

Abstract
Diabetic nephropathy ensues from events involving earliest changes in the glomeruli and podocytes, followed by accumulation of extracellular matrix in the mesangium. Postulated mechanisms include roles for vascular endothelial growth factor (VEGF), produced by podocytes and contributing to enhanced excretion of urinary albumin and recruitment/activation of inflammatory cells, and transforming growth factor-β (TGF-β), elicited largely from mesangial cells and driving production of extracellular matrix. RAGE, a receptor for advanced glycation endproducts (AGEs) and S100/calgranulins, displays enhanced expression in podocytes of genetically diabetic db/db mice by age 13 weeks. RAGE-bearing podocytes express high levels of VEGF by this time, in parallel with enhanced recruitment of mononuclear phagocytes to the glomeruli; events prevented by blockade of RAGE. By age 27 weeks, soluble RAGE-treated db/db mice displayed diminished albuminuria and glomerulosclerosis, and improved renal function. Diabetic homozygous RAGE null mice failed to develop significantly increased mesangial matrix expansion or thickening of the glomerular basement membrane. We propose that activation of RAGE contributes to expression of VEGF and enhanced attraction/activation of inflammatory cells in the diabetic glomerulus, thereby setting the stage for mesangial activation and TGF-β production; processes which converge to cause albuminuria and glomerulosclerosis.
Diabetes is emerging as the leading cause of end-stage renal failure and the requirement for chronic dialysis throughout the world. 1 The striking reduction in survival of diabetic subjects on commencement of dialysis 2 suggests that disruption of cellular homeostasis induced by renal failure in diabetes may not be overcome by this form of replacement therapy. Although the pathological and pathophysiologic indices of diabetic nephropathy are well-described, the precise molecular events that lead to the development of hyperpermeability and leakage of albumin and other proteins into the urine, glomerulosclerosis, and the decline in renal function are only recently being uncovered. Multiple studies have implicated activation of the transforming growth factor-β (TGF-β) system in the pathogenesis of diabetic nephropathy, based on the properties of TGF-β, as well as the observation that levels of TGF-β mRNA and protein are significantly increased in the glomeruli and tubulointerstitium in human diabetes and in animal models. 3-5 Short-term administration of anti-TGF-β antibodies to mice with streptozotocin-induced diabetes suppressed enhanced expression of genes encoding extracellular matrix molecules and the development of glomerular hypertrophy. 6 Indeed, previous studies have definitively linked TGF-β to altered cellular properties that eventuate in diabetic renal functional and pathological damage; long-term administration of monoclonal anti-TGF-β antibody to genetically diabetic db/db mice prevented the development of glomerular mesangial matrix expansion and renal insufficiency. 7 However, treatment with anti-TGF-β antibody failed to prevent excess albuminuria. These observations suggested that events distinct to activation of TGF-β in the diabetic kidney contributed to derangements in glomerular permeability and the development of proteinuria.
In this context, vascular endothelial growth factor (VEGF) is up-regulated early in diabetes, specifically within podocytes, cells importantly involved in maintenance of glomerular basement membrane structure and integrity. 8 In vivo, blockade of VEGF by administration of neutralizing antibodies to diabetic rats abolished hyperfiltration and partially suppressed increased urinary albumin excretion, without affecting control of hyperglycemia. 9 Importantly, in addition to its effects on vascular permeability, VEGF mediates recruitment/activation of mononuclear phagocytes (MPs) in a receptor-dependent fashion; 10,11 indeed, increased numbers of mononuclear inflammatory cells have been identified in the glomeruli in early experimental diabetes, 12,13 as well as in human diabetic kidney. 14
Taken together, these studies suggested that activation of distinct VEGF and TGF-β axes may contribute importantly to the functional and morphological alterations that result in diabetic nephropathy. It was in this context that we speculated a role for receptor for AGE (RAGE). Expression of receptor for AGE (RAGE) is enhanced in human diabetic kidney. Specifically, in the glomerulus, RAGE is expressed especially at the base of podocytes, but not to an appreciable degree in mesangium nor or in the endothelium. 15 RAGE, a multiligand member of the immunoglobulin superfamily, 16,17 engages ligands implicated in diabetic complications and inflammation, advanced glycation endproducts (AGEs), and members of the S100/calgranulin family. 18 Together with these observations, and the emerging role for podocyte activation in the pathogenesis of diabetic nephropathy, 13,19-27 it was logical to test the hypothesis that activation of RAGE contributed to increased excretion of urinary albumin and attraction of MPs to diabetic glomeruli driven by enhanced expression of podocyte VEGF and other chemoattractants at early times in diabetes, and expansion of mesangial matrix driven by TGF-β; events that intersect in the development of diabetes-associated renal insufficiency. To test these concepts, we used the insulin-resistant hyperglycemic db/db mouse, in which diabetes is uniformly established by 8 weeks of age, and the age-dependent decline in renal function and structural abnormalities of the kidney, parallel, at least in part, those observed in human diabetic nephropathy. 28,29 In addition, we performed studies in homozygous RAGE null mice and strain-matched controls. 30 Diabetes was induced with streptozotocin (stz) and the molecular mediators leading to glomerulosclerosis were examined after 12 weeks of diabetes.
Materials and Methods
Cell Culture
Murine podocytes were generously provided by Dr. Paul Klotman (Mt. Sinai School of Medicine, New York, NY) and cultured as described. 31,32 Undifferentiated cells were grown on tissue culture dishes in RPMI 1640 containing fetal bovine serum (10%), penicillin/streptomycin (Invitrogen, Carlsbad, CA) and γ-interferon (10 units/ml; Sigma, St. Louis, MO) maintained at 33° in the presence of CO2 (5%). To induce differentiation of the cells into mature podocytes, cells were grown at 37°C on dishes coated with type 1 collagen (0.2 mg/ml; Sigma) in the same medium as above except that γ-interferon was omitted.
Animal Studies
All animal studies were conducted with the review and approval of the Institutional Animal Care and Use Committee of Columbia University. Male db/db mice (catalog no. 000642) and heterozygote lean non-diabetic mice (catalog no. 100433) were obtained from the Jackson Laboratories (Bar Harbor, ME) and housed in a standard non-pathogen-free environment; water and chow diet were provided ad libitum and mice were exposed to 12 hour light/dark cycles. At age 8 weeks, db/db mice were divided into two groups. The first group received daily intraperitoneal injections of sterile, endotoxin-free murine soluble RAGE, 18,33,34 50 μg per day. The second group received equal volumes of vehicle, sterile, endotoxin-free phosphate-buffered saline (PBS), pH 7.4. Mice were sacrificed at age 13 weeks and 27 weeks. Non-diabetic m/db mice were used as controls and treated either with vehicle, PBS or murine sRAGE, 50 μg per day as indicated. At sacrifice, body weight was assessed, and blood and both kidneys were retrieved for analysis. In other studies, male RAGE null mice (129/B6) were used. RAGE null mice are viable and display normal reproductive fitness (30; Liliensiek B, Weigand MA, Bierhaus A, Nicklas W, Kaspar M, Hofer S, Plachky J, Grone HJ, Schmidt AM, Yan SD, Martin E, Stern DM, Hammerling GJ, Nawroth PP, Arnold B, manuscript in preparation). Controls included male 129/B6 animals (Jackson Laboratories). Diabetes was induced in control and RAGE null mice using stz, 65 mg/kg, in citrate buffer for 5 consecutive days at age 8 weeks. 33 Control mice received vehicle, citrate buffer. Mice were sacrificed after 12 weeks of diabetes (serum glucose ≥250 mg/dl on two separate occasions) or control state.
Analyses
Blood
At sacrifice, blood was obtained from the retro-orbital sinus. Serum creatinine and blood glucose were determined by Analytics (Gaithersburg, MD). The percentage of glycohemoglobin was determined on lysates prepared from red blood cells using a kit from Wallac (Akron, OH) according to the manufacturer’s protocol.
Urine
Twenty-four hour urine collections were obtained from each animal using metabolic cages. Urine albumin and creatinine and plasma creatinine were determined using Albuwell M and creatinine assays from Exocell (Philadelphia, PA) according to the manufacturer’s instructions. Creatinine clearance was determined by the following formula: (urine creatinine in 24 hours) × (volume of urine in 24 hours)/(serum creatinine). Results are expressed as (ml/hour per 100 gram body weight).
