

Abstract
Advanced glycation end products (AGEs) may be involved in either amyloidogenesis or complications related to amyloid. We hypothesized that AGEs may influence the pathogenesis of AA amyloidosis, and investigated the spatial and temporal relationship between AGEs, carboxy methyl lysine (CML), the AGE receptor (RAGE), and AA amyloid in humans and mice. Specimens from patients with AL and ATTR amyloidosis served as a control. Using immunohistochemistry, AGEs, CML, and RAGE were found within amyloid deposits, more commonly in AA amyloid than in AL amyloid and not in ATTR amyloid. Western blotting showed that multiple proteins (between 12 and >60 kd) are modified, but not the AA amyloid fibril protein itself. In the murine model of AA amyloidosis, we found a marked interindividual variability with respect to local and systemic CML levels, as well as to splenic RAGE transcription. Serum levels of CML correlated with the duration of the inflammatory response but not with amounts of splenic RAGE mRNA. Other as yet unidentified variables, especially of the heterogeneous group of AGEs, probably modulate transcription of RAGE and influence amyloidogenesis. CML serum levels, in turn, may prove useful in predicting patients at risk.
Advanced glycation end products (AGEs) formed by nonenzymatic glycoxidation of proteins and lipids have been implicated in complications contributing to the increased morbidity and mortality of patients suffering from diabetes and uremia. Hyperglycemia in diabetic patients, and oxidative stress and carbonyl stress in uremic patients, contribute to the formation of AGEs, which are a chemically heterogeneous group of stable covalently bound and cross-linked adducts. 1-4 The detection of AGEs in prion plaques, 5 deposits of Aβ amyloid from Alzheimer patients, 6 hemodialysis-related Aβ2M amyloidosis, 7 and murine AApoAII amyloidosis 8 has indicated that nonenzymatic glycoxidation may also be involved in either amyloidogenesis or complications related to the deposition of amyloid. Amyloidoses are characterized by proteinaceous deposits of autologous origin that show specific structural and tinctorial properties. In AA amyloidosis, the acute-phase protein serum amyloid A (SAA) is the precursor of the AA fibril protein deposited in tissues. In the West, AA amyloidosis is most commonly related to rheumatoid arthritis. 9 Patients suffering from rheumatoid arthritis have significantly elevated serum and urine levels of AGEs, which correlate with parameters of disease activity such as C-reactive peptide, erythrocyte sedimentation rate, rheumatoid factor, and Lansbury index. 10-12 The activity of the inflammatory disease has also a major impact on amyloidogenesis, 13-15 and increased levels of AGEs and the possibility of developing AA amyloidosis are associated with the same risk factors. This raises the question whether AGEs may influence the pathology of AA amyloidosis.
The formation of AGEs is irreversible and the degree of modification correlates with the life span of the modified protein. AGEs are biologically active and may initiate a range of cellular responses including stimulation of monocyte chemotaxis, osteoclast-induced bone resorption, proliferation of vascular smooth muscle cells, aggregation of platelets, and stimulation of secretion of inflammatory cytokines, collagenase, and several growth factors. 4,16 The biological effect of AGEs is mediated, at least partly, by the receptor of advanced glycation end products (RAGE). RAGE is a multiligand, signal transduction receptor belonging to the immunoglobulin superfamily and it is expressed by a variety of cell types including endothelial cells, smooth muscle cells, lymphocytes, monocytes, and neurons. 16,17 Binding of ligands to RAGE 17,18 stimulates expression of RAGE itself, 17,18 and generates oxidative stress, synthesis and secretion of proinflammatory cytokines, and chemotaxis. 16-18 Thus, activation of RAGE propagates a chronic inflammatory disease state, that may further support the generation of AGEs. Yan and colleagues 19 have shown that canceling out the activation of cellular RAGE delayed the onset of reactive amyloidosis in mice, thus describing a putative pathophysiological pathway by which AGEs may influence amyloidogenesis.
To provide further evidence for the hypothesis that AGEs and RAGE may influence the pathogenesis of AA amyloidosis, we investigated the spatial and temporal relationship between AGEs, carboxy methyl lysine (CML), RAGE, and AA amyloid in humans and mice. Specimens from patients with light chain-associated (AL) amyloidosis and senile cardiovascular (ATTR) amyloidosis served as a control.
Materials and Methods
Patient Selection
Fifty-five archived, formalin-fixed, paraffin-embedded, autopsy specimens from a series of 25 patients were used in this study. These patients have been described previously when histological examination demonstrated the presence of generalized amyloidosis, either AA, AL, or ATTR amyloidosis. 20,21 Age, sex distribution, and underlying diseases are summarized in Table 1 ▶ . The classification of amyloid was based on immunohistochemistry and clinical history as described elsewhere. 21-23 Nine patients had suffered from AA amyloidosis, eight from AL amyloidosis, and eight from senile cardiovascular ATTR amyloidosis. 20,21 The following organ specimens were available for use in this study: liver (from 20 patients), kidney (17 patients), spleen (14 patients), and heart (4 patients). Variable amounts of amyloid were present in all of the specimens as vascular and interstitial deposits.
Table 1.
