

Abstract
The human cathelicidin LL-37 is a cationic host defense peptide (antimicrobial peptide) expressed primarily by neutrophils and epithelial cells. This peptide, up-regulated under conditions of inflammation, has immunomodulatory and antimicrobial functions. We demonstrate that LL-37 is a potent inhibitor of human neutrophil apoptosis, signaling through P2X7 receptors and G-protein-coupled receptors other than the formyl peptide receptor-like-1 molecule. This process involved modulation of Mcl-1 expression, inhibition of BID and procaspase-3 cleavage, and the activation of phosphatidylinositol-3 kinase but not the extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase pathway. In contrast to the inhibition of neutrophil apoptosis, LL-37 induced apoptosis in primary airway epithelial cells, demonstrating alternate consequences of LL-37-mediated modulation of apoptotic pathways in different human primary cells. We propose that these novel immunomodulatory properties of LL-37 contribute to peptide-mediated enhancement of innate host defenses against acute infection and are of considerable significance in the development of such peptides and their synthetic analogs as potential therapeutics for use against multiple antibiotic-resistant infectious diseases.
Keywords: cathelicidin, antimicrobial peptide, neutrophil, epithelial cell, FPRL-1, P2X7
INTRODUCTION
Cationic host defense peptides (CHDP; also known as antimicrobial peptides) are key, evolutionarily conserved components of host defenses [1]. The importance of CHDP to human immunity is indicated by the increased susceptibility to infection of individuals with specific granule deficiency [2] and morbus Kostmann patients [3] (conditions in which neutrophil deficiency in defensins and cathelicidin, respectively, constitute key components of the disease) and by the up-regulation of peptide expression in lung [4], skin [5], and other sites in response to inflammation. Furthermore, studies using knockout mouse models, transgenics, and gene therapy augmentation clearly demonstrate the significance of CHDP to host defense against infections [6-10].
Recently, various CHDP have been shown to have immunomodulatory properties [11], with a growing number of these demonstrated in vivo, including chemokine production, angiogenesis, anti-endotoxin activity, and chemotaxis [12-15]. In addition, multiple studies of CHDP have demonstrated broad-spectrum antimicrobial activities in vitro. Consequently, development of CHDP and their synthetic derivatives promises future antimicrobial therapeutics relatively unaffected by common bacterial resistance mechanisms and potentially combining microbicidal activities with immunomodulatory properties [16].
The CHDP demonstrating the most significant immunomodulatory potential to date is LL-37, the predominant, mature peptide fragment of the sole human cathelicidin, human cationic antimicrobial protein (hCAP)-18. This peptide is found in neutrophil-specific granules, produced by a range of epithelial cells and to a lesser extent, by lymphocytes and macrophages [17]. LL-37 expression has been detected at ∼5 μg/ml in bronchoalveolar lavage (BAL) from healthy infants and is up-regulated by inflammation, detected in BAL from infants with pulmonary infections and in individuals with cystic fibrosis lung disease at up to ∼30 μg/ml and ∼15 μg/ml, respectively [4, 18], and at levels of ∼1.5 mg/ml in psoriatic skin lesions [5].
The full extent of the immunomodulatory functions of LL-37 and the underlying mechanisms involved remain undetermined and are of considerable significance in the development of therapeutic, synthetic analogs. We hypothesized that the potential for LL-37 to modulate the function and survival of key innate-immune effector cells could be critical to enhancing the clearance of infection and resolution of inflammation. In particular, neutrophils, a cell type for which LL-37 is directly and indirectly chemotactic [12, 19-22], represent a key component of innate defense against infection. These cells have a short half-life, “programmed” to die by apoptosis, and exert a defensive role by intracellular killing and the release of defensins and cathelicidins, lytic enzymes, proteases, and inflammatory mediators [23, 24]. In this study, we demonstrate that LL-37 acted as a potent inhibitor of human neutrophil apoptosis, signaling through P2X7 receptors and G-protein-coupled receptors other than formyl peptide receptor-like-1 molecule (FPRL-1) and modulating Mcl-1 expression, inhibiting cleavage of BID and procaspase-3, and involving the activation of phosphatidylinositol-3 kinase (PI-3K, but not the extracellular signal-regulated kinase (ERK) 1/2 mitogen-activated protein kinase (MAPK) pathway. In contrast, LL-37 induced apoptosis in primary airway epithelial cells, demonstrating alternate consequences of LL-37-mediated modulation of apoptotic pathways in different human primary innate-immune effector cells exposed to this CHDP.