Morphometry
Dissected kidneys were fixed in buffered formalin (10%) overnight and then routinely processed for light microscopy. Fixed paraffin-embedded tissues were cut (3 μm thick) and mounted on slides coated with 3-aminopropyltriethoxy silane (Sigma) followed by incubation at 37°C overnight. Light microscopic views after staining with periodic acid Schiff (PAS) (Sigma) were scanned into a computer and the quantification of areas of mesangial matrix and glomerulus was performed using a Zeiss microscope and image analysis system (MediaCybernetics). To calculate mesangial area, only nuclei-free regions were included. Forty glomeruli from each animal were selected at random on the stained sections (20 from the outer region and 20 from the inner region). Thickness of the glomerular basement membrane (GBM) was determined using small pieces of kidney tissue fixed with glutaraldehyde (2.5%) and processed for electron microscopy. Thickness of the GBM was measured in at least five capillaries for each mouse/condition. Morphometry was performed by investigators blinded to the experimental protocol.
Immunohistochemistry
Polyclonal antibodies against RAGE, 33,34 S100/calgranulins, 18 and affinity-purified antibodies against CML-modified adducts 15 were used as previously described. Antibodies to CML-adducts displayed weak immunoreactivity against carboxy(ethyl)lysine (CEL) adducts in ELISA; however, the affinity of the antibody for CML-adducts was >1000 times that for CEL-adducts in these assays. Antibodies to VEGF (Zymed Labs, South San Francisco, CA), monoclonal antibodies to synaptopodin (reactive with the murine epitope) (Maine Biotechnology, Portland, ME), and antibodies against murine Mac-3 (Sigma) were used and immunostaining performed using kits from Vector (Burlingame, CA). In each case, the immunoreaction was visualized using 3,3′-diaminobenzidine (Sigma) and sections were lightly counterstained with hematoxylin. In all cases, the respective nonimmune IgG for each antibody was used at the same concentration as the primary antibody; no specific immunostaining was observed using nonimmune IgG (data not shown).
Immunoblotting
Snap-frozen kidney cortical tissue was homogenized in PBS containing protease inhibitor mixture (Roche Molecular Biochemicals, Indianapolis, IN). Cultured podocytes were washed three times in ice-cold PBS and harvested into lysis buffer (Tris-HCl, 0.05 mol/L (pH 8.0); NaCl, 0.15 mol/L; Triton X, 1.0%; and PMSF, 0.0005 mol/L). Protein concentrations were measured using the Bio-Rad assay system (Bio-Rad Laboratories, Hercules, CA). Equal amounts of protein were subjected to electrophoresis onto Tris-glycine gels (Invitrogen/Novex, Carlsbad, CA) and the contents of the gels were transferred to nitrocellulose membranes (Invitrogen/Novex). Immunoblotting was performed using rabbit anti-RAGE IgG (5 μg/ml), rabbit anti-S100/calgranulin IgG (5 μg/ml), rabbit anti-murine VEGF IgG (1 μg/ml; Santa Cruz Biotechnology, Santa Cruz, CA), or goat anti-murine VCAM-1 IgG (0.4 μg/ml; Santa Cruz Biotechnology). Horseradish peroxidase-conjugated goat anti-rabbit antibody (1:12,000 dilution) (Sigma) was used to identify the sites of primary antibody binding. The enhanced chemiluminescence (ECL) detection system (Amersham-Pharmacia Biotech, Piscataway, NJ) was used to visualize the immunoreaction. Quantitative analysis of the band density was performed using ImageQuant (Molecular Dynamics, Foster City, CA).
Northern Blotting
Northern blots were performed from RNA extracted from renal cortices and hybridized with 32P-labeled cDNA encoding murine transforming growth factor-β1 (cDNA was generously provided by Dr. F.N. Ziyadeh (University of Pennsylvania)). 6 Total RNA was extracted from snap-frozen tissues using Trizol Reagent (Invitrogen). Total RNA (30 μg) was electrophoresed onto an agarose gel (1%), transferred onto a nylon membrane (Schleicher & Schuell, Keene, NH) and cross-linked by ultraviolet light. The cDNA probes were first labeled with Redivue deoxycytidine-5′-[α-32P]triphosphate (∼6000 ci/mmol; Amersham Pharmacia Biotech) using random-prime labeling cDNA kit (Stratagene, La Jolla, CA), and the membrane was hybridized with the labeled cDNA probe. After hybridization overnight at 68°C, the blots were washed twice for 15 minutes in 2X standard saline citrate (SSC) containing sodium dodecyl sulphate (SDS) (0.1%) at room temperature, followed by two washes for 15 minutes in SSC (0.1X) and SDS (0.1%) at 60°C. The membranes were autoradiographed at −70°C for up to 5 days. The blots were stripped twice (total of 30 minutes) at 100°C in SSC (0.1X) containing SDS (0.1%) and rehybridized with cDNA fragment encoding β-actin. Quantitative analysis of the band density was performed using ImageQuant and levels of TGF-β1 were calculated relative to radioactivity measured for β-actin.
Statistical Analysis
The mean ± SE (SE) of the mean is reported. Statistical significance (defined as P < 0.05) was determined by analysis of variance. Where indicated, post-hoc analysis was used using Dunnett’s t-test using StatView 4.0 (Abacus Concepts, Inc., Berkeley, CA). Results that did not achieve statistical significance are designated in the figures as “NS.”
Results
RAGE and Its Ligands Are Expressed in Murine Diabetic Kidney
In contrast to biological settings in which excess accumulation of ligand is accompanied by diminished levels of cellular receptors, expression of RAGE is enhanced in diabetic tissues in a manner overlapping with that of AGEs and S100/calgranulins, such as in atherosclerotic lesions and infected periodontium. 33,34 In human diabetic kidney, we previously demonstrated enhanced expression of RAGE in diabetic glomeruli, particularly within podocytes, compared with age-matched euglycemic controls. 15 Consistent with the hypothesis that early activation of RAGE in diabetic kidney may modulate processes involved in the development of nephropathy, renal cortical tissue retrieved from the kidneys of db/db mice at age 13 weeks displayed increased RAGE antigen by immunoblotting compared with non-diabetic, m/db mice, of the same age (Figure 1a ▶ ; P < 0.05). Immunohistochemistry revealed that the principal site of RAGE expression in the glomerulus of the db/db mouse was the podocyte (Figure 1, b and c) ▶ , as confirmed by co-localization experiments using anti-synaptopodin IgG (data not shown). In contrast, no appreciable staining for RAGE epitopes was detected in mesangium or other cellular structures within the glomerulus. To further demonstrate that podocytes express RAGE, we studied the expression of podocyte RAGE in vitro, using a conditionally immortalized murine podocyte cell line derived from a transgenic mouse expressing a temperature-sensitive SV 40 large T antigen. 31,32 Although RAGE antigen was detectable in undifferentiated podocytes, differentiation of these cells on incubation at 37°C in the absence of γ-interferon resulted in an ∼4-fold increase in RAGE antigen (Figure 1d ▶ ; P < 0.05). Further, we assessed expression of RAGE antigen in non-permeabilized differentiated cultured podocytes by immunohistochemistry. Figure 1e ▶ depicts a single podocyte with arborized processes; RAGE immunoreactivity was evident on the cell surface and appeared to be largely localized to podocyte processes. Although this localization in vitro does not indicate the precise subcellular localization in vivo in the mature podocytes of intact glomeruli, these findings demonstrate that RAGE antigen is detectable by immunoblotting and immunohistochemistry in cultured podocytes.
Figure 1.

Expression of RAGE antigen is enhanced in db/db kidney and localizes to the podocyte at age 13 weeks. a: Immunoblotting. Male control (m/db) and diabetic (db/db) mice were studied. At age 8 weeks, db/db mice were treated once daily with murine sRAGE (50 μg) by intraperitoneal route, or with equal volumes of PBS. Mice were sacrificed at age 13 weeks and renal cortical tissue retrieved for SDS/PAGE and immunoblotting with rabbit anti-RAGE IgG. The approximate Mr of standard molecular weight markers is shown. *, P < 0.05. b and c: Immunohistochemistry. Renal cortical tissue was subjected to immunohistochemistry using anti-RAGE IgG. RAGE antigen was principally expressed in the podocyte as confirmed by co-localization using anti-synaptopodin IgG (not shown). The illustrated immunostaining is representative of n = 5 mice/condition. Bar, 16 μm. d and e: In vitro studies of podocyte RAGE. d: Immunoblotting. Cultured murine podocytes were grown under conditions for propagation vs. differentiation. Cells were harvested and subjected to SDS/PAGE and immunoblotting with anti-RAGE IgG. The approximate Mr of standard molecular weight markers is shown. *, P < 0.05. e: Immunohistochemistry. Cultured murine podocytes, under differentiating conditions, were subjected to immunostaining with anti-RAGE IgG. Part e displays a single differentiated cultured murine podocyte with arborized processes; RAGE antigen appears to be localized to the peripheral processes. In both a and d, representative bands from each condition are shown; immunoblots from n = 3 mice or n = 3 experiments per condition were performed. Mean ± SE of densitometric analysis is reported.