Age and Sex Distribution, Underlying Disease, and Cause of Death of the Patients Studied
| AA amyloidosis | AL amyloidosis | ATTR amyloidosis | |
|---|---|---|---|
| Number of cases | 9 | 8 | 8 |
| Total number of amyloidotic organs investigated | 25 | 20 | 10 |
| Age (years, range) | 56 (35–70) | 63 (50–73) | 85 (78–99) |
| Sex (m:f) | 6:3 | 3:5 | 5:3 |
| Underlying disease (n) | Rheumatoid arthritis (5) | Plasmacytoma (3) | Senile cardiovascular amyloidosis (8) |
| Rosai-Dorfman disease (1) | Immunocytoma (1) | ||
| Unknown (3) | Primary amyloidosis (4) | ||
| Cause of death (n) | Cardiac failure (5) | Cardiac failure (4) | Intestinal infarction (1) |
| Pulmonary thromboembolism (2) | Myocardial infarction (2) | Myocardial infarction (2) | |
| Renal failure (1) | Septic shock (1) | Pulmonary embolism (2) | |
| Acute appendicitis (1) | Gastrointestinal bleeding (1) | Panarteriitis nodosa (1) | |
| Cardiac failure (2) |
In addition, unfixed splenic tissue containing amyloid was available from three of the above-mentioned patients with generalized AA amyloidosis 14,24 and this was used for the preparation of amyloid fibril proteins as described previously 25 using ∼6 g of amyloidotic tissue. 24
Induction of AA Amyloidosis in Mice
Six- to 8-week-old BALB/c female mice were purchased from Harlan Winkelmann GmbH (Borchen, Germany). The mice were fed a normal chow diet and water ad libitum. AA amyloidosis was induced by daily subcutaneous injections of 7% azocasein for 20 days. The animals were sacrificed in an atmosphere of CO2 after 5 (five animals), 10 (five animals), 15 (five animals), and 20 (five animals) injections. Five mice received no injections and served as negative controls. The mice were bled by cardiac puncture and serum was obtained by centrifugation for 10 minutes at 2500 × g (4°C) before storage at −80°C until required. The spleens were removed and half of each organ was frozen immediately in liquid nitrogen and stored at −80°C until further use. The other half of the organ was fixed in 5% paraformaldehyde, pH 7.4, in phosphate-buffered saline (PBS), for 24 to 48 hours and then embedded in paraffin.
Preparation of AGE-Modified Proteins
AGE-modified keyhole limpet hemocyanin (AGE-KLH) was produced by incubating KLH (20 mg/ml; Sigma, Deisenhofen, Germany) with 1 mol/L of glucose in 200 mmol/L of PBS, pH 7.4, containing 0.025% NaN3 at 37°C for 3 months. Bovine serum albumin, human fibronectin (0.5 μg/ml), and human laminin (1 mg/ml; all from Sigma) were glycosylated nonenzymatically in a similar way for a period of 1 month. Collagen IV (Sigma) was dissolved in 10 mmol/L of sodium acetate, pH 5.5, and diluted with 10 mmol/L of Tris-HCl, pH 7.5, containing 100 mmol/L of NaCl, and then at a concentration of 1 mg/ml incubated with glucose as described above for 1 month. Incubation of KLH at 37°C in the absence of glucose served as a control. Surplus glucose was removed by dialysis in PBS (pH 7.4) using a cellulose dialysis membrane Spectra Por 4, MW 12,000 to 14,000 (Spectrum, Gardena, CA). Successful glycosylation was tested using fluorospectrometry. Fluorescence was measured at excitation and emission wavelengths of 398 and 450 nm, respectively, with a LS-5 Perkin-Elmer spectrofluorimeter (Perkin-Elmer, Munich, Germany).
Generation and Characterization of Polyclonal Anti-AGE Antibodies
Rabbits were immunized with 20 mg/ml of AGE-KLH according to a standardized immunization protocol (Eurogentec, Herstal, Belgium). IgG was obtained by using HiTrap protein G columns (Amersham Pharmacia Biotech, Freiburg, Germany). The specificity was tested using an enzyme-linked immunosorbent assay (ELISA) using either AGE-modified bovine serum albumin, collagen type IV, fibronectin, KLH, or laminin. Anti-AGE recognized KLH, AGE-bovine serum albumin, AGE-collagen, AGE-KLH, and AGE-laminin, but not native bovine serum albumin, collagen, or laminin (data not shown).
Histochemistry and Immunohistochemistry
Paraffin sections were stained with hematoxylin and eosin. The presence of amyloid was demonstrated by the appearance of green birefringence from alkaline alcoholic Congo red staining under polarized light. 26 Immunostaining was performed with antibodies directed against AGE (polyclonal, anti-AGE, dilution 1:20), CML (monoclonal, clone 4G9, dilution 1:1000; Roche Diagnostics GmbH, Penzberg, Germany), 27 human RAGE (monoclonal, dilution 1:20), 28 and human AA amyloid (monoclonal, clone mc1, dilution 1:500; DAKO, Hamburg, Germany). Before immunostaining with anti-AGE the specimens were pretreated with 10 mmol/L of ethylenediaminetetraacetic acid (2 × 10 minutes, 450 W microwave oven). Before immunostaining with anti-CML the specimens were pretreated with Proteinase K (30 minutes at 37°C). Immunostaining was performed as described elsewhere. 24 Specimens of formalin-fixed and paraffin-embedded arteriosclerotic vessels served as positive controls for AGE, CML, and RAGE. The specificity of immunostaining was controlled using specimens containing known classes of amyloid (anti-AA amyloid). Incubation with preimmune serum and omission of primary antibodies served as negative controls.
