

Abstract
Helix packing is important in the folding, stability, and association of membrane proteins. Packing analysis of the helical portions of 7 integral membrane proteins and 37 soluble proteins show that the helices in membrane proteins have higher packing values (0.431) than in soluble proteins (0.405). The highest packing values in integral membrane proteins originate from small hydrophobic (G and A) and small hydroxyl-containing (S and T) amino acids, whereas in soluble proteins large hydrophobic and aromatic residues have the highest packing values. The highest packing values for membrane proteins are found in the transmembrane helix–helix interfaces. Glycine and alanine have the highest occurrence among the buried amino acids in membrane proteins, whereas leucine and alanine are the most common buried residue in soluble proteins. These observations are consistent with a shorter axial separation between helices in membrane proteins. The tight helix packing revealed in this analysis contributes to membrane protein stability and likely compensates for the lack of the hydrophobic effect as a driving force for helix–helix association in membranes.
Keywords: helix interactions, occluded surface
Internal residues determine in large part the way proteins fold and function. Interiors of soluble proteins are tightly packed with densities approaching those of crystals of small organic molecules (1, 2). The stability of both native (3) and designed proteins (4) is closely correlated with the packing of core residues. Increased packing appears to be one mechanism by which the extremely stable hyperthermophilic proteins gain increased stability over their mesophilic counterparts (5). Packing analyses have been critical for evaluating structural models (6–8), designing novel proteins (4, 9), and generally understanding how the final tertiary structure of a protein is encoded in its primary sequence (2). The recent structure determinations of several large membrane protein complexes (10–20) provide a reasonable set of data to investigate the packing properties of helical integral membrane proteins.
The internal packing of membrane proteins is in many ways simpler than that of soluble proteins. The hydrophobic core of polytopic integral membrane proteins is most often formed by well-packed membrane spanning α-helices. Only in a few known cases does the transmembrane (TM) region consist of an antiparallel bundle of β-strands, as in the porins (21) or a combination of both helix and β-strands, as has been suggested for the acetylcholine receptor (22, 23). Helices spontaneously form on insertion of a hydrophobic sequence into a membrane bilayer because of the negative free energy associated with hydrogen bonding of the polar backbone carbonyls and amide groups. In the two-stage model of membrane protein folding, insertion of hydrophobic TM helices is followed by helix association (24, 25). As a result, helix-to-helix packing is a key element in defining the tertiary and quaternary structure of most membrane proteins and membrane protein complexes.
The folding of soluble proteins is thought to be driven by the hydrophobic effect and the increase in entropy associated with burying hydrophobic residues in the protein interior. Within the hydrophobic protein core, van der Waals interactions contribute significantly to the tight packing geometries that are associated with final folded protein structures. For TM helices of integral membrane proteins, the hydrophobic effect is lost as a driving force for helix association once the helices are inserted into hydrophobic bilayers. Helix association occurs through a combination of hydrogen-bonding, electrostatic, and van der Waals interactions. Unlike soluble proteins, membrane proteins rely on internal polar pockets for protein function; catalytic residues and ligand binding sites are often buried in the protein interior. This geometry then raises the question as to whether the interiors of membrane proteins are loosely packed or whether different amino acids are responsible for the packing of the helical segments of membrane and soluble proteins. The method of occluded surface (OS) (5, 8) (Fig. 1) was chosen to address these questions because it provides a direct measure of molecular packing and allows the fractionation of the atomic or molecular surface. Of importance is that the packing interactions can be quantified at the atomic, amino acid, or molecular level and both buried and surface exposed residues may be analyzed directly in contrast to the more commonly used Voronoi procedure (1). Moreover, the OS analysis promises to be a useful tool for probing the functional role of polar and conserved residues in the interior of membrane proteins by revealing how the neighboring and most closely associated residues (and atoms) pack.
Figure 1.

Schematic of the OS calculation for the methyl group of threonine. The diagram depicts the normals extending from the molecular surface associated with the methyl group. The surface normals terminate if they encounter the van der Waals surface of residues within 2.8 Å.
Materials and Methods
Methods.
The protein structures analyzed were obtained from the Protein Data Bank (PDB). The PDB codes and the corresponding protein names for the membrane proteins are listed in Table 1. For Table 2, nine polytopic membrane proteins having a total of 2230 amino acids in 83 TM helices were analyzed. For Table 3, seven polytopic membrane proteins and 37 helix-containing (α-bundle, α-nonbundle, αβ) (26) soluble proteins were studied. The set of membrane proteins contains a total of 69 TM helices and 1679 amino acids, whereas the soluble proteins contain a total of 311 helices (α-bundle, 95; α-nonbundle, 118; αβ, 98) and 3708 amino acids (α-bundle, 1415; α-nonbundle, 1367; αβ, 926).