These studies established that expression of RAGE was increased in diabetic murine kidney, especially in the podocyte. As activation of RAGE occurs on engagement of its ligands, we sought to identify ligand expression and accumulation in db/db kidney. AGEs are a heterogenous class of structures. Recent studies suggested that a principal and specific AGE found in vivo, carboxymethyl (lysine) (CML) adducts of proteins/lipids, 35-37 are signal transduction ligands of RAGE. 37 Consistent with previous observations in human diabetic nephropathy, 36,38 immunohistochemistry revealed increased accumulation of CML-epitopes in db/db kidney, especially within the mesangium, glomerular basement membranes and vascular structures compared with control, m/db mice at age 13 weeks (Figure 2, b and a ▶ , respectively).
Figure 2.
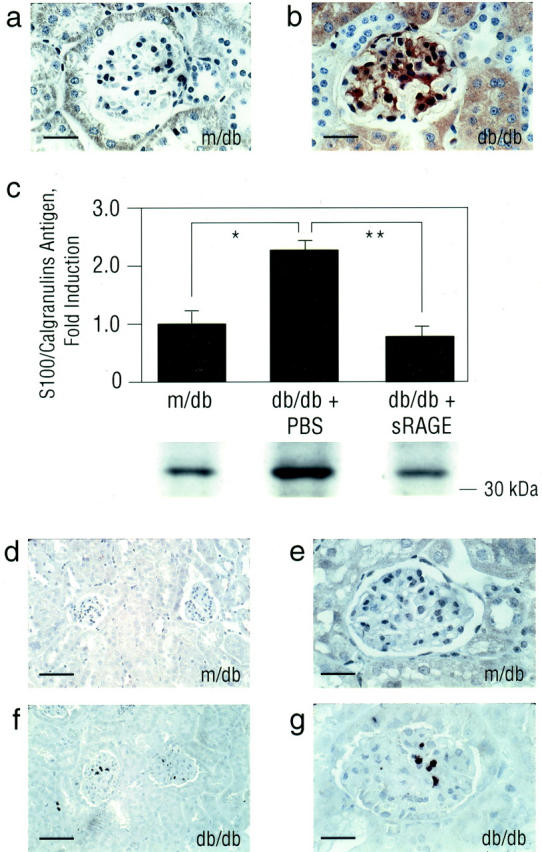
Expression of CML epitopes and S100/calgranulins is enhanced in db/db kidney at age 13 weeks. a and b: Immunohistochemistry. Renal tissue from m/db or db/db mice at age 13 weeks was subjected to immunohistochemistry using affinity-purified anti-CML IgG. Bar, 16 μm. c: Immunoblotting. M/db and PBS- vs. sRAGE-treated db/db mice were sacrificed at age 13 weeks and renal cortical tissue retrieved for SDS/PAGE and immunoblotting using rabbit anti-S100/calgranulin IgG. The approximate Mr of standard molecular weight markers is shown. Representative bands from each condition are shown; immunoblots from n = 3 mice per condition were performed. Densitometric analysis is indicated, and mean ± SE is reported. *, P < 0.05; and **, P < 0.01. d–g: Immunohistochemistry. Renal tissue from m/db or db/db mice was subjected to immunohistochemistry using anti-S100/calgranulin IgG. S100/calgranulins were principally expressed in mononuclear phagocytes infiltrating the glomerulus/interstitium as confirmed by co-localization studies using anti-Mac 3 IgG (not shown). Bar; d and f, 40 μm; and bar; e and g, 16 μm. In a, b, d, e, f, and g, immunostaining is representative of n = 7 mice/condition.
RAGE also interacts with S100/calgranulins, members of a family of proinflammatory cytokines. These molecules are principally located intracellularly, in inflammatory cells such as polymorphonuclear leukocytes, mononuclear phagocytes and lymphocytes, and neuronal cells. 39,40 However, on cellular activation, S100/calgranulins may be released from these cells, thus freeing them to engage cell surface receptors such as RAGE. 18 Immunoblotting of renal cortical tissue retrieved at age 13 weeks revealed an ∼2.3-fold increase in expression of S100/calgranulin antigen in db/db mice compared with m/db controls (Figure 2c ▶ ; P < 0.05). S100/calgranulin antigens were principally expressed in inflammatory cells infiltrating the glomerulus and interstitium in db/db mice compared with non-diabetic m/db animals (Figure 2, f and g, and 2, d and e ▶ , respectively). Co-localization studies using anti-Mac 3 IgG indicated that these S100/calgranulin-expressing cells were MPs (data not shown).
Taken together, these observations suggested that RAGE and its ligands, AGEs (CML) and S100/calgranulins, although detectable only at low levels in the absence of diabetes, were present to enhanced degrees in the kidney after 5 weeks of diabetes. As the principal site of increased glomerular RAGE expression was in the podocyte, we tested the hypothesis that engagement of RAGE by AGEs and S100/calgranulins within the glomerulus early in diabetes caused podocyte activation, thereby triggering a series of events leading, ultimately, to altered renal function and glomerulosclerosis. A first test was to determine whether increased expression of VEGF, principally expressed in the podocyte in diabetic kidney, 8 was RAGE-dependent. To study this, m/db and db/db mice were treated with murine soluble (s) RAGE, the extracellular ligand-binding domain of the receptor which acts as a decoy to intercept ligand and block access to cell surface RAGE. 18,33,34 Mice received once daily intraperitoneal administration of murine sRAGE, 50 μg, or equal volumes of vehicle, phosphate-buffered saline (PBS). Treatment commenced at age 8 weeks, at which time hyperglycemia was documented in all animals, and was continued until the time of sacrifice. At age 13 weeks, renal cortical tissue retrieved from db/db mice displayed VEGF antigen by immunoblotting (Figure 3a) ▶ . Consistent with previous observations in experimental diabetes, the principal site of VEGF expression in diabetic kidney was the podocyte (Figure 3, b and c) ▶ , as demonstrated by studies using anti-synaptopodin IgG (Figure 3d) ▶ . Importantly, treatment of db/db mice treated with sRAGE from age 8 to 13 weeks prevented the increase in expression of VEGF antigen in the kidney at 13 weeks (Figure 3a ▶ ; P < 0.01). However, treatment of m/db mice with sRAGE had no effect on levels of VEGF antigen (Figure 3e) ▶ . We extended these studies to cultured podocytes to determine whether activation of RAGE in these cells modulated expression of VEGF. Compared to incubation with irrelevant protein (IgG), incubation of differentiated podocytes with S100B 18 resulted in increased expression of VEGF antigen; P < 0.05. That podocyte RAGE was importantly involved in S100B-mediated effects was demonstrated by suppression of VEGF expression in the presence of higher doses of either excess sRAGE (P < 0.05) or anti-RAGE IgG (P < 0.05), but not by lower doses of excess sRAGE or anti-RAGE IgG, nor by nonimmune IgG; P > 0.05 (Figure 3f) ▶ .
Figure 3.