Gel Electrophoresis and Western Blotting
Quantities of between 50 and 200 mg of tissue, which had been stored at −80°C, were homogenized in 0.5 to 2.0 ml of lysis buffer (4 mol/L urea, 0.5% sodium dodecyl sulfate, 62.5 mmol/L Tris, pH 6.8, 1 mg/ml protease inhibitor cocktail; Sigma) using an Ultra-Turrax T8 (IKA-Labortechnik, Staufen, Germany). The homogenate was centrifuged at 11,000 × g for 10 minutes at 4°C. Aliquots of 25 μg or 50 μg were resolved in 10 to 16.5% two-phase polyacrylamide gels and visualized by staining with Coomassie blue. Proteins on unstained polyacrylamide gels were transferred onto a polyvinylidene difluoride membrane (Immobilon-PSQ; Millipore, Bedford, MA) and blocked overnight with Roti-Block (Carl ROTH GmbH, Karlsruhe, Germany). Immunostaining was performed using anti-AA amyloid (monoclonal, clone mc1, dilution 1:500; DAKO), anti-AGE (dilution 1:500), anti-CML (1:250), or biotinylated anti-human serum albumin (monoclonal, 1:1000; Roche Diagnostics GmbH). Alkaline phosphatase-conjugated second or anti-rabbit/anti-mouse antibodies (dilution 1:1000, room temperature, 60 minutes; DAKO) or alkaline phosphatase-conjugated streptavidin (1:3000, room temperature, 60 minutes; Rockland, PA) served as bridging reagents and 1-Step Tetranitroblue tetrazolium/5-bromo-4-chloro-3-indolyphosphate (Pierce, Bonn, Germany) as chromogen.
Competitive ELISA for the Quantification of CML
The concentration of CML in murine serum and spleen homogenates was quantified using a competitive ELISA developed by Roche Diagnostics GmbH. 29 Samples from serum or tissue homogenate were diluted (serum, 1:20; tissue homogenate, 1:200) in washing buffer and pretreated with Proteinase K (1 mg/ml, 37°C for 2 hours; Roche Diagnostics GmbH).
Polymerase Chain Reaction of Murine RAGE mRNA
Total RNA was isolated from small pieces (<10 mg) of frozen spleen. After homogenization using an Ultra-Turrax T8 (IKA-Labortechnik), the homogenate was prepared with the RNeasy mini kit according to manufacturer’s instructions (Qiagen, Hilden, Germany). The reverse transcription into cDNA was performed using the Omniscript RT Kit (Qiagen), oligo(dT)12-18 primers (Promega, Mannheim, Germany), RNaseOUT recombinant ribonuclease inhibitor (LifeTechnologies, Karlsruhe, Germany), and 1 μg of RNA per sample as a template. Two μl of the cDNA template were used for the polymerase chain reaction (PCR) analysis. The master mix used contained the following per reaction (50 μl): 1.5 mmol/L MgCl2, 200 μmol/L of each dNTP, 50 pmol of each primer, and 2.5 U TaqDNA polymerase (TaqPCR core kit from Qiagen). The sequences of the primers for the murine RAGE-mRNA were: 5′-CAGGGTCACAGAAACCGG-3′ (upstream), and 5′-ATTCAGCTCTGCACGTTCCT-3′ (downstream). The standard temperature profile included an initial denaturation for 2 minutes at 92°C, followed by 35 cycles of denaturation at 92°C for 30 seconds, annealing at 60°C for 90 seconds, and an extension at 72°C for 60 seconds. The products were transferred onto a 1.5% agarose gel labeled with ethidium bromide, and analyzed by transillumination. The PCR product was isolated from the gel using the MinElute extraction kit from Qiagen and sequenced on the capillary sequencer 310 from ABI Prism (Foster City, CA).
Quantification of Murine RAGE mRNA by Real-Time Reverse Transcriptase-PCR
Assays were performed on the LightCycler (Roche Diagnostics GmbH). Several dilutions of plasmids containing the cDNA fragments of RAGE were used as internal controls. For plasmid construction, the cDNA fragments were amplified using the primer described above and inserted into the pCR2.1-TOPO vector (Invitrogen, Groningen, The Netherlands). The copy number of the resulting plasmids was calculated after DNA quantification. All reactions contained 2 mmol/L of MgCl2, 10 pmol of each primer, 2 μl 10× LightCycler-DNA Master SYBRGreen I (Roche Diagnostics GmbH), and 13.2 μl of distilled water to adjust the final volume (20 μl). Two μl of the cDNA template were added to the prepared capillaries. The standard temperature profile started with an initial denaturation at 95°C for 30 seconds, followed by 40 cycles of denaturation at 95°C for 1 second, annealing at 62°C for 5 seconds, and an extension at 72°C for 15 seconds.
The quantification of RAGE mRNA was performed as follows: β-actin (upstream primer: 5′-TGACGGGGTCACCCACACTGTGCCCATCTA-3′, downstream primer: 5′-CTAGAAGCATTTGCGGTGGACGATGGAGGG-3′) was used as a housekeeping gene, and the RAGE mRNA/β-actin mRNA ratio was calculated for all samples. Each sample was measured in triplicate.
Statistical Analysis
All values are expressed as mean ± SD. The chi-square test and the unpaired Student’s t-test were used where appropriate. The Pearson correlation coefficient was used to determine the relationship between metric parameters. A value of P < 0.05 was considered to be statistically significant.
Results
Prevalence of AGEs and RAGE in Human AA Amyloidosis
AGEs were found immunohistochemically in every patient (100%) with AA amyloidosis within amyloid deposits (Figure 1) ▶ . It was present in deposits of the kidney, liver, and spleen. AGEs were often present only in some deposits, whereas others within the same specimen were negative (Figure 1) ▶ . Only a single liver specimen showed amyloid deposits completely devoid of AGEs (Table 2) ▶ . AGEs were also present in tubulus epithelium of kidneys, on erythrocytes, in the interstitium, and within vessel walls. In seven (78%) patients AGEs were identified as CML by using a specific antibody (Figure 1) ▶ . CML was found most commonly within the amyloid deposits of the kidney (100%), followed by spleen (67%) and liver (11%; Table 2 ▶ ). It was interesting to note that CML was also strongly expressed by cells lining AA amyloid deposits (Figure 1 ▶ , inset).