Table 1.
PDB codes of the proteins analyzed
| PDB code | Description | Ref. |
|---|---|---|
| 1aij | Bacterial photosynthetic reaction center from Rhodobacter spheroides | 46 |
| 6prc | Bacterial photosynthetic reaction center from Rhodopseudomonas viridis | 47 |
| 1bcc | Ubiquinol cytochrome c oxidoreductase from chicken heart mitochondria | 11 |
| 1be3 | Ubiquinol cytochrome c oxidoreductase from bovine heart mitochondria | 17 |
| 1bl8 | Potassium channel from Streptomyces lividans | 12 |
| 2brd | Bacteriorhodopsin from Halobacterium salinarium | 14 |
| 1fum | Fumarate reductase from Escherichia coli | 20 |
| 1msl | Mechanosensitive ion channel from Mycobacterium tuberculosis | 19 |
| 1occ | Cytochrome c oxidase from bovine heart | 13 |
Table 2.
Average packing values of amino acids in α-helices of membrane proteins*
| 1aij | 6prc | 1bcc | 1be3 | 1bl8 | 2brd | 1fum | 1msl | 1occ-α | 1occ | |
|---|---|---|---|---|---|---|---|---|---|---|
| Gly | 0.533 | 0.541 | 0.553 | 0.497 | 0.431 | 0.459 | 0.533 | 0.607 | 0.567 | 0.534 |
| Ala | 0.548 | 0.519 | 0.438 | 0.410 | 0.500 | 0.448 | 0.456 | 0.421 | 0.477 | 0.485 |
| Ser | 0.522 | 0.519 | 0.465 | 0.445 | 0.533 | 0.398 | 0.443 | 0.493 | 0.491 | 0.484 |
| Cys | 0.613 | 0.495 | 0.573 | 0.470 | 0.294 | — | 0.590 | — | — | 0.478 |
| Val | 0.451 | 0.447 | 0.426 | 0.377 | 0.392 | 0.347 | 0.346 | 0.445 | 0.452 | 0.442 |
| Thr | 0.502 | 0.420 | 0.431 | 0.410 | 0.376 | 0.460 | 0.465 | 0.415 | 0.468 | 0.459 |
| Asp | 0.249 | 0.510 | 0.421 | 0.425 | — | 0.529 | — | 0.330 | 0.434 | 0.503 |
| Asn | 0.564 | 0.507 | 0.394 | 0.357 | — | 0.367 | 0.461 | 0.392 | 0.450 | 0.473 |
| Leu | 0.441 | 0.402 | 0.396 | 0.402 | 0.366 | 0.384 | 0.414 | 0.326 | 0.431 | 0.423 |
| Ile | 0.430 | 0.460 | 0.418 | 0.385 | 0.325 | 0.411 | 0.39 | 0.424 | 0.437 | 0.428 |
| Lys | 0.242 | — | 0.333 | 0.366 | — | 0.285 | 0.226 | 0.295 | 0.405 | 0.389 |
| Arg | 0.48 | 0.424 | 0.384 | 0.339 | 0.339 | 0.408 | 0.414 | — | 0.393 | 0.378 |
| Glu | 0.445 | 0.251 | 0.327 | 0.328 | 0.543 | 0.442 | 0.370 | — | 0.487 | 0.412 |
| Gln | 0.391 | 0.486 | 0.403 | 0.404 | — | 0.160 | — | — | 0.433 | 0.461 |
| Met | 0.538 | 0.524 | 0.409 | 0.406 | 0.550 | 0.510 | 0.471 | — | 0.422 | 0.438 |
| Pro | 0.527 | 0.570 | 0.439 | 0.404 | — | 0.515 | 0.381 | — | 0.489 | 0.512 |
| His | 0.551 | 0.459 | 0.429 | 0.416 | — | — | 0.399 | — | 0.484 | 0.453 |
| Phe | 0.407 | 0.444 | 0.429 | 0.407 | 0.337 | 0.393 | 0.433 | 0.309 | 0.447 | 0.44 |
| Tyr | 0.377 | 0.381 | 0.451 | 0.419 | 0.322 | 0.427 | 0.360 | 0.261 | 0.485 | 0.402 |
| Trp | 0.447 | 0.450 | 0.480 | 0.397 | — | 0.436 | 0.384 | — | 0.429 | 0.432 |
| Average | 0.469 | 0.469 | 0.427 | 0.408 | 0.417 | 0.413 | 0.412 | 0.389 | 0.461 | 0.451 |
The occluded surface calculations were carried out on full protein structures, but the packing values reported represent the helical residues only.
Table 3.