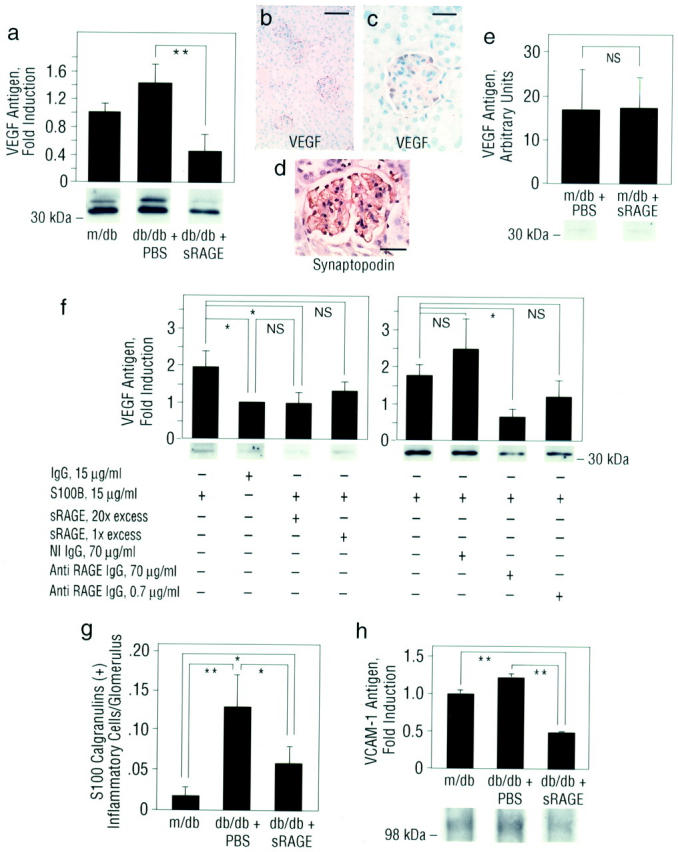
Expression of VEGF and VCAM-1 antigens is enhanced in db/db kidney at age 13 weeks: prevention by blockade of RAGE. a, e, and h: Immunoblotting. Control m/db and db/db mice were sacrificed at age 13 weeks and renal cortical tissue retrieved for SDS/PAGE and immunoblotting using anti-murine VEGF IgG (a and e) or anti-murine VCAM-1 IgG (h). The approximate Mr of standard molecular weight markers is shown. Representative bands from each condition are shown; immunoblots from n = 3 mice per condition were performed. Densitometric analysis is indicated, and mean ± SE is reported. **, P < 0.01. b–d: Immunohistochemistry. Renal cortical tissue from db/db mice was subjected to immunohistochemistry using anti-murine VEGF IgG. VEGF antigen (b and c) was specifically expressed in the podocytes as confirmed by co-localization using anti-synaptopodin IgG (d). The illustrated immunostaining is representative of n = 3 mice/condition. Bar: b, 40 μm; c and d, 15 μm. f: Cultured podocytes. Differentiated cultured murine podocytes were exposed to irrelevant IgG or S100B (15 μg/ml) for 6 hours. Where indicated, S100B was preincubated with molar excesses of sRAGE (20- or onefold) for 2 hours, or podocytes were preincubated with anti-RAGE IgG or nonimmune IgG (70 or 0.7 μg/ml) for 2 hours before addition of S100B. Cells were lysed and SDS/PAGE and immunoblotting performed using anti-VEGF IgG. The approximate Mr of standard molecular weight markers is shown. These experiments were performed at least three times; *, P < 0.05. g: Immunohistochemistry and quantification of infiltrating inflammatory cells. The mean number of S100/calgranulin-expressing inflammatory cells ± SE infiltrating the glomeruli in randomly chosen 30 glomeruli per condition (n = 3 to 4 mice/condition) is reported. Co-localization studies using anti-Mac 3 IgG indicated that the infiltrating cells were MPs (not shown). *, P < 0.05; and **, P < 0.01.
In addition to modulation of permeability, VEGF mediates attraction/activation of MPs. 10,11 Consistent with earlier observations in experimental models and human diabetes, an ∼6.5-fold increase in S100/calgranulin expressing inflammatory cells per glomerulus was noted in db/db mice compared with controls (Figure 3g ▶ ; P < 0.01). Immunostaining using anti-Mac 3 IgG confirmed that MPs were the principal cells expressing S100/calgranulin within the glomerulus (data not shown). That RAGE was centrally involved in attracting increased numbers of inflammatory cells to the glomerulus was shown by the significant attenuation in numbers of MPs infiltrating diabetic glomeruli from sRAGE-treated mice compared with those mice receiving PBS (Figure 3g ▶ ; P < 0.05). It was likely, however, that VEGF was not the sole trigger in diabetic kidney mediating increased attraction and infiltration of inflammatory cells. As previous studies demonstrated that engagement of RAGE by AGE/CML epitopes or S100/calgranulins resulted in increased expression of vascular cell adhesion molecule-1 (VCAM-1), 18,41 we assessed levels of this proinflammatory adhesion molecule in diabetic glomeruli. Immunoblotting revealed that VCAM-1 epitopes in db/db mice treated with sRAGE were significantly lower than those levels observed in vehicle-treated diabetic mice at age 13 weeks (Figure 3h ▶ ; P < 0.01). In contrast, treatment of control m/db mice with sRAGE did not affect levels of VCAM-1 antigen (data not shown). These observations support the premise that early up-regulation of RAGE contributes to enhanced influx of inflammatory cells in diabetic kidney, as blockade of RAGE from the time of initial development of hyperglycemia prevented these phenomena. Importantly, in parallel with decreased expression of VEGF and VCAM-1 epitopes, and decreased numbers of infiltrating MPs, levels of RAGE (Figure 1a ▶ ; P < 0.05) and S100/calgranulin antigens (Figure 2c ▶ ; P < 0.01) were decreased in renal cortical tissue retrieved from sRAGE-treated db/db mice at age 13 weeks compared with vehicle-treated controls. In contrast, treatment of m/db mice with sRAGE did not result in decreased levels of RAGE or S100/calgranulins in m/db animals vs. those treated with PBS at age 13 weeks (data not shown).
These results suggested that blockade of RAGE in db/db mice prevented enhanced expression of factors capable of mediating, at least in part, glomerular hyperpermeability and MP infiltration/activation. An important test of these concepts, however, was the extent to which long-term blockade of RAGE might impact on activation of the TGF-β axis, a critical mediator of mesangial expansion and glomerulosclerosis in diabetic kidney. To study this, additional groups of db/db mice were treated once daily with sRAGE, or vehicle, PBS, beginning at age 8 weeks and continued until age 27 weeks. In parallel with decreased levels of RAGE (Figure 4a ▶ ; P < 0.05) and S100 calgranulin antigens (Figure 4b ▶ ; P < 0.05) in renal cortical tissue retrieved from sRAGE-treated db/db mice compared with mice receiving PBS, increased expression of mRNA encoding TGF-β1 was significantly suppressed in cortical tissue retrieved from sRAGE-treated db/db mice at 27 weeks (Figure 4c ▶ ; P < 0.01). We performed PAS-staining of renal cortical tissue retrieved from md/b and db/db animals; consistent with our observations regarding expression of TGF-β1, although glomerular and mesangial area, as well as mesangial matrix fraction were significantly increased in db/db mice compared with m/db animals (Figure 4, d, e, and f ▶ and Figure 4, h and g ▶ , respectively; P < 0.00001), those db/db mice treated with sRAGE displayed decreased glomerular and mesangial area, in parallel with mesangial matrix fraction compared with db/db mice treated with vehicle (Figure 4, d, e, and f ▶ ; and Figure 4, i and h ▶ , respectively; P < 0.00001). In addition, expression of VEGF antigen, although increased in PBS-treated db/db mice compared to m/db controls, was significantly suppressed in diabetic mice treated with sRAGE; P < 0.05 (Figure 5a) ▶ . PBS-treated db/db mice displayed increased thickness of the GBM compared with non-diabetic controls (Figure 5, b, d, and c ▶ , respectively). Importantly, treatment of diabetic mice with sRAGE prevented the increase in GBM thickness at 27 weeks in db/db mice compared with mice treated with vehicle, PBS (Figure 5, b, e, and d ▶ ; P < 0.05).
Figure 4.

Expression of RAGE and S100/calgranulin antigens, and mRNA for TGF-β1 is enhanced in db/db kidney at age 27 weeks: suppression by blockade of RAGE. a and b: Immunoblotting. Renal cortical tissue was retrieved from m/db or PBS- vs. sRAGE-treated db/db mice at age 27 weeks. SDS/PAGE and immunoblotting were performed using rabbit anti-RAGE IgG (a) or anti-S100/calgranulin IgG (b). The approximate Mr of standard molecular weight markers is shown. Representative bands from each condition are shown; in a, immunoblots from n = 6 mice per condition were performed, and in b, n = 3 mice/condition were used. Densitometric analysis is indicated, and mean ± SE is reported. *, P < 0.05, and **, P < 0.01. c: Northern blotting. At age 27 weeks, renal cortical tissue was retrieved from the indicated mice and Northern blotting using labeled probes to either TGF-β1 or β-actin performed. Representative bands from n = 3 mice per condition are shown. Densitometric analysis is indicated, and mean ± SE is reported. **, P < 0.01. d–i: Morphometric analysis. Glomeruli were examined from PAS-stained sections (g, h, and i) and morphometric analysis performed for glomerular area (d), mesangial area (e), and mesangial matrix fraction (f). Numbers of mice per condition were: 3 m/db, 5 db/db + PBS, and 4 db/db + sRAGE. Mean ± SE is reported. ****, P < 0.0001. In g, a representative PAS-stained section from a non-diabetic m/db mouse is shown; in h and i, representative PAS-stained sections from db/db mice treated with PBS vs. sRAGE, respectively, is displayed. Note that PAS-positive material in PBS-treated db/db mice exceeds that seen in m/db mice and in sRAGE-treated db/db animals of the same age. Bar, 25 μm.
Figure 5.

Levels of VEGF antigen and GBM thickness are enhanced in db/db kidney at age 27 weeks: suppression by blockade of RAGE. a: Immunoblotting. At age 27 weeks, renal cortical tissue was retrieved from the indicated mice and immunoblotting performed using anti-VEGF IgG. Representative bands from n = 3 mice per condition are shown. Densitometric analysis is indicated, and mean ± SE is reported. *, P < 0.05. b–e: GBM thickness. At age 27 weeks, kidneys were retrieved from the indicated mice and electron microscopy performed for measurement of GBM thickness (c, d, and e). N = 6 to 7 mice/condition. The mean ± SE is reported. *, P < 0.05, **, P < 0.01; and ****, P < 0.0001. In d, note that the thickness of the GBM in PBS-treated db/db mice exceeds that observed in either non-diabetic m/db mice (c) or sRAGE-treated db/db mice of the same age (e). Magnification, ×5000.