Figure 1.
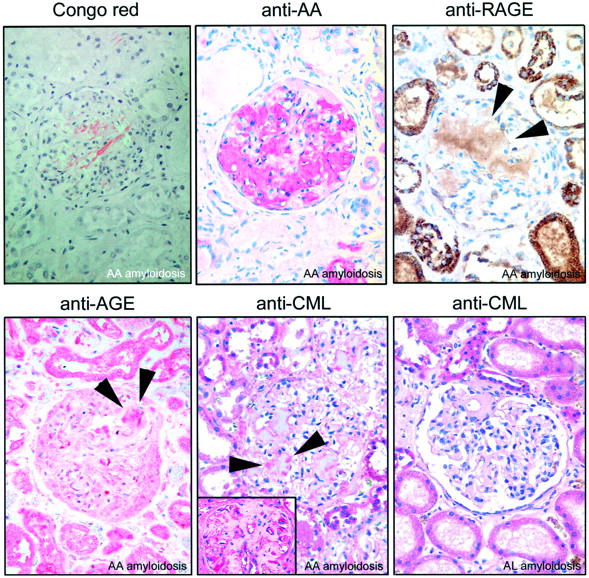
Kidney specimens from a patient with generalized AA amyloidosis show AGE, CML, and RAGE within amyloid deposits (arrowheads). CML was also found in cells lining amyloid deposits (inset). CML was found less commonly in AL amyloid. Congo red staining in polarized light. Immunostaining with anti-AA amyloid, anti-AGE, anti-CML, and anti-RAGE; hematoxylin counterstain. Original magnifications: ×20; ×40 (inset).
Table 2.
Distribution Pattern of AGEs, CML, and the RAGE within AA, AL, and ATTR Amyloid Deposits
| AGE n (%) | CML n (%) | RAGE n (%) | |
|---|---|---|---|
| AA amyloidosis | |||
| Kidney (n = 7) | 7 (100) | 7 (100) | 6 (86) |
| Liver (n = 9) | 8 (89) | 1 (11) | 7 (77) |
| Spleen (n = 9) | 9 (100) | 6 (67) | 7 (77) |
| Total (n = 25) | 24 (96) | 14 (56) | 20 (80) |
| AL amyloidosis | |||
| Kidney (n = 7) | 4 (57) | 3 (43) | 2 (29) |
| Liver (n = 8) | 5 (63) | 0 | 2 (25) |
| Spleen (n = 5) | 2 (40) | 4 (80) | 0 |
| Total (n = 20) | 11 (55) | 7 (20) | 4 (20) |
| ATTR amyloidosis | |||
| Kidney (n = 3) | 0 | 0 | 0 |
| Liver (n = 3) | 0 | 0 | 0 |
| Heart (n = 4) | 0 | 0 | 0 |
| Total (n = 10) | 0 | 0 | 0 |
RAGE displayed a spatial relationship with amyloid in all of the cases studied. RAGE was found either within the amyloid deposits (Figure 1) ▶ or adjacent to the deposits where it was extracellular or intracellular. Intracellular immunostaining was confined mainly to myocytes of vessel walls, endothelial cells, and macrophages, which were either attached to or enclosed by amyloid deposits. Within amyloid deposits, RAGE again was found most commonly in the kidney (86%), followed by spleen (77%) and liver (77%; Table 2 ▶ ). Immunostaining for RAGE was more extended and more even as compared to AGE and CML (Figure 1) ▶ .
Detection of AGEs in Homogenates of Human AA Amyloidosis
Western blots were performed for further analysis of the nature of the AGE-modified compounds associated with AA amyloid. Unfixed samples of amyloidotic spleens obtained from three of the nine patients with AA amyloidosis studied immunohistochemically were homogenized and submitted to electrophoresis and Western blotting. Using an antibody directed against AA amyloid, immunoreactive bands were detected in the crude tissue homogenates of all three patients (Figure 2) ▶ . All three patients showed bands at ∼6 kd, which are consistent with the molecular weight of the AA amyloid fibril proteins (Figure 2) ▶ , whereas patients 2 and 3 showed additional bands at ∼12 kd representing the precursor protein serum amyloid A. Immunoblotting of the homogenates with anti-AGE showed multiple immunoreactive bands in all cases (Figure 2) ▶ ranging from 17 kd to >60 kd were immunoreactive for AGE (Figure 2) ▶ . None of these bands had the same size as the fibril proteins. Immunoblotting of the homogenates with anti-CML showed similar bands in all cases ranging from 12 kd to >60 kd (Figure 2) ▶ , some of these bands were attributable to nonenzymatically glycosylated human serum albumin (data not shown).
Figure 2.

Unfixed, crude amyloidotic spleen homogenates from three patients with generalized AA amyloidosis were homogenized and separated by gel electrophoresis. Western blotting with an antibody directed against human AA amyloid identified bands of ∼6 and 12 kd. Immunolabeling with anti-AGE and anti-CML showed multiple bands ranging from 12 to >60 kd. Molecular weight standards in kd (MW). CB, Coomassie blue staining; Pt.-No., patient number; AA, Western blotting with anti-AA amyloid; AGE, anti-AGE; and CML, anti-CML.