Average packing values and the percent occurrence of amino acids in the helices of membrane and soluble proteins*
| Membrane proteins
|
α-Bundle proteins†
|
α-Nonbundle proteins†
|
αβ proteins†
|
|||||
|---|---|---|---|---|---|---|---|---|
| Packing value | Occurrence (%) | Packing value | Occurrence (%) | Packing value | Occurrence (%) | Packing value | Occurrence (%) | |
| Gly | 0.512 | 7.35 | 0.451 | 2.61 | 0.432 | 4.39 | 0.441 | 2.81 |
| Ala | 0.471 | 11.12 | 0.455 | 10.18 | 0.443 | 12.58 | 0.449 | 13.28 |
| Ser | 0.484 | 5.26 | 0.402 | 4.24 | 0.388 | 4.61 | 0.379 | 6.26 |
| Cys | 0.463 | 0.69 | 0.516 | 2.69 | 0.543 | 2.41 | 0.525 | 1.08 |
| Val | 0.408 | 9.44 | 0.450 | 5.58 | 0.463 | 7.17 | 0.464 | 6.80 |
| Thr | 0.441 | 6.93 | 0.406 | 4.81 | 0.396 | 5.56 | 0.388 | 4.43 |
| Asp | 0.417 | 1.37 | 0.352 | 4.10 | 0.340 | 3.37 | 0.321 | 6.91 |
| Asn | 0.432 | 1.75 | 0.366 | 3.60 | 0.382 | 3.80 | 0.368 | 3.35 |
| Leu | 0.402 | 15.80 | 0.482 | 13.22 | 0.461 | 13.46 | 0.449 | 11.23 |
| Ile | 0.412 | 8.30 | 0.474 | 4.88 | 0.454 | 6.73 | 0.476 | 6.37 |
| Lys | 0.322 | 1.94 | 0.288 | 6.29 | 0.293 | 5.12 | 0.276 | 6.70 |
| Arg | 0.381 | 2.44 | 0.324 | 6.86 | 0.339 | 5.63 | 0.296 | 5.62 |
| Glu | 0.418 | 1.52 | 0.323 | 8.34 | 0.305 | 6.07 | 0.300 | 8.32 |
| Gln | 0.406 | 1.14 | 0.364 | 6.71 | 0.337 | 4.68 | 0.302 | 3.89 |
| Met | 0.454 | 4.34 | 0.427 | 2.40 | 0.461 | 2.78 | 0.408 | 2.38 |
| Pro | 0.461 | 2.51 | 0.344 | 1.84 | 0.356 | 2.56 | 0.302 | 1.73 |
| His | 0.453 | 2.63 | 0.448 | 2.19 | 0.437 | 2.12 | 0.322 | 1.51 |
| Phe | 0.403 | 9.25 | 0.471 | 3.53 | 0.497 | 2.71 | 0.449 | 3.13 |
| Tyr | 0.387 | 2.97 | 0.464 | 4.31 | 0.415 | 2.41 | 0.388 | 0.76 |
| Trp | 0.427 | 3.24 | 0.507 | 1.63 | 0.466 | 1.83 | 0.472 | 3.46 |
| Average‡ | 0.431 | 0.411 | 0.411 | 0.388 | ||||
The occluded surface calculations were carried out on full protein structures, but the packing values reported represent the helical residues only.
† The soluble proteins were classified according to Michie et al. (26).
‡ The average packing value for each protein class was determined by summing up the packing values of all amino acids in their helical sections and dividing the sum by the total number of amino acids in the helical section of each protein class.
The method of OS (Fig. 1) for the analysis of packing interactions in proteins has been previously described (5, 8). Briefly, a packing value is composed of two parameters, the OS area and the distribution of distances to occluded atoms. A molecular dot surface of each residue is calculated with a 1.4-Å probe. The dot density was chosen such that each dot represents ≈0.215 Å2 of the surface area. A normal is extended radially from each dot until it either intersects the van der Waals surface of a neighboring atom or reaches a length of 2.8 Å (the diameter of a water molecule). The OS, So, is defined as that molecular surface area on the originating atom associated with normals that intersect with another atom surface as opposed to reaching the 2.8-Å limit. All other molecular surface area is considered nonoccluded or exposed. The packing value (PV) for each residue is defined as:
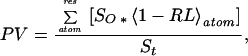 |
where St is the total surface of the residue (sum of occluded and nonoccluded areas) and RL is the length of the extended normal from one surface to the other divided by 2.8 (actual length in Å/2.8 Å). Division by the total molecular surface area normalizes the packing value to account for various sizes of amino acid residues. The average OS packing value for a protein is simply the average of all residue packing values for that protein. An important aspect of the OS method is that because packing is estimated only for the residue surface which is occluded by other atoms, the method works equally well for both buried residues and surface residues.