Taken together, these observations suggested that RAGE blockade in db/db mice suppressed enhanced expression of factors driving hyperpermeability, MP infiltration/activation, and mesangial and GBM expansion. The key test of these concepts, however, was whether blockade of the receptor and these mediators was linked to preservation of renal function. At age 27 weeks, increased urinary albumin excretion observed in PBS-treated db/db mice, 0.20 ± 0.6 μg albumin/μg creatinine, was largely prevented in the presence of sRAGE, 0.11 ± 0.3 μg albumin/μg creatinine (Figure 6a ▶ ; P < 0.05). Indeed, the amount of urinary albumin observed in db/db mice treated with sRAGE was not significantly different from that seen in control m/db animals (0.08 ± 0.01 albumin/μg creatinine) (Figure 6a ▶ ; P > 0.05). Furthermore, although creatinine clearance was decreased in PBS-treated db/db mice compared with control m/db animals (3.03 ± 0.47 vs. 6.75 ± 0.31 ml/hour/100 × g body weight, respectively; P < 0.001), significantly increased creatinine clearance was observed in db/db mice treated with sRAGE (5.16 ± 0.50 ml/hour/100 × g body weight; P < 0.05 vs. db/db + PBS) (Figure 6b) ▶ . Importantly, the effects of RAGE blockade in db/db mice were independent of changes in body weight, urine volume, serum glucose, or glycosylated hemoglobin (Table 1) ▶ .
Figure 6.

Renal function is diminished in db/db mice at 27 weeks: prevention by blockade of RAGE. At age 27 weeks, mice were placed in metabolic cages and urine/serum collected. Albuminuria (a) and creatinine clearance (b) were determined. In both a and b, n = 5 mice/condition. The mean ± SE is reported. *, P < 0.05, **, P < 0.01, and ***, P < 0.001.
Table 1.
Characteristics of m/db and db/db Mice: Age 27 Weeks
| m/db | db/db (PBS) | db/db (sRAGE) | |
|---|---|---|---|
| Body weight (gm) | 29.3 ± 0.3 (6) | 59.5 ± 0.8* (7) | 58.4 ± 0.7* (7) |
| Serum/Red blood cell | |||
| Serum creatinine (mg/dl) | 0.5 ± 0.04 (5) | 0.6 (5) | 0.4 ± 0.04† (5) |
| Serum glucose (mg/dl) | 178.2 ± 12.4 (5) | 623.8 ± 79.0‡ (5) | 447.2 ± 96.9§ (5) |
| Glycosylated hemoglobin (%) | 5.6 ± 0.3 (3) | 9.6 ± 1.0‡ (3) | 9.4 ± 0.9§ (3) |
| Urine | |||
| Urine volume (ml) | 0.6 ± 0.1 (5) | 1.2 ± 0.2§ (5) | 1.2 ± 0.2§ (5) |
At age 8 weeks, db/db mice were treated with either sRAGE (50 μg/day) or vehicle (PBS). At age 27 weeks, control m/db, db/db (vehicle) and sRAGE-treated db/db mice were assessed for body weight and the following indices in serum, red blood cells and urine. Values are expressed as mean ± SE. (The number of mice studied per condition is indicated in parentheses after each value).
*, p < 0.001 vs. m/db mice.
†, p < 0.05 vs. PBS-treated db/db mice.
‡, p < 0.01 vs. m/db mice.
§, p < 0.05 vs. m/db mice.
To confirm a central role for RAGE in the diabetes-associated nephropathy, we used homozygous RAGE null mice. 30 Homozygous RAGE null mice are viable and display normal fertility and lifespan. These mice provide a means to test the effects of global deletion of RAGE on the molecular mediators underlying the pathogenesis of diabetic nephropathy. RAGE null mice (129/B6) and strain-matched controls were rendered diabetic with streptozotocin or treated with vehicle. Mice displaying serum glucose ≥250 mg/dl were studied; >95% of RAGE null mice and strain-matched controls became diabetic. Mice were sacrificed after 12 weeks of diabetes to identify the early effects of RAGE deletion in the diabetic kidney. Although diabetic strain-matched controls displayed an ∼1.6-fold increase in kidney/body weight ratio (P < 0.00001) compared to their non-diabetic littermates after 12 weeks of diabetes, no significant increase in this parameter was evident in diabetic RAGE null mice vs. euglycemic mice lacking RAGE; P > 0.05 (Figure 7) ▶ .Consistent with our earlier studies using sRAGE in db/db mice, diabetic RAGE null mice failed to demonstrate increased VEGF antigen in the kidney cortex compared to non-diabetic mice of the same genotype; P > 0.05 (Figure 7b) ▶ . In contrast, strain-matched control mice displayed an ∼1.9-fold increase in VEGF antigen after 12 weeks of diabetes compared to non-diabetic strain-matched controls; P < 0.05 (Figure 7b) ▶ .
Figure 7.

Diabetic RAGE null mice do not display increased VEGF antigen or mRNA for TGF-β in the renal cortex. a: Kidney weight. Diabetes (or control) was induced in RAGE null mice (129/B6) and strain-matched controls. The numbers of mice used for these studies were as follows: 8 RAGE null (without diabetes), 9 RAGE null (with diabetes), 14 strain-matched control (without diabetes), and 13 strain-matched control (with diabetes). After 12 weeks of diabetes, mice were sacrificed. Kidney weight/body weight ratio is reported. *****, P < 0.00001. b: VEGF antigen. At sacrifice, renal cortical tissue was prepared from the indicated mice and immunoblotting performed using anti-VEGF IgG. *, P < 0.05. c: Northern blotting. Renal cortical tissue was prepared from RAGE null mice and strain-matched controls using labeled probes to TGF-β or β-actin; *, P < 0.05 and **, P < 0.01. In b and c, representative bands from n = 3 mice per condition are shown. Densitometric analysis is indicated, and mean ± SE is reported.
Northern blot studies revealed that diabetic strain-matched controls displayed an ∼1.5-fold increase in mRNA for TGF-β1 in the cortex of the kidney compared to non-diabetic controls after 12 weeks; P < 0.01 (Figure 7c) ▶ . Levels of TGF-β1 mRNA were not significantly different, however, in diabetic vs. euglycemic RAGE null mice; P > 0.05 (Figure 7c) ▶ . Consistent with these observations, although diabetic strain-matched controls (n = 13) demonstrated an ∼18% increase in glomerular area, and an ∼61% increase in mesangial area compared to non-diabetic controls (n = 14); P < 0.001, diabetic RAGE null mice (n = 9) displayed an ∼5% increase in glomerular area, and a −6% change in mesangial area compared to non-diabetic RAGE null animals (n = 8); P > 0.05.
Furthermore, after 12 weeks of diabetes in wild-type mice (n = 4), GBM thickness was increased 14% compared to non-diabetic controls (n = 4); P < 0.01. In contrast, induction of diabetes in RAGE null mice (n = 4) resulted in a ∼3% increase in GBM thickness compared to non-diabetic mice lacking RAGE (n = 4); P > 0.05. Importantly, mean levels of blood glucose in diabetic RAGE null mice and diabetic strain-matched controls were not significantly different. Mean levels of blood glucose at sacrifice in diabetic RAGE null vs. diabetic strain-matched controls were 441.4 ± 139.9 and 473.0 ± 76.7 mg/dl, respectively; P > 0.05. Similarly, mean levels of blood glucose did not differ between citrate-treated non-diabetic RAGE null and control mice; 106.7 ± 20.8 and 96.2 ± 18.1 mg/dl, respectively; P > 0.05.