Prevalences of AGEs and RAGE in Human AL and ATTR Amyloidosis
AGEs were found significantly less commonly in AL amyloidosis (P < 0.01). They were present within the deposits of five patients (63%), including liver (63%), kidney (57%), and spleen (40%; Table 2 ▶ ). The staining pattern of anti-AGE within AL amyloid was similar to AA amyloid. In five patients AGEs were specified as CML (Table 2) ▶ and was found most commonly in the spleen (80%) followed by kidney (43%; Table 2 ▶ ). CML was not found in the liver. In contrast to AA amyloid, CML was not observed in cells lining AL amyloid deposits (Figure 1) ▶ . The different prevalences of CML between AA and AL amyloid were statistically not significant.
RAGE was also present in AL amyloid deposits, but significantly less common than in AA amyloid (P < 0.01; Table 2 ▶ ). Neither AGE, CML, nor RAGE were found in deposits of ATTR amyloid (Table 2) ▶ .
CML and RAGE in the Murine Model of Reactive Amyloidosis
The immunohistochemical and biochemical studies provide evidence for high prevalences of AGEs, CML, and RAGE in human AA amyloid deposits. However, they provide no information about the order of events and a putative temporal correlation. To investigate the occurrences and expression profile of CML and RAGE, respectively, during amyloidogenesis, AA amyloidosis was induced in amyloid-susceptible BALB/c-mice. A standard induction protocol with daily subcutaneous injections of azocasein resulted in the formation of amyloid. Amyloid occurred in the spleen as early as after 15 injections (two of five mice) and was present in all mice after 20 injections. A competitive ELISA was performed to measure the relative amounts of CML in serum and spleen during amyloidogenesis. The amount of CML in the serum showed a V-shaped pattern with the lowest level after 15 injections and the highest after 20 injections (Table 3) ▶ . The difference in the serum levels of CML between 15 and 20 injections proved to be significant (P < 0.05). In contrast to the serum, the concentration of CML in the spleen showed only mild variations throughout time (Table 3) ▶ and it did not correlate with the number of injections given (P = 0.88) or with the serum level of CML (P = 0.68).
Table 3.
AA Amyloidosis Was Induced in BALB/c Mice Using Daily Injections of 7% Azocasein
| Number of injections | CML serum (ng/mg protein) | CML spleen (ng/mg protein) | Copies RAGE mRNA/copies β-actin mRNA × 105 |
|---|---|---|---|
| 0 (n = 5) | 8.25 ± 2.87 | 10.03 ± 3.14 | 31.14 ± 65.33 |
| 5 (n = 5) | 6.65 ± 1.82 | 9.17 ± 2.70 | 1.91 ± 1.10† |
| 10 (n = 5) | 7.83 ± 2.29 | 8.13 ± 3.31 | 10.25 ± 5.80† |
| 15 (n = 5) | 4.86 ± 2.21* | 9.35 ± 2.38 | 60.06 ± 46.98† |
| 20 (n = 5) | 8.84 ± 2.10* | 9.14 ± 1.27 | 52.86 ± 57.88 |
*Fifteen versus 20 injections; P < 0.05.
†Five versus 10 injections; P < 0.01; 5 versus 20 injections; P < 0.05.
Levels of CML in serum and spleen homogenates as well as the relative amount of mRNA of the RAGE were measured as indicated.
The amount of RAGE mRNA was quantified in murine spleens using real-time PCR and showed a V-shaped pattern: the amount of mRNA decreased after five injections and increased significantly thereafter (5 versus 10 injections, P < 0.01) with a peak after 15 injections (5 versus 15 injections, P < 0.05) (Table 3) ▶ . No further significant correlations were found between the relative amount of RAGE mRNA and the number of injections given. The relative amount of RAGE mRNA did not correlate with the levels of CML in the serum (P = 0.901) or spleen (P = 0.226).
It is important to note that CML levels in serum and spleen as well as the amount of RAGE mRNA showed great, nonsystematic interindividual variability, as documented by the high SD, irrespective of the number of injections applied.
Discussion
Prevalence and Pathogenesis of AGEs in Amyloidosis
In the present study we investigated the spatial and temporal relationships between the presence of AGEs and AA amyloid in human and murine tissues and compared it with AL and ATTR amyloidosis. Using immunohistochemistry, we found AGEs that were spatially related to human amyloid deposits and occurred significantly more commonly in AA than AL or ATTR amyloidosis. Contradicting results have been obtained regarding the occurrence of AGEs in human AA amyloidosis, which is related most likely to the heterogeneity of AGEs. Using monoclonal antibodies Niwa and colleagues 30 were unable to detect imidazolone in AA amyloid deposits. Uesugi and colleagues 31 applied antibodies directed against CML and pyrraline and found only CML in renal amyloid deposits. We applied polyclonal antibodies, which are more likely to recognize a broad spectrum of different AGEs in AA amyloidosis. 32 We also found CML in AA and AL amyloidosis, confirming the observation made by Uesugi and colleagues. 31 However, CML was less common than AGEs in general, as demonstrated here by comparing the staining of polyclonal and monoclonal antibodies. Altogether these observations show that AGEs in AA and AL amyloid constitute a heterogeneous group, and that CML contributes only partly to AGEs formed in AA and AL amyloid.