The OS calculations were carried out on full protein structures, but the analysis and therefore the packing values reported in Table 2–4 represent the helical residues only. Prosthetic groups were included into calculations, whereas detergent, lipid, and water molecules were excluded. The calculations were done on monomers except for the ion channels, where the functional tetramer (1bl8) and pentamer (1msl) were used. For membrane proteins, we assigned the hydrophobic boundaries based on the position of basic and acidic residues, which bracketed the central hydrophobic portion of their TM helices. The helices in soluble proteins were assigned as described in the corresponding PDB file.
Table 4.
Distribution of average packing values in helical membrane and soluble proteins*
| Position‡ | Membrane proteins
|
α-Bundle proteins†
|
α-Nonbundle proteins†
|
αβ proteins†
|
||||
|---|---|---|---|---|---|---|---|---|
| Packing value | Occurrence (%) | Packing value | Occurrence (%) | Packing value | Occurrence (%) | Packing value | Occurrence (%) | |
| ≤0.3 (exposed) | 0.250 | 15.61 | 0.234 | 24.17 | 0.231 | 23.48 | 0.228 | 29.26 |
| >0.3–0.55 (interm.) | 0.432 | 68.28 | 0.431 | 58.37 | 0.437 | 61.45 | 0.419 | 56.05 |
| >0.55 (buried) | 0.598 | 16.11 | 0.590 | 17.46 | 0.591 | 15.07 | 0.591 | 14.69 |
The occluded surface calculations were carried out on full protein structures, but the packing values reported represent the helical residues only.
†The soluble proteins were classified according to Michie et al. (26).
‡See text.
Results and Discussion
Packing Analysis of Helices in Integral Membrane Proteins.
The packing analysis of the helices in nine integral membrane proteins is summarized in Table 2 which lists (i) the average packing value of each amino acid type, and (ii) the average packing value for each protein. The proteins include ion channels (1bl8, 1msl), a proton pump (2brd), and electron transport proteins (1aij, 1bcc, 1be3, 1fum, 1occ, 6prc). The PDB codes and the corresponding protein names for the membrane proteins are listed in Table 1. For cytochrome c oxidase the values for all TM subunits (1occ) and for the α-subunit (1occ-α) alone are given. This was done because 7 of 10 TM subunits have only a single TM helix with potentially different packing constraints. The packing values range from 0.613 for cysteine in the bacterial photosynthetic reaction center (1aij) to 0.16 for glutamine in bacteriorhodopsin (2brd). The average packing values range from 0.389 to 0.469 for the proteins reported in Table 2, giving an average of 0.431 (Table 3). The packing values for the helices of 37 soluble proteins solved by crystallographic methods have packing values between 0.333 and 0.456, resulting in an average of 0.405 (data not shown). The soluble proteins studied contain α-bundle, α-nonbundle, and αβ proteins as defined by Michie et al. (26).
Comparison of the Packing for Amino Acids in the Helical Segments of TM and Soluble Proteins.
Packing of the helical segments of 7 integral membrane proteins and 37 soluble proteins is summarized in Table 3, which lists (i) the average packing values for the 20 amino acid residues averaged in four protein classes, (ii) the average packing value for each protein class, and (iii) the percent occurrence of the 20 amino acids for each protein class. The average packing values range from 0.431 for membrane proteins to 0.411 for α-bundle proteins and α-nonbundle proteins, and 0.388 for αβ proteins. Only one of the two bacterial photosynthetic reaction centers (1aij) and one of the two ubiquinol cytochrome c oxidoreductases (1be3) in Table 2 were included to determine the average. The α-subunit of cytochrome c oxidase (1occ-α) separately was also not included. The data in Table 3 show that the helical core of membrane proteins is more tightly packed than that of soluble proteins.
The breakdown of the packing values for each amino acid type in Table 3 provides insights into their role in helix packing. The positively charged residues, Lys and Arg, populate the low end of the packing value range for the protein classes studied. The negatively charged residues, Asp and Glu, as well as the amides, Asn and Gln, have low packing values in soluble proteins, but significantly higher values in membrane proteins (Table 3). The charged and polar residues in soluble proteins are located in exposed positions. In membrane proteins, Arg (0.381) and Lys (0.322) tend to be oriented toward the polar head groups of the phospholipids in membrane proteins (27). The negatively charged residues, Asp (0.417) and Glu (0.418), have higher packing values than Arg and Lys. Asp and Glu, as well as Asn (0.432), Gln (0.406), and His (0.453), tend to be found at conserved positions where they interact with polar sites on adjacent helices (28). Ligand interactions explain the high packing value of histidine compared with the other charged amino acids in all proteins that have α-helices as their major structural motif. All other amino acids differ in the ranking of their packing values between membrane and soluble proteins. The packing values of cysteine are skewed because of disulfide bond formation and its interaction with prosthetic groups.