Discussion
The observation by Ziyadeh and colleagues 7 that long-term treatment of db/db mice with blocking antibody to TGF-β suppressed excess matrix gene expression, glomerular mesangial matrix expansion, and prevented the development of renal insufficiency in the absence of significant modulation of albuminuria suggests that axes distinct from TGF-β and its receptors may contribute to the events leading to diabetes-associated renal disease. Here, we propose that hyperglycemia triggers AGE-mediated enhanced expression of podocyte VEGF via RAGE, thereby providing a stimulus for the development of hyperpermeability, as well as attraction of inflammatory cells such as MPs to the glomerulus. We propose that ligands of RAGE, as well as products of activated inflammatory cells, such as low molecular weight cytokines and growth factors, may be driven across the glomerular capillary wall by outwardly directed filtration forces, facilitating their interaction with podocytes, thereby promoting increased GBM synthesis and altered glomerular permselectivity. Previous studies indicated that AGEs mediated increased expression of VEGF in both in vitro and in vivo analyses. 42-45 In the present experiments, we have shown that incubation of RAGE-bearing cultured murine podocytes with a prototypic RAGE ligand resulted in increased expression of VEGF, in a manner suppressed by sRAGE or blocking antibodies to the receptor. Indeed, in their study, Ziyadeh and colleagues 7 found that in db/db mice treated with blocking antibody to TGF-β, levels of VEGF mRNA in the kidney were only slightly decreased, supporting the concept that distinct effector axes may regulate enhanced expression of this permeability and chemoattractant factor. Together with RAGE-mediated increased expression of VCAM-1, as well, perhaps, as other chemoattractant molecules, we propose that enhanced attraction/activation of MPs drives the generation of a number of toxic and growth-promoting substances such as proteolytic enzymes, cytokines, reactive oxygen species (ROS), cyclooxygenase and lipoxygenase products, platelet activating factors, and growth factors, that may activate the mesangium, thereby leading to further damage of glomerular structure and impingement on renal function. 46,47 Figure 8 ▶ illustrates a proposed scheme by which hyperglycemia-mediated generation of AGEs leads to up-regulation and activation of podocyte RAGE; processes linked to enhanced expression of VEGF and its consequences for permeability as well as MP attraction/activation.
Figure 8.

Hypothetical schema of consequences of podocyte RAGE activation in the pathogenesis of diabetic nephropathy. We hypothesize that in hyperglycemia, accelerated generation of ligands of RAGE, AGEs, and S100/calgranulins, leads to up-regulation of podocyte RAGE; an important consequence of which is increased expression/activity of VEGF. Increased VEGF activity leads to both hyperpermeability and proteinuria, as well as attraction and activation of MP within the diabetic glomerulus. In addition, engagement of podocyte RAGE by its ligands further leads to increased expression of VCAM-1. We propose that increased expression of VEGF and VCAM-1 leads to increased migration and activation of MPs within the glomerulus. Activated MP may then release a range of mediators and species linked to mesangial activation; a critical consequence of which is enhanced generation of TGF-β, culminating in glomerular sclerosis. In addition, activated MP may release further S100/calgranulins, thereby setting up a chronic cascade of ligand up-regulation and receptor activation. Taken together, we propose that RAGE-mediated proteinuria and glomerular sclerosis contribute to diabetes-associated renal dysfunction.
In this context, it is important to note that recent studies in cultured murine podocytes suggest a complementary/alternate hypothesis; Iglesias-De La Cruz and colleagues 48 reported that differentiated cultured podocytes grown in the presence of either high levels of glucose (25 mmol/L) or TGF-β1 (2.0 ng/ml) resulted in increased expression of α1 and α5(IV) collagen and VEGF. These observations underscore the concept that sustained hyperglycemia may contribute to mechanisms by which podocyte perturbation contributes to diabetic nephropathy. In vivo, however, the complexity of the interplay of the TGF-β/VEGF axes is demonstrated by the observation that despite blockade of the TGF-β axis in db/db mice using blocking monoclonal antibodies, levels of mRNA for VEGF were only slightly decreased, 7 thus suggesting that at least in part, up-regulation of VEGF may be independent of the action of TGF-β.
Evidence suggests that, once set in motion, the inexorable increase in proteinuria in diabetes itself is a potent trigger for ongoing glomerular damage and renal dysfunction, as enhanced exposure of tubular cells to urinary protein has been linked to activation of NF-κB, as well as increased expression of chemokines such as RANTES and monocyte chemoattractant protein-1 (MCP-1). 49-52 Although these processes are likely mediated, at least in part via TGF-β, our findings suggest that earlier in the events that typify diabetic nephropathy, activation of RAGE sets the stage for both excess albuminuria as well as glomerulosclerosis and renal insufficiency.
Supportive of an important role for RAGE in induction of pathways leading to tubulointerstitital sclerosis in the diabetic kidney is the recent observation that AGEs mediated epithelial-myofibroblast transdifferentiation via RAGE in cultured renal proximal tubular epithelial cells. 53 In addition, Yamamoto and colleagues 54 prepared transgenic mice expressing full-length RAGE in endothelial cells (and to some extent in mononuclear phagocytes) to show that when bred with a diabetes-susceptible strain, albuminuria and glomerulosclerosis ensued. Although specific molecular indices of permeability and fibrosis were not examined in the latter study, these observations, together with the present findings, suggest key roles for RAGE in the development of albuminuria and glomerulosclerosis and highlight new insights into early and global mechanisms underlying scarring of the renal cortex in diabetes. Indeed, the recent observation that long-term treatment of normotensive subjects with type 1 diabetes and albuminuria with inhibitors of angiotensin converting enzyme inhibitors or nifedipine did not prevent disease progression, in terms of renal structure, albuminuria, or decline in glomerular filtration rate, 55 suggests that fundamental elements of cellular activation that portend advancement of disease are set in motion long before the appearance of albuminuria. 56,57
Together with our findings in db/db mice, in which blockade of RAGE was accomplished using soluble receptor, our studies in RAGE null mice support an important role for this molecule in the pathogenesis of diabetic nephropathy. Diabetic RAGE null animals did not display increased expression of VEGF or TGF-β in the renal cortex. Studies to assess the effects of deletion of RAGE on renal function will be best accomplished by breeding RAGE null animals into the db/db background. These studies, although underway, will require extended periods of time to generate animals sufficiently back-crossed for accurate interpretation of the results. In addition, a limitation of our studies in RAGE null mice is that we did not distinguish the specific cellular contributions of RAGE-bearing macrophages or podocytes (or, perhaps, other cell types) in these processes. Studies are underway to generate transgenic mice bearing dominant-negative RAGE 18,37 selectively in cells of mononuclear phagocyte lineage, or in podocytes. As RAGE is primarily expressed by the podocyte in diabetic glomeruli, it will be especially critical to generate the latter group of mice to rigorously test these hypotheses. Thus, in comparison to the studies of Yamamoto, 54 targeted expression of RAGE mutants in cells endogenously expressing the receptor in human/murine diabetes (podocytes and MPs) should mimic as closely as possible the naturally occurring setting.
Lastly, important roles for activation of podocytes in other forms of nephropathy have been suggested. For example, in rats with experimentally induced passive Heymann nephritis (PHN) and proteinuria in the glomerulus, podocytes selectively expressed cytochrome b558, an essential component of an NADPH-oxidoreductase enzymatic complex. In PHN, podocytes undergo an oxidative respiratory burst with release of ROS into GBMs. These events were linked to the development of proteinuria, 58 as treatment with scavengers of ROS assuaged the development of proteinuria. 59,60 These observations have striking parallels to the biology of RAGE. Engagement of RAGE activates NADPH oxidase species in endothelial cells and MPs, thereby triggering signal transduction mechanisms such as p21ras, erk 1/2 MAP kinases, and NF-κB in an oxidant-sensitive manner. 61-63 It is possible that the podocyte is not an innocent bystander amid distinct derangements in the diabetic glomerulus, but, rather, a key inciting participant, at least in part driven by its enhanced expression of RAGE in a ligand-enriched environment, in the events that lead to scarring and failure of the kidney in diabetes.
Acknowledgments
We thank Drs. Gabriel Godman and Giuseppe Andres for the invaluable advice and suggestions in this work.
Footnotes
Address reprint requests to Dr. Ann Marie Schmidt, Departmentof Surgery, College of Physicians & Surgeons, Columbia University, 630 West 168th Street, P&S 17–401, New York, NY 10032. E-mail: ams11@columbia.edu.
Supported, in part, by the Surgical Research Fund of the College of Physicians & Surgeons, Columbia University, and by grants from Juvenile Diabetes Research Fund International (no. 4200–945) to D.M.S. and A.M.S. T.M.W. is a recipient of a grant from Deutsche Forschungsgemeinschaft (DFG) (We 2450/1–1). P.P.N. was supported by grants from the DFG (Na 138). A.M.S. is a recipient of a Burroughs Wellcome Fund Clinical Scientist Award in Translational Research (APP no. 2602).
Thoralf M. Wendt and Nozomu Tanji contributed equally to this work.
