T, Lynch JF: Molecular genetics and clinical-pathology features of hereditary nonpolyposis colorectal carcinoma (Lynch syndrome): historical journey from pedigree anecdote to molecular genetic confirmation. Oncology 1998, 55:103-108 [DOI] [PubMed] [Google Scholar]
- 45.Prelich G, Tan CK, Kostura M, Mathews MB, So AG, Downey KM, Stillman B: Functional identity of proliferating cell nuclear antigen and a DNA polymerase-δ auxiliary protein. Nature 1987, 326:517-520 [DOI] [PubMed] [Google Scholar]
- 46.Umar A, Buermeyer AB, Simon JA, Thomas DC, Clark AB, Liskay RM, Kunkel TA: Requirement for PCNA in DNA mismatch repair at a step preceding DNA resynthesis. Cell 1996, 87:65-73 [DOI] [PubMed] [Google Scholar]
- 47.Gary R, Ludwig DL, Cornelius HL, MacInnes MA, Park MS: The DNA repair endonuclease XPG binds to proliferating cell nuclear antigen (PCNA) and shares sequence elements with the PCNA-binding regions of FEN-1 and cyclin-dependent kinase inhibitor p21. J Biol Chem 1997, 272:24522-24529 [DOI] [PubMed] [Google Scholar]
- 48.Lala PK, Chakraborty C: Role of nitric oxide in carcinogenesis and tumour progression. Lancet Oncol 2001, 2:149-156 [DOI] [PubMed] [Google Scholar]
- 49.Jaiswal M, LaRusso NF, Gores GJ: Nitric oxide in gastrointestinal epithelial cell carcinogenesis: linking inflammation to oncogenesis. Am J Physiol 2001, 281:G626-G634 [DOI] [PubMed] [Google Scholar]
- 50.Phoa N, Epe B: Influence of nitric oxide on the generation and repair of oxidative DNA damage in mammalian cells. Carcinogenesis 2002, 23:469-475 [DOI] [PubMed] [Google Scholar]
- 51.Tamura G: Genetic and epigenetic alterations of tumor suppressor and tumor-related genes in gastric cancer. Histol Histopathol 2002, 17:323-329 [DOI] [PubMed] [Google Scholar]