Small residues contribute to tight packing in all helical proteins. They are the major reason for the high packing values in membrane proteins. Membrane proteins have the highest content of glycine when only helical secondary structure is considered (Table 3). In contrast, the percentage of alanine is nearly the same in the helices of all proteins studied (Table 3). The high abundance of glycine and alanine in TM helices is also seen in a statistical analysis of membrane protein sequences, which includes both bi- and polytopic membrane proteins (29). In soluble proteins the large hydrophobic residues Leu, Ile, and Val provide the highest packing values, which is a result of the fact that these residues are frequently found in the buried core of soluble proteins. The aromatic residues Phe and Trp also have high packing values in soluble proteins. Except for Trp, the packing values of these residues in membrane proteins are below the protein average.
Proline has a high packing value in helical membrane proteins, whereas it has a low packing value in soluble proteins, where it is known as a helix breaker. Proline residues are often associated with kinks in helical secondary structure (30–32). Both rhodopsin and bacteriorhodopsin have several prolines in their TM helices, and it has been suggested that these function to open up internal cavities for binding of the retinal prosthetic group (33, 34). The packing value of 0.515 for the helical prolines in bacteriorhodopsin (Table 2) is inconsistent with the idea of proline only being used for introducing kinks that would result in low packing values. In contrast, mutational studies (35) and model peptides (36, 37) lead to the conclusion that the structural propensity of proline is strongly dependent on its environment, and that under certain conditions proline can even stabilize helical structures (37). In fact, as shown before, proline residues in cytochrome c oxidase occur only in buried and intermediately exposed locations (38) and their Φ and Ψ dihedral angles fall in the standard α-helical region of a Ramachandran plot (39). Bends are more often observed in helices which contain more than one proline residue or a combination of proline and glycine residues that are spaced four residues apart (39).
Small hydroxyl-containing amino acids, serine and threonine, have higher packing values in membrane proteins than in soluble proteins. We measured the distance between the hydroxyl group and the protein backbone in membrane proteins to establish how the polar side chain is hydrogen bonded. We found that the hydroxyl oxygen in 59% of the serines and 72% of the threonines is 2.4–3.4 Å from the i-4 or i-3 backbone nitrogen, consistent with hydrogen bonding back to the polar backbone. These percentages are remarkably similar to those obtained for helical soluble proteins (40). In soluble proteins, the majority of the serines and threonines are exposed to the solvent (40), whereas in membrane proteins serines and threonines prefer to face the protein interior (41). The interior location is the likely reason for higher packing values observed in membrane proteins.
Packing Values for Different Amino Acid Classes.
The packing values were combined for different amino acid classes: small (A,G), hydroxyl (S,T), amide (N,Q), charged (D,E,R,H,K), hydrophobic (I,L,M,V), and aromatic (F,W,Y). Fig. 2A highlights the contribution of each amino acid class to the average protein packing value in each protein class. The packing values for each amino acid class were calculated separately for membrane proteins, α-bundle, α-nonbundle, and αβ proteins by using the data from Table 3. For instance, the average packing value for small residues (A and G) in membrane proteins was determined by summing up the packing values of all alanines and glycines in the helices of membrane proteins and dividing this value by the total number of alanines and glycines in these helices. Fig. 2A plots the difference between the average packing value for the amino acid class and the average packing value for the protein class (e.g., 0.431 for membrane proteins). The data in Fig. 2 reflect only the helical portions of the proteins studied. Except for aromatic, cysteine, and large hydrophobic residues, all amino acids have higher packing values in helical membrane proteins. For all proteins studied, small residues have higher packing values than the protein average, whereas amides and charged residues have lower packing values than the protein average.
Figure 2.

Comparison of helix packing between membrane and soluble proteins. The soluble proteins were classified according to Michie et al. (26). (A) Contributions to helix packing by different amino acid classes. The packing values in Table 3 were combined for small (A,G), hydroxyl (S,T), amide (N,Q), charged (D,E,R,H,K), hydrophobic (I,L,M,V), and aromatic (F,W,Y) amino acids. The contribution of each amino acid class is plotted by taking the difference between the average packing value for the amino acid class and the average packing value for the protein class as described in the text. These data reflect only the helical portions of the proteins studied. (B) Contribution of amino acid classes to the overall packing in different classes of helical proteins. The packing value differences plotted in A are multiplied by the percent occurrence of the amino acid class for each protein class.