References
- 1.Ritz E, Rychilik I, Locatelli F, Halimi S: End-stage renal failure in type 2 diabetes: a medical catastrophe of worldwide dimension. Am J Kidney Dis 1999, 34:795-808 [DOI] [PubMed] [Google Scholar]
- 2.Koch M, Kutkuhn B, Grabensee B, Ritz E: Apolipoprotein A, fibrinogen, age, and history of stroke are predictors of death in dialyzed diabetic subjects: prospective study of 412 subjects. Nephrol Dial Transplant 1997, 12:2603-2611 [DOI] [PubMed] [Google Scholar]
- 3.Yamamoto T, Nakamura T, Noble NA, Ruoslahti E, Border WA: Expression of transforming growth factor β is elevated in human and experimental diabetic nephropathy. Proc Natl Acad Sci USA 1993, 90:1814-1818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Border WA, Noble NA: Transforming growth factor β in tissue fibrosis. N Engl J Med 1994, 331:1286-1292 [DOI] [PubMed] [Google Scholar]
- 5.Wolf G, Hamann A, Han DC, Helmchen U, Thaiss F, Ziyadeh FN, Stahl RA: Leptin stimulates proliferation and TGF-β expression in renal glomerular endothelial cells: potential role in glomerulosclerosis. Kidney Int 1999, 56:860-872 [DOI] [PubMed] [Google Scholar]
- 6.Sharma K, Jin Y, Guo J, Ziyadeh FN: Neutralization of TGF-β by anti-TGF-β antibody attenuates kidney hypertrophy and the enhanced extracellular matrix gene expression in stz-induced diabetic mice. Diabetes 1996, 45:522-530 [DOI] [PubMed] [Google Scholar]
- 7.Ziyadeh FN, Hoffman BB, Han DC, Iglesias-De La Cruz MC, Hong SW, Isono M, Chen S, McGowan TA, Sharma K: Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-β antibody in db/db diabetic mice. Proc Natl Acad Sci USA 2000, 97:8015-8020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cooper ME, Vranes D, Youssef S, Stacker SA, Cox AJ, Rizkalla B, Casley DJ, Bach LA, Kelly DJ, Gilbert RE: Increased renal expression of vascular endothelial growth factor (VEGF) and its receptor VEGFR-2 in experimental diabetes. Diabetes 1999, 48:2229-2239 [DOI] [PubMed] [Google Scholar]
- 9.De Vriese A, Tilton R, Vanholder R: Hyperfiltration and albuminuria in diabetes: role of vascular endothelial growth factor (VEGF). J Am Soc Nephrol 1999, 10:A3434(Abstract) [Google Scholar]
- 10.Clauss M, Gerlach M, Gerlach H, Brett J, Wang F, Familletti PC, Pan YC, Olander JV, Connolly DT, Stern D: Vascular permeability factor: a tumor-derived polypeptide that induces endothelial cell and monocyte procoagulant activity, and promotes monocyte migration. J Exp Med 1990, 172:1535-1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barleon B, Sozzani S, Zhou D, Weich HA, Mantovani A, Marme D: Migration of monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood 1996, 87:3336-3343 [PubMed] [Google Scholar]
- 12.Sassy-Prigent C, Heudes D, Mandet C, Belair MF, Michel O, Perdereau B, Bariety J, Bruneval P: Early glomerular macrophage recruitment in streptozotocin-induced diabetic rats. Diabetes 2000, 49:466-475 [DOI] [PubMed] [Google Scholar]
- 13.Coimbra TM, Janssen U, Grone HJ, Ostendorf T, Kunter U, Schmidt H, Brabant G, Floege J: Early events leading to renal injury in obese Zucker (fatty) rats with type II diabetes. Kidney Int 2000, 57:167-182 [DOI] [PubMed] [Google Scholar]
- 14.Furata T, Saito T, Ootaka T, Soma J, Obara K, Abe K, Yoshinaga K: The role of macrophages in diabetes glomerulosclerosis. Am J Kidney Dis 1993, 21:180-185 [DOI] [PubMed] [Google Scholar]
- 15.Tanji N, Markowitz GS, Fu C, Kislinger T, Taguchi A, Pischetsrieder M, Stern D, Schmidt AM, D’Agati VD: The expression of advanced glycation endproducts and their cellular receptor RAGE in diabetic nephropathy and non-diabetic renal disease. J Am Soc Nephrol 2000, 11:1656-1666 [DOI] [PubMed] [Google Scholar]
- 16.Schmidt AM, Vianna M, Gerlach M, Brett J, Ryan J, Kao J, Esposito C, Hegarty H, Hurley W, Clauss M, Wang F, Pan YC, Tsang TC, Stern D: Isolation and characterization of binding proteins for advanced glycosylation endproducts from lung tissue which are present on the endothelial cell surface. J Biol Chem 1992, 267:14987-14997 [PubMed] [Google Scholar]
- 17.Neeper M, Schmidt AM, Brett J, Yan SD, Wang F, Pan YC, Elliston KD, Stern D, Shaw A: Cloning and expression of RAGE: a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem 1992, 267:14998-15004 [PubMed] [Google Scholar]
- 18.Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, Neurath MF, Slattery T, Beach D, McClary J, Morser JD, Stern D, Schmidt AM: RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell 1999, 97:889-901 [DOI] [PubMed] [Google Scholar]
- 19.Regoli M, Bendayan M: Alterations in the expression of alpha 3 beta 1 integrin in certain membrane domains of the glomerular epithelial cells (podocytes) in diabetes mellitus. Diabetologia 1997, 40:15-22 [DOI] [PubMed] [Google Scholar]
- 20.Pagtalunan ME, Miller PL, Jumping-Eagle S, Nelson RG, Myers BD, Rennke HG, Coplon NS, Sun L, Meyer TM: Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest 1997, 99:342-348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williams B: A potential role for angiotensin II-induced vascular endothelial growth factor expression in the pathogenesis of diabetic nephropathy? Miner Electrolyte Metab 1998, 24:400-404 [DOI] [PubMed] [Google Scholar]
- 22.Meyer TW, Bennett PH, Nelson RG: Podocyte number predicts long-term urinary albumin excretion in Pima Indians with type II diabetes and microalbuminuria. Diabetologia 4 1999, 2:1341-1344 [DOI] [PubMed] [Google Scholar]
- 23.Phillips A, Janssen U, Floege J: Progression of diabetic nephropathy: insights from cell culture studies and animal models. Kidney Blood Press Res 1999, 22:81-97 [DOI] [PubMed] [Google Scholar]
- 24.Pavenstadt H: Roles of the podocyte in glomerular function. Am J Physiol 2000, 278:F173-F179 [DOI] [PubMed] [Google Scholar]
- 25.Nakamura T, Ushiyama C, Osada S, Hara M, Shimada N, Koide H: Pioglitazone reduces urinary podocyte excretion in type 2 diabetes patients with microalbuminuria. Metabolism 2001, 50:1193-1196 [DOI] [PubMed] [Google Scholar]
- 26.Hoshi S, Shu Y, Yoshida F, Inagaki T, Sonoda J, Watanable T, Nomoto K, Nagata M: Podocyte injury promotes progressive nephropathy in Zucker diabetic fatty rats. Lab Invest 2002, 82:25-35 [DOI] [PubMed] [Google Scholar]
- 27.Mifsud SA, Allen TJ, Bertram JF, Hulthen UL, Kelly DJ, Cooper ME, Wilkinson-Berka JL, Gilbert RE: Podocyte foot process broadening in experimental diabetic nephropathy: amelioration with renin-angiotensin blockade. Diabetologia 2001, 44:878-882 [DOI] [PubMed] [Google Scholar]
- 28.Cohen MP, Clements RS, Hud E, Cohen JA, Ziyadeh FN: Evolution of renal functional abnormalities in the db/db mouse that parallels the development of human diabetic nephropathy. Exp Nephrol 1996, 4:166-171 [PubMed] [Google Scholar]
- 29.Cohen MP, Clements RS, Cohen JA, Shearman CW: Prevention of the decline in renal function in the diabetic db/db mouse. Diabetologia 1996, 39:270-274 [DOI] [PubMed] [Google Scholar]
- 30.Sakaguchi T, Sousa M, Yan SD, Yan SF, Duda S, Arnold B, Nawroth PP, Schmidt AM, Stern DM, Naka Y: Restenosis: central role of RAGE-dependent neointimal expansion. Circulation (Suppl) 2001, 104:2471II-522 [Google Scholar]
- 31.Mundel P, Reiser J, Zuniga Mejia Borja A, Pavenstadt H, Davidson GR, Kriz W, Zeller R: Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines. Exp Cell Res 1997, 236:248-258 [DOI] [PubMed] [Google Scholar]
- 32.Mundel P, Reiser J, Kriz W: Induction of differentiation in cultured rat and human podocytes. J Am Soc Nephrol 1997, 8:699-705 [DOI] [PubMed] [Google Scholar]
- 33.Park L, Raman KG, Lee KJ, Lu Y, Ferran LJ, Jr, Chow WS, Stern D, Schmidt AM: Suppression of accelerated diabetic atherosclerosis by soluble receptor for AGE (sRAGE). Nat Med 1998, 4:1025-1031 [DOI] [PubMed] [Google Scholar]