In our series of specimens, the increased prevalences of AGEs and CML in AA amyloid are attributable to the underlying disease and not to patient’s age: most patients with AA amyloidosis had suffered from a chronic or recurrent inflammatory disease known to be associated with an increased serum concentration of AGEs, including CML, and, on average, patients with AA amyloidosis were younger than those with AL and ATTR amyloidosis. The presence of AGEs in some patients with AL amyloidosis may also be related to the underlying disease; the majority had suffered from either multiple myeloma or primary amyloidosis disease conditions that are known to cause chronic renal failure and chronic recurrent infections that may result in the generation of AGEs. We did not find AGEs in ATTR amyloid, again indicating that patient’s age cannot account for the occurrence of AGEs in AA and AL amyloidosis. Patients with ATTR amyloid had the highest mean age in our series.
Pathophysiological Role of AGEs and RAGE in AA Amyloidosis
AGEs may influence the pathogenesis of an amyloid disease in different ways. Modification of the precursor or fibril protein may result in altered biological and biophysical properties for the propagation of fibril formation. Using tissue homogenates and Western blotting, we were able to show that neither the precursor nor the fibril protein of human AA amyloidosis were modified by AGEs or CML. Thus, if AGEs or CML do have an impact on the pathogenesis of AA amyloidosis, this effect is not mediated by the modification of SAA or AA fibril proteins.
AGEs may influence amyloidogenesis by activating RAGE. Binding of ligands to RAGE leads to translocation of nuclear factor-kappa B via transient activation of tyrosine phosphorylation, extracellular signal-regulated kinases 1 and 2, and p38 mitogen-activated protein kinase. 19,33,34 Yan and colleagues 19 have shown that RAGE is able to influence the pathogenesis of AA amyloidosis; canceling out the activation of cellular RAGE delayed the onset of reactive amyloidosis in mice. They also demonstrated that AA fibril proteins and the amyloidogenic variant of murine SAA (SAA1.1) are ligands for RAGE. 19 We found RAGE to be spatially related to amyloid deposits in all patients with AA amyloidosis, which confirms a previous observation made in a single case. 19 Again, it was found significantly more commonly in AA than in AL or ATTR amyloid. RAGE was not only cell bound, but also present extracellularly within the deposits. Because RAGE was shown to bind AA fibril proteins it may become enriched within AA amyloid deposits. 19 Similar to AGEs, the presence of RAGE may also be related to the underlying disease.
CML binds to cellular RAGE and may influence the expression profile of RAGE 33 and, indeed, we found CML highly expressed in cells lining amyloid deposits. By using a murine model, we investigated the temporal relationship between CML levels in serum and spleen, and the expression profile of RAGE mRNA during amyloidogenesis. Interestingly, CML was present in healthy animals and CML-levels in the serum decreased after an inflammatory stimulus was set. Thus, during the early course of a chronic inflammatory stimulus, adaptive mechanisms seem to reduce CML load and RAGE expression. The decreased amount of CML, in turn, may be related to increased clearance mediated by RAGE. It is interesting to note that the low serum levels of CML after 15 injections were associated with the highest amounts of RAGE mRNA in the spleen. However, with an ongoing inflammatory stimulus, which then leads to de novo formation of AGEs, the serum CML level increases significantly while amyloid is deposited in the spleen. Thus serum levels of CML may indicate duration of the disease. Future studies are required to show whether serum levels of AGE or CML can be used to predict severity or duration of a chronic inflammatory disease and thus predict the occurrence or progression of AA amyloidosis, as it was demonstrated for Aβ2M amyloidosis. 35
Statistical analyses failed to show any correlation between serum or splenic CML levels and amounts of RAGE mRNA, indicating that transcription of RAGE does not depend on CML levels only. At least three different ligands, which occur during amyloidogenesis, compete for binding to RAGE: SAA1.1, AA fibril protein, and CML. 19 Indeed, AA fibril proteins and CML have similar kd values (between 60 and 76 nmol/L) 16,19,33 for binding to RAGE. Apart from CML, other AGEs are likely to bind to RAGE and may modulate its expression, 33 which, altogether, may explain why the transcription levels of RAGE demonstrate such a variability. Furthermore, different receptors are capable of binding and/or clearing AGEs, including the macrophage scavenger receptor classes A and B, 80 K-H phosphoprotein, oligosaccharyl transferase-48, and galectin-3, 36,37 which all may influence local and systemic concentrations of AGEs. However, it is important to note that the highest amount of RAGE mRNA was found when amyloid started depositing in the spleen.
In our series, immunostaining of amyloid with anti-AGE was sparse, heterogeneous, and present in only some deposits, whereas others, within the same specimen, were immunonegative. Thus, AGEs constitute just a minor component of AA amyloid deposits and it is even not the fibril protein being modified. These observations suggest that the presence of AGEs, per se, is no prerequisite for the formation of AA amyloid fibrils themselves. Similarly, Niwa 38 proposed that modification of β2-microglobluin in dialysis related Aβ2M amyloidosis occurs after amyloid has been formed.
In summary, we show that AGEs and RAGE are spatially related to AA amyloid, and that high-molecular weight compounds are modified in AA amyloid, but not the AA fibril protein itself. We found a marked interindividual variability with respect to local and systemic CML levels as well as to local RAGE transcription, making it difficult to find simple explanations for a putative pathophysiological link between CML, RAGE, and AA amyloidosis. Other as yet unidentified variables, particularly belonging to the heterogeneous group of AGEs, may modulate transcription of RAGE and influence amyloidogenesis. Nevertheless, CML serum levels may prove useful in predicting patients at risk.
Footnotes
Address reprint requests to PD Dr. med. Christoph Röcken, Department of Pathology, Otto-von-Guericke-University, Leipziger Str. 44, D-39120 Magdeburg, Germany. E-mail: christoph.roecken@medizin.uni-magdeburg.de.