If the occurrence of individual amino acids is taken into account, the highest contribution to overall packing in membrane proteins results from small amino acids. This result is illustrated in Fig. 2B which weights the results in Fig. 2A to the amino acid composition. In contrast to membrane proteins, large hydrophobic residues and to a smaller extent aromatic residues provide the highest packing for soluble proteins (Fig. 2B). Fig. 2B also illustrates the differences in packing values for proline, serine, and threonine between membrane proteins and soluble proteins. Finally, the variation between the average packing values of the different amino acid classes in membrane proteins is much smaller than in soluble proteins suggesting more homogeneous packing in membrane proteins (Fig. 2B).
To further investigate the reason for the observed differences in packing between soluble and membrane proteins, we compared the occurrence and distribution of residues with packing values of 0.3 and less, between 0.3 and 0.55, and greater than 0.55 (Table 4). Visual inspection of the protein crystal structures show that the packing cutoffs (≤0.3 and >0.55) correspond to clearly exposed and buried residues, respectively. Membrane proteins have the highest packing values and the lowest occurrence for exposed residues with packing values of 0.3 and less. The majority of these loosely packed residues in membrane proteins are large hydrophobic amino acids (Leu, 23%; Val, 12%; and Ile, 9%) and phenylalanine (12%). These residues contribute <5% each to the exposed amino acids in soluble proteins (data not shown). αβ proteins have the lowest packing values for exposed (packing values ≤0.3) residues. They also have the lowest percentage of buried amino acids, consistent with their low average packing. The most tightly packed buried residues (packing values >0.55) in membrane proteins, α-helical bundle, and α-nonbundle proteins have similar average packing values and occurrences (Table 4). This argues that packing densities in the helical cores of membrane and soluble proteins are roughly comparable and that helix–helix interactions generally lead to high packing values. In membrane proteins, helix–helix contacts are maximized in “two-dimensional” bilayer environments.
The most striking difference between membrane and soluble proteins with respect to buried residues is that membrane proteins have at least double the number of Gly and less than half of the number of Leu residues than soluble helical proteins (data not shown). To look for possible consequences of this observation, we measured the axial separation in the membrane and α bundle proteins studied by using the program define_s (42) (Fig. 3). The average axial separation for membrane proteins is 9.0 Å, whereas the separation in α-bundle proteins is 9.6 Å. This finding is consistent with the observation that membrane helix interactions are dominated by small residues which are tightly packed. Moreover, in Fig. 3 there appears to be two major distributions of interhelical distances, one distribution centered at ≈7.3 Å and a second distribution centered at ≈10.8 Å.
Figure 3.

Distribution of the axial separation in membrane and α-bundle proteins determined by using the program define_s (42). The average axial separation for membrane proteins is 9.0 Å, whereas the separation in α-bundle proteins is 9.6 Å.
The high packing values observed for membrane proteins contradict the proposal that TM helices may be more loosely packed than the interiors of soluble proteins because of the “inside-out” nature of membrane protein packing. The simple idea is that membrane proteins fold with polar and conserved residues facing the interior of a helical bundle. These residues comprise the functional or catalytic sites. Nonconserved residues are generally oriented out toward the lipids and are considerably more hydrophobic than the interior residues (43). The model that has emerged from our previous analysis of four polytopic membrane proteins is that residues with small side chains serve to guide and stabilize the tight association of TM helices (39). This result is confirmed in the present study by the high amount of Gly among the buried residues and the shorter axial separation of membrane proteins compared with α bundle proteins. In single pass membrane proteins the packing features of residues with small side chains facilitate dimerization (44), whereas in polytopic membrane proteins it appears to create an internal architecture of tightly packed helix bundles and more loosely packed interhelical spaces.
Conclusions
The observation that membrane proteins generally pack more tightly than soluble proteins has important implications for their stability and function. The stability of membrane-embedded and soluble proteins is comparable despite the fact that membrane proteins do not rely on the hydrophobic effect to drive protein folding (45). Detailed packing interactions clearly contribute a significant energetic component to membrane protein stability and are likely to play a leading role in guiding helix association in membrane protein folding (25). The same strategy for increasing protein stability (i.e., increasing packing interactions) appears to be used in some hyperthermophilic proteins (5).
The tight packing of membrane proteins must also have functional consequences. Tightly packed helices in polytopic membrane proteins open up interhelical regions that are more loosely packed. In the structures studied here, these regions often contain polar residues and provide the binding sites for substrates and ligands. This geometry is consistent with the occurrence of two distributions of interhelical distances (Fig. 3). The OS analysis promises to be a useful tool for probing the functional role of polar and conserved residues in the interior of membrane proteins by revealing how the neighboring and most closely associated residues (and atoms) pack. The method of OS will be useful for refining the packing interactions in membrane proteins as the number of solved structures increases.