- 34.Lalla E, Lamster IB, Feit M, Huang L, Spessot A, Qu W, Kislinger T, Lu Y, Stern DM, Schmidt AM: Blockade of RAGE suppresses periodontitis-associated alveolar bone loss in diabetic mice. J Clin Invest 2000, 105:1117-1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reddy S, Bichler J, Wells-Knecht KJ, Thorpe SR, Baynes JW: N ε-(carboxymethyl)lysine is a dominant advanced glycation end product (AGE) antigen in tissue proteins. Biochemistry 1995, 34:10872-10878 [DOI] [PubMed] [Google Scholar]
- 36.Schleicher ED, Wagner E, Nerlich AG: Increased accumulation of the glycoxidation product N(ε)-(carboxymethyl)lysine in human tissues in diabetes and aging. J Clin Invest 1997, 99:457-468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kislinger T, Fu C, Huber B, Qu W, Taguchi A, Du Yan S, Hofmann M, Yan SF, Pischetsrieder M, Stern D, Schmidt AM: Nε (carboxymethyl)lysine modifications of proteins are ligands for RAGE that activate cell signalling pathways and modulate gene expression. J Biol Chem 1999, 274:31740-31749 [DOI] [PubMed] [Google Scholar]
- 38.Horie K, Miyata T, Maeda K, Miyata S, Sugiyama S, Sakai H, Strihou CY, Monnier VM, Witztum JL, Kurokawa K: Immunohistochemical colocalization of glycoxidation products and lipid peroxidation products in diabetic renal glomerular lesions: implication for glycoxidative stress in the pathogenesis of diabetic nephropathy J Clin Invest 1997, 100:2995-3004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schafer BW, Heinzmann CW: The S100 family of EF-hand calcium-binding proteins: functions and pathology. Trends Biochem Sci 1996, 21:134-140 [DOI] [PubMed] [Google Scholar]
- 40.Zimmer DB, Cornwall EH, Landar A, Song W: The S100 protein family: history, function, and expression. Brain Res Bull 1995, 37:417-429 [DOI] [PubMed] [Google Scholar]
- 41.Schmidt AM, Hori O, Chen JX, Li JF, Crandall J, Zhang J, Cao R, Yan SD, Brett J, Stern D: Advanced glycation endproducts interacting with their endothelial receptor induce expression of vascular cell adhesion molecule-1 (VCAM-1): a potential mechanism for the accelerated vasculopathy of diabetes. J Clin Invest 1995, 96:1395-1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamagishi S, Yonekura H, Yamamoto Y, Katsuno K, Sato F, Mita I, Ooka H, Satozawa N, Kawakami T, Nomura M, Yamamoto H: Advanced glycation end products-driven angiogenesis in vitro: induction of the growth and tube formation of human microvascular endothelial cells through autocrine vascular endothelial growth factor. J Biol Chem 1997, 272:8723-8730 [DOI] [PubMed] [Google Scholar]
- 43.Stitt AW, Bhadur T, McMullen CB, Gardiner TA, Archer DB: Advanced glycation end products induce blood-retinal barrier dysfunction in normoglycemic rats. Mol Cell Biol Res Commun 2000, 3:380-388 [DOI] [PubMed] [Google Scholar]
- 44.Tsuchida K, Makita Z, Yamagishi S, Atsumi T, Miyoshi H, Obara S, Ishida M, Ishikawa S, Yasumura K, Koike T: Suppression of transforming growth factor β and vascular endothelial growth factor in diabetic nephropathy in rats by a novel advanced glycation endproduct inhibitor, OPB-9195. Diabetologia 1999, 42:579-588 [DOI] [PubMed] [Google Scholar]
- 45.Lu M, Kuroki M, Amano S, Tolentino M, Keough K, Kim I, Bucala R, Adamis AP: Advanced glycation end products increase retinal vascular endothelial growth factor expression. J Clin Invest 1998, 101:1219-1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rovin BH, Schreiner GF: Cell-mediated immunity in glomerular disease. Annu Rev Med 1991, 42:25-33 [DOI] [PubMed] [Google Scholar]
- 47.Nathan CF: Secretory products of macrophages. J Clin Invest 1987, 79:319-326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Iglesias-De La Cruz MC, Ziyadeh FN, Isono M, Kouahou M, Han DC, Kalluri R, Mundel P, Chen S: Effects of high glucose and TGF-β1 on the expression of collagen IV and vascular endothelial growth factor in mouse podocytes. Kidney Int 2002, 62:901-913 [DOI] [PubMed] [Google Scholar]
- 49.Remuzzi G, Bertani T: Pathophysiology of progressive nephropathies. N Engl J Med 1998, 339:1448-1456 [DOI] [PubMed] [Google Scholar]
- 50.Abbate M, Zoja C, Corna D, Capitano M, Bertani T, Remuzzi G: In progressive nephropathies, overload of tubular cells with filtered proteins translates glomerular permeability dysfunction into cellular signals of interstitial inflammation. J Am Soc Nephrol 1998, 9:1213-1224 [DOI] [PubMed] [Google Scholar]
- 51.Wang Y, Chen J, Chen L, Tay YC, Rangan GK, Harris DC: Induction of monocyte chemoattractant protein-1 in proximal tubule cells by urinary protein. J Am Soc Nephrol 1997, 8:1537-1545 [DOI] [PubMed] [Google Scholar]
- 52.Zoja C, Donadelli R, Colleoni S, Figliuzzi M, Bonazzola S, Morigi M, Remuzzi G: Protein overload stimulates RANTES production by proximal tubular cells depending on NF-κ B activation. Kidney Int 1998, 53:1608-1615 [DOI] [PubMed] [Google Scholar]
- 53.Oldfield MD, Bach LA, Forbes JM, Nikolic-Paterson D, McRobert A, Thallas V, Atkins RC, Osicka T, Jerums G, Cooper ME: AGEs cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation endproducts (RAGE). J Clin Invest 2001, 108:1853-1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yamamoto Y, Kato I, Doi T, Yonekura H, Ohashi S, Watanabe T, Yamagishi S, Sakurai S, Takasawa S, Okamoto H, Yamamoto H: Development and prevention of advanced diabetic nephropathy in RAGE-overexpressing mice. J Clin Invest 2001, 108:261-268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.: The European Study for the Prevention of Renal Disease in Type 1 Diabetes (ESPRIT) Study Group: Effect of 3 years of antihypertensive therapy on renal structure in type 1 diabetic subjects with albuminuria. Diabetes 2001, 50:843-850 [DOI] [PubMed] [Google Scholar]
- 56.Viberti GC, Jarrett RJ, Keen H: Microalbuminuria as a predictor of clinical nephropathy in insulin-dependent diabetes mellitus. Lancet 1982, 1:1430-1432 [DOI] [PubMed] [Google Scholar]
- 57.Mogensen CE: Microalbuminuria predicts clinical proteinuria and early mortality in maturity-onset diabetes. N Engl J Med 1984, 310:356-360 [DOI] [PubMed] [Google Scholar]
- 58.Neale TJ, Ullrich R, Ojha P, Poczewski H, Verhoeven AJ, Kerjaschki D: Reactive oxygen species and neutrophil respiratory burst cytochrome b558 are produced by kidney glomerular cells in passive Heymann nephritis. Proc Natl Acad Sci USA 1993, 90:3645-3649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lotan D, Kaplan BS, Fong JS, Goodyer PR, de Chadarevian JP: Reduction of protein excretion by dimethyl sulfoxide in rats with passive Heymann nephritis. Kidney Int 1984, 25:778-788 [DOI] [PubMed] [Google Scholar]
- 60.Kaplan BS, Milner LS, Lotan D, Mills M, Goodyer PR, Fong JS: Interactions of dimethyl sulfoxide and non-steroidal anti-inflammatory agents in passive Heymann’s nephritis. J Lab Clin Med 1986, 107:425-430 [PubMed] [Google Scholar]
- 61.Yan SD, Schmidt AM, Anderson GM, Zhang J, Brett J, Zou YS, Pinsky D, Stern D: Enhanced cellular oxidant stress by the interaction of advanced glycation endproducts with their receptors/binding proteins. J Biol Chem 1994, 269:9889-9897 [PubMed] [Google Scholar]
- 62.Lander HL, Tauras JM, Ogiste JS, Moss RA, Schmidt: Activation of the receptor for advanced glycation endproducts triggers a MAP kinase pathway regulated by oxidant stress. J Biol Chem 1997, 272:17810-17814 [DOI] [PubMed] [Google Scholar]
- 63.Wautier MP, Chappey O, Corda S, Stern DM, Schmidt AM, Wautier JL: Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am J Physiol 2001, 280:E685-E694 [DOI] [PubMed] [Google Scholar]