Supported by grants from the Deutsche Forschungsgemeinschaft, Bonn Bad-Godesberg, Germany (grant no. RO 1173/3); the German Government (grant no. BMBF-01ZZ9604); the Pinguin Stiftung, Düsseldorf, Germany; and the Wilhelm Vaillant-Stiftung, München, Germany.
References
- 1.Vlassara H: Advanced glycation end-products and atherosclerosis. Ann Med 1996, 28:419-426 [DOI] [PubMed] [Google Scholar]
- 2.Vlassara H: Recent progress in advanced glycation end products and diabetic complications. Diabetes 1997, 46:S19-S25 [DOI] [PubMed] [Google Scholar]
- 3.Miyata T, Ypersele de Strihou C, Kurokawa K, Baynes JW: Alterations in nonenzymatic biochemistry in uremia: origin and significance of “carbonyl stress” in long-term uremic complications. Kidney Int 1999, 55:389-399 [DOI] [PubMed] [Google Scholar]
- 4.Miyata T, Kurokawa K, Destrihou CV: Advanced glycation and lipoxidation end products: role of reactive carbonyl compounds generated during carbohydrate and lipid metabolism. J Am Soc Nephrol 2000, 11:1744-1752 [DOI] [PubMed] [Google Scholar]
- 5.Sasaki N, Takeuchi M, Chowei H, Kikuchi S, Hayashi Y, Nakano N, Ikeda H, Yamagishi S, Kitamoto T, Saito T, Makita Z: Advanced glycation end products (AGE) and their receptor (RAGE) in the brain of patients with Creutzfeldt-Jakob disease with prion plaques. Neurosci Lett 2002, 326:117-120 [DOI] [PubMed] [Google Scholar]
- 6.Münch G, Schinzel R, Loske C, Wong A, Durany N, Li JJ, Vlassara H, Smith MA, Perry G, Riederer P: Alzheimer’s disease—synergistic effects of glucose deficit, oxidative stress and advanced glycation endproducts. J Neural Transm 1998, 105:439-461 [DOI] [PubMed] [Google Scholar]
- 7.Miyata T, Oda O, Inagi R, Iida Y, Araki N, Yamada N, Horiuchi S, Taniguchi N, Maeda K, Kinoshita T: Beta 2-microglobulin modified with advanced glycation end products is a major component of hemodialysis-associated amyloidosis. J Clin Invest 1993, 92:1243-1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoshii Y, Kawano H, Gondo T, Takahashi M, Ishihara T, Higuchi K, Horiuchi S: Immunohistochemical study with anti-advanced glycation end-products antibody in murine amyloidosis. Pathol Int 1996, 46:738-742 [DOI] [PubMed] [Google Scholar]
- 9.Röcken C, Shakespeare A: Pathology, diagnosis and pathogenesis of AA amyloidosis. Virchows Arch 2002, 440:111-122 [DOI] [PubMed] [Google Scholar]
- 10.Miyata T, Ishiguro N, Yasuda Y, Ito T, Nangaku M, Iwata H, Kurokawa K: Increased pentosidine, an advanced glycation end product, in plasma and synovial fluid from patients with rheumatoid arthritis and its relation with inflammatory markers. Biochem Biophys Res Commun 1998, 244:45-49 [DOI] [PubMed] [Google Scholar]
- 11.Rodriguez-Garcia J, Requena JR, Rodriguez-Segade S: Increased concentrations of serum pentosidine in rheumatoid arthritis. Clin Chem 1998, 44:250-255 [PubMed] [Google Scholar]
- 12.Takahashi M, Suzuki M, Kushida K, Miyamoto S, Inoue T: Relationship between pentosidine levels in serum and urine and activity in rheumatoid arthritis. Br J Rheumatol 1997, 36:637-642 [DOI] [PubMed] [Google Scholar]
- 13.Gillmore JD, Lovat LB, Persey MR, Pepys MB, Hawkins PN: Amyloid load and clinical outcome in AA amyloidosis in relation to circulating concentration of serum amyloid A protein. Lancet 2001, 358:24-29 [DOI] [PubMed] [Google Scholar]
- 14.Röcken C, Wieker K, Grote HJ, Müller G, Franke A, Roessner A: Rosai-Dorfman disease and generalized AA amyloidosis. A case report. Hum Pathol 2000, 31:621-624 [DOI] [PubMed] [Google Scholar]
- 15.de Beer FC, Nel AE, Gie RP, Donald PR, Strachan AF: Serum amyloid A protein and C-reactive protein levels in pulmonary amyloidosis: relationship to amyloidosis. Thorax 1984, 39:196-200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmidt AM, Yan SD, Yan SF, Stern DM: The biology of the receptor for advanced glycation end products and its ligands. Biochim Biophys Acta 2000, 1498:99-111 [DOI] [PubMed] [Google Scholar]
- 17.Yeh CH, Sturgis L, Haidacher J, Zhang XN, Sherwood SJ, Bjercke RJ, Juhasz O, Crow MT, Tilton RG, Denner L: Requirement for p38 and p44/p42 mitogen-activated protein kinases in RAGE-mediated nuclear factor-kappaB transcriptional activation and cytokine secretion. Diabetes 2001, 50:1495-1504 [DOI] [PubMed] [Google Scholar]
- 18.Schwedler S, Schinzel R, Vaith P, Wanner C: Inflammation and advanced glycation end products in uremia: simple coexistence, potentiation or causal relationship? Kidney Int 2001, 59(Suppl 78):S32-S36 [DOI] [PubMed] [Google Scholar]