Acknowledgments
We thank Wei Liu and Adriana Gonis for their contributions in determining the interaxial separation and the Ser/Thr hydrogen bonding, respectively. This work was supported by grants from the National Institutes of Health (GM 46732 and GM 41412).
Abbreviations
- OS
occluded surface, TM, transmembrane
- PDB
Protein Data Bank
References
- 1.Richards F M. J Mol Biol. 1974;82:1–14. doi: 10.1016/0022-2836(74)90570-1. [DOI] [PubMed] [Google Scholar]
- 2.Varadarajan R, Richards F M, Connelly P R. Curr Sci. 1990;59:819–824. [Google Scholar]
- 3.Richards F M. Cell Mol Life Sci. 1997;53:790–802. doi: 10.1007/s000180050100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang L, Veenstra D L, Radmer R J, Kollman P A. Proteins Struct Funct Genet. 1998;32:438–458. [PubMed] [Google Scholar]
- 5.DeDecker B S, O'Brien R, Fleming P J, Geiger J H, Jackson S P, Sigler P B. J Mol Biol. 1996;264:1072–1084. doi: 10.1006/jmbi.1996.0697. [DOI] [PubMed] [Google Scholar]
- 6.Ponder J W, Richards F M. Cold Spring Harbor Symp Quant Biol. 1987;52:421–428. doi: 10.1101/sqb.1987.052.01.049. [DOI] [PubMed] [Google Scholar]
- 7.Gregoret L M, Cohen F E. J Mol Biol. 1990;211:959–974. doi: 10.1016/0022-2836(90)90086-2. [DOI] [PubMed] [Google Scholar]
- 8.Pattabiraman N, Ward K B, Fleming P J. J Mol Recognit. 1995;8:334–344. doi: 10.1002/jmr.300080603. [DOI] [PubMed] [Google Scholar]
- 9.Kono H, Nishiyama M, Tanokura M, Doi J. Protein Eng. 1998;11:47–52. doi: 10.1093/protein/11.1.47. [DOI] [PubMed] [Google Scholar]
- 10.Feher G, Allen J P, Okamura M Y, Rees D C. Nature (London) 1989;339:111–116. [Google Scholar]
- 11.Zhang Z, Huang L, Shulmeister V M, Chi Y I, Kim K K, Hung L W, Crofts A R, Berry E A, Kim S H. Nature (London) 1998;392:677–684. doi: 10.1038/33612. [DOI] [PubMed] [Google Scholar]
- 12.Doyle D A, Cabral J M, Pfuetzner R A, Kuo A, Gulbis J M, Cohen S L, Chait B T, Mackinnon R. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 13.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. Science. 1996;272:1136–1144. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- 14.Grigorieff N, Ceska T A, Downing K H, Baldwin J M, Henderson R. J Mol Biol. 1996;259:393–421. doi: 10.1006/jmbi.1996.0328. [DOI] [PubMed] [Google Scholar]
- 15.Kimura Y, Vassylyev D G, Miyazawa A, Kidera A, Matsushima M, Mitsuoka K, Murata K, Hirai T, Fujiyoshi Y. Nature (London) 1997;389:206–211. doi: 10.1038/38323. [DOI] [PubMed] [Google Scholar]
- 16.Koepke J, Hu X, Muenke C, Schulten K, Michel H. Structure. 1996;4:581–597. doi: 10.1016/s0969-2126(96)00063-9. [DOI] [PubMed] [Google Scholar]
- 17.Iwata S, Lee J W, Okada K, Lee J K, Iwata M, Rasmussen B, Link T A, Ramaswamy S, Jap B K. Science. 1998;281:64–71. doi: 10.1126/science.281.5373.64. [DOI] [PubMed] [Google Scholar]
- 18.Deisenhofer J, Michel H. Science. 1989;245:1463–1473. doi: 10.1126/science.245.4925.1463. [DOI] [PubMed] [Google Scholar]
- 19.Chang G, Spencer R H, Lee A T, Barclay M T, Rees D C. Science. 1998;282:2220–2226. doi: 10.1126/science.282.5397.2220. [DOI] [PubMed] [Google Scholar]
- 20.Iverson T M, Luna-Chavez C, Cecchini G, Rees D C. Science. 1999;284:1961–1966. doi: 10.1126/science.284.5422.1961. [DOI] [PubMed] [Google Scholar]
- 21.Cowan S W. Curr Opin Struct Biol. 1993;3:501–507. [Google Scholar]
- 22.Unwin N. J Mol Biol. 1993;229:1101–1124. doi: 10.1006/jmbi.1993.1107. [DOI] [PubMed] [Google Scholar]
- 23.Galzi J L, Changeux J P. Curr Opin Struct Biol. 1994;4:554–565. [Google Scholar]
- 24.Popot J L, Engelman D M. Biochemistry. 1990;29:4031–4037. doi: 10.1021/bi00469a001. [DOI] [PubMed] [Google Scholar]
- 25.Fleming K G, Ackerman A L, Engelman D M. J Mol Biol. 1997;272:266–275. doi: 10.1006/jmbi.1997.1236. [DOI] [PubMed] [Google Scholar]
- 26.Michie A D, Orengo C A, Thornton J M. J Mol Biol. 1996;262:168–185. doi: 10.1006/jmbi.1996.0506. [DOI] [PubMed] [Google Scholar]
- 27.Ballesteros J A, Weinstein H. Biophys J. 1992;62:107–109. doi: 10.1016/S0006-3495(92)81794-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang D, Weinstein H. FEBS Lett. 1994;337:207–212. doi: 10.1016/0014-5793(94)80274-2. [DOI] [PubMed] [Google Scholar]
- 29.Arkin I T, Brunger A T. Biochim Biophys Acta. 1998;1429:113–128. doi: 10.1016/s0167-4838(98)00225-8. [DOI] [PubMed] [Google Scholar]
- 30.Brandl C J, Deber C M. Proc Natl Acad Sci USA. 1986;83:917–921. doi: 10.1073/pnas.83.4.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deber C M, Glibowicka M, Woolley G A. Biopolymers. 1990;29:149–157. doi: 10.1002/bip.360290120. [DOI] [PubMed] [Google Scholar]
- 32.Polinsky A, Goodman M, Williams K A, Deber C M. Biopolymers. 1992;32:399–406. doi: 10.1002/bip.360320416. [DOI] [PubMed] [Google Scholar]
- 33.Deber C M, Sorrell B J, Xu G Y. Biochem Biophys Res Commun. 1990;172:862–869. doi: 10.1016/0006-291x(90)90755-c. [DOI] [PubMed] [Google Scholar]
- 34.Williams K A, Deber C M. Biochemistry. 1991;30:8919–8923. doi: 10.1021/bi00101a001. [DOI] [PubMed] [Google Scholar]
- 35.Strehlow K G, Robertson A D, Baldwin R L. Biochemistry. 1991;30:5810–5814. doi: 10.1021/bi00237a026. [DOI] [PubMed] [Google Scholar]
- 36.Vogel H. Q Rev Biophys. 1992;25:433–457. doi: 10.1017/s0033583500004364. [DOI] [PubMed] [Google Scholar]
- 37.Li S C, Goto N K, Williams K A, Deber C M. Proc Natl Acad Sci USA. 1996;93:6676–6681. doi: 10.1073/pnas.93.13.6676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wallin E, Tsukihara T, Yoshikawa S, Von Heijne G, Elofsson A. Protein Sci. 1997;6:808–815. doi: 10.1002/pro.5560060407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Javadpour M M, Eilers M, Groesbeek M, Smith S O. Biophys J. 1999;77:1609–1618. doi: 10.1016/S0006-3495(99)77009-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gray T M, Matthews B W. J Mol Biol. 1984;175:75–81. doi: 10.1016/0022-2836(84)90446-7. [DOI] [PubMed] [Google Scholar]
- 41.Pilpel Y, Ben-Tal N, Lancet D. J Mol Biol. 1999;294:921–935. doi: 10.1006/jmbi.1999.3257. [DOI] [PubMed] [Google Scholar]
- 42.Richards F M, Kundrot C E. Proteins. 1988;3:71–84. doi: 10.1002/prot.340030202. [DOI] [PubMed] [Google Scholar]
- 43.Rees D C, DeAntonio L, Eisenberg D. Science. 1989;245:510–513. doi: 10.1126/science.2667138. [DOI] [PubMed] [Google Scholar]
- 44.Russ W P, Engelman D M. J Mol Biol. 2000;296:911–919. doi: 10.1006/jmbi.1999.3489. [DOI] [PubMed] [Google Scholar]
- 45.Stowell M H B, Rees D C. Adv Protein Chem. 1995;46:279–311. doi: 10.1016/s0065-3233(08)60338-1. [DOI] [PubMed] [Google Scholar]
- 46.Stowell M H B, McPhillips T M, Rees D C, Soltis S M, Abresch E, Feher G. Science. 1997;276:812–816. doi: 10.1126/science.276.5313.812. [DOI] [PubMed] [Google Scholar]
- 47.Lancaster C R, Michel H. J Mol Biol. 1999;286:883–898. doi: 10.1006/jmbi.1998.2532. [DOI] [PubMed] [Google Scholar]