- 19.Yan SD, Zhu HJ, Zhu AP, Golabek A, Du H, Roher A, Yu J, Soto C, Schmidt AM, Stern D, Kindy M: Receptor-dependent cell stress and amyloid accumulation in systemic amyloidosis. Nat Med 2000, 6:643-651 [DOI] [PubMed] [Google Scholar]
- 20.Müller D, Roessner A, Röcken C: Distribution pattern of matrix metalloproteinases (MMP-1, -2, -3, and -9), tissue inhibitors of matrix metalloproteinases (TIMP-1 and -2), and a2-macroglobulin in cases of generalized AA- and AL amyloidosis. Virchows Arch 2000, 437:521-527 [DOI] [PubMed] [Google Scholar]
- 21.Strege RJ, Saeger W, Linke RP: Diagnosis and immunohistochemical classification of systemic amyloidoses. Report of 43 cases in an unselected autopsy series. Virchows Arch 1998, 433:19-27 [DOI] [PubMed] [Google Scholar]
- 22.Röcken C, Schwotzer E, Linke RP, Saeger W: The classification of amyloid deposits in clinicopathological practice. Histopathology 1996, 29:325-335 [DOI] [PubMed] [Google Scholar]
- 23.Röcken C, Roessner A: An evaluation of antigen retrieval procedures for immunoelectron microscopical classification of amyloid deposits. J Histochem Cytochem 1999, 47:1385-1394 [DOI] [PubMed] [Google Scholar]
- 24.Röcken C, Stix B, Brömme D, Ansorge S, Roessner A, Bühling F: A putative role for cathepsin K in degradation of AA and AL amyloidosis. Am J Pathol 2001, 158:1029-1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stix B, Kähne T, Sletten K, Raynes J, Roessner A, Röcken C: Proteolysis of AA amyloid fibril proteins by matrix metalloproteinases-1, -2, and -3. Am J Pathol 2001, 159:561-570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Puchtler H, Sweat F, Levine M: On the binding of Congo red by amyloid. J Histochem Cytochem 1962, 10:355-364 [Google Scholar]
- 27.Mellinghoff AC, Reininger AJ, Wuerth JP, Founds HW, Landgraf R, Hepp KD: Formation of plasma advanced glycosylation end products (AGEs) has no influence on plasma viscosity. Diabet Med 1997, 14:832-836 [DOI] [PubMed] [Google Scholar]
- 28.Srikrishna G, Huttunen HJ, Johansson L, Weigle B, Yamaguchi Y, Rauvala H, Freeze HH: N-Glycans on the receptor for advanced glycation end products influence amphoterin binding and neurite outgrowth. J Neurochem 2002, 80:998-1008 [DOI] [PubMed] [Google Scholar]
- 29.Misselwitz J, Franke S, Kauf E, John U, Stein G: Advanced glycation end products in children with chronic renal failure and type 1 diabetes. Pediatr Nephrol 2002, 17:316-321 [DOI] [PubMed] [Google Scholar]
- 30.Niwa T, Katsuzaki T, Miyazaki S, Momoi T, Akiba T, Miyazaki T, Nokura K, Hayase F, Tatemichi N, Takei Y: Amyloid beta(2)-microglobulin is modified with imidazolone, a novel advanced glycation end product, in dialysis-related amyloidosis. Kidney Int 1997, 51:187-194 [DOI] [PubMed] [Google Scholar]
- 31.Uesugi N, Sakata N, Nagai R, Jono T, Horiuchi S, Takebayashi S: Glycoxidative modification of AA amyloid deposits in renal tissue. Nephrol Dial Transplant 2000, 15:355-365 [DOI] [PubMed] [Google Scholar]
- 32.Al-Abed Y, Kapurniotu A, Bucala R: Advanced glycation end products: detection and reversal. Methods Enzymol 1999, 309:152-172 [DOI] [PubMed] [Google Scholar]
- 33.Kislinger T, Fu C, Huber B, Qu W, Taguchi A, Du YS, Hofmann M, Yan SF, Pischetsrieder M, Stern D, Schmidt AM: N(epsilon)-(carboxymethyl)lysine adducts of proteins are ligands for receptor for advanced glycation end products that activate cell signaling pathways and modulate gene expression. J Biol Chem 1999, 274:31740-31749 [DOI] [PubMed] [Google Scholar]
- 34.Sousa MM, DuYan S, Stern D, Saraiva MJ: Interaction of the receptor for advanced glycation end products (RAGE) with transthyretin triggers nuclear transcription factor kB (NF-kB) activation. Lab Invest 2000, 80:1101-1110 [DOI] [PubMed] [Google Scholar]
- 35.Motomiya Y, Iwamoto H, Uji Y, Higashi T, Uchimura T, Maruyama I: Potential value of CML-Hb in predicting the progression of bone cysts in dialysis-related amyloidosis. Nephron 2001, 89:286-290 [DOI] [PubMed] [Google Scholar]
- 36.Thornalley PJ: Cell activation by glycated proteins. AGE receptors, receptor recognition factors and functional classification of AGEs. Cell Mol Biol 1998, 44:1013-1023 [PubMed] [Google Scholar]
- 37.Ohgami N, Nagai R, Ikemoto M, Arai H, Kuniyasu A, Horiuchi S, Nakayama H: Cd36, a member of the class b scavenger receptor family, as a receptor for advanced glycation end products. J Biol Chem 2001, 276:3195-3202 [DOI] [PubMed] [Google Scholar]
- 38.Niwa T: Dialysis-related amyloidosis: pathogenesis focusing on AGE modification. Semin Dial 2001, 14:123-126 [DOI] [PubMed] [Google Scholar]