MATERIALS AND METHODS
Reagents
Recombinant human tumor necrosis factor α (TNF-α), interleukin (IL)-1β, and granulocyte macrophage-colony stimulating factor (GM-CSF) were purchased from Research Diagnostics Inc. (Flanders, NJ). H-Trp-Arg-Trp-Trp-Trp-Trp-CONH2 (WRW4) and H-Trp-Lys-Tyr-Met-Val-D-Met-CONH2 (WKYMVm; both reconstituted in dimethyl sulfoxide, according to the manufacturer's instructions), caspase inhibitor I [Z-Val-Ala-Asp-fluoromethylketone (Z-VAD-FMK)], and pertussis toxin (PTX) were purchased from Calbiochem, Merk Biosciences (Nottingham, UK). Oxidized adenosine 5′-triphosphate (ATP), PD098059, and wortmannin were purchased from Sigma-Aldrich (Poole, UK, or Oakville, Ontario, Canada). Rabbit polyclonal antibodies against Mcl-1, BID, and cleaved caspase-3 were purchased from Cell Signaling Technology (Mississauga, Ontario, Canada). Mouse polyclonal antibody against caspase-3 was purchased from Alexis Biochemicals (Axxora, San Diego, CA). Horseradish peroxidase (HRP)-conjugated goat antirabbit and antimouse immunoglobulin G antibodies were purchased from Cell Signaling Technology (Beverly, MA) and Amersham Biosciences (Piscataway, NJ), respectively. Lipopolysaccharide (LPS) from Pseudomonas aeruginosa Strain H103 was highly purified free of proteins and lipids using the Darveau-Hancock method as described previously [25]. Briefly, P. aeruginosa was grown overnight in Luria-Bertani broth at 37°C. Cells were collected and washed, and the isolated LPS pellets were extracted with a 2:1 chloroform:methanol solution to remove contaminating lipids. Purified LPS samples were quantitated using an assay for the specific sugar 2-keto-3-deoxyoctosonic acid assay and then resuspended in endotoxin-free water (Sigma-Aldrich). LL-37 was synthesized by N-(9-fluorenyl) methoxycarbonyl chemistry at the Nucleic Acid/Protein Service unit at the University of British Columbia (UBC; Vancouver, Canada), as described previously [26]. Peptides were purified by reverse-phase high-performance liquid chromatography and were at least 98% pure. LL-37 was dissolved in endotoxin-free water (Sigma-Aldrich) and stored at −20°C until further use. The concentration of the peptides in solution was determined by amino acid analysis. All reagents were tested to ensure that they were free of endotoxin and reconstituted in endotoxin-free water.
Isolation of human blood neutrophils
Fresh human venous blood was collected from volunteers, according to University of Edinburgh (Scotland) Research Ethics Committee approval #1702/95/4/72 or UBC Clinical Research Ethics Board protocol C02-0091, using sodium citrate solution (Phoenix Pharma Ltd., Gloucester, UK) as an anticoagulant or Vacutainer® collection tubes containing sodium heparin (BD Biosciences, Mississauga, Ontario, Canada, or Oxford, UK).
For neutrophil isolation, blood was centrifuged at 300 g for 20 min at room temperature, platelet-rich plasma was removed, and cells were suspended gently in 1% Dextran T-500 (Amersham Pharmacia Biotech, Buckingham, UK) in 0.9% saline and sedimented for 30 min at room temperature. The leukocyte-rich upper layer was then fractionated by using three-step discontinuous, isotonic Percoll gradients as described previously [27]. Briefly, cells were centrifuged at 300 g for 6 min, resuspended in 55% isotonic Percoll (Amersham Pharmacia Biotech), layered on top of 68% and 81% isotonic Percoll layers, and centrifuged at 700 g for 20 min at room temperature. Neutrophils were collected, washed in phosphate-buffered saline (PBS) without calcium or magnesium, and resuspended in Iscoves's Dulbecco's modified Eagle's medium (IDMEM; Invitrogen, Paisley, UK, or Burlington, Ontario, Canada) with 10% (v/v) heat-inactivated fetal bovine serum (FBS). Alternatively, neutrophils were purified by Ficoll-Paque gradient centrifugation as described previously [28]. Briefly, cells were centrifuged at 200 g for 7 min, remaining erythrocytes were lysed hypotonically with ice-cold, distilled water for 30 s, followed by restoration of tonicity with 2.5% saline, and neutrophils were separated by centrifugation over a Ficoll-Paque Plus (Amersham Pharmacia Biotech) density gradient at 400 g for 25 min at 4°C. The cells were washed with Krebs-Ringer phosphate buffer (pH 7.3), containing glucose (10 mM) and Mg2+ (1.5 mM), and resuspended in RPMI-1640 media (Invitrogen), supplemented with 10% (v/v) heat-inactivated FBS, 1% (v/v) L-glutamine, and 1 nM sodium pyruvate. Polymorphonuclear leukocytes isolated were 95–98% neutrophils using morphological criteria, and viability was assessed by trypan blue exclusion. Dose-dependent LL-37-mediated inhibition of neutrophil apoptosis was observed by fluorescent-activated cell sorter (FACS), irrespective of the anticoagulant or methodology used to isolate cells. The former method was used in receptor inhibitor studies and the latter method, in analyses by Western immunoblot and enzyme-linked immunosorbent assay (ELISA).
Neutrophil apoptosis
Freshly isolated human blood neutrophils were incubated at 37°C, 5% CO2, for 0, 1, 4, or 20 h, at 6 × 105 cells in 200 μl IDMEM with 10% (v/v) heat-inactivated FBS in flexible, 96-well culture plates in the presence of LL-37, GM-CSF, or WKYMVm at the stated concentrations or a vehicle control, at least in duplicate. Where inhibitors were used, these (or vehicle control) were added 30 min before the addition of LL-37. Apoptosis was assessed by flow cytometry using fluorescein isothiocyanate (FITC)-labeled Annexin V (Roche Applied Sciences, Lewes, East Sussex, UK), diluted 1:500 with binding buffer (Hanks' balanced salt solution with 5 mM CaCl) and 5 μg/ml propidium iodide (Molecular Probes/Invitrogen) at 4°C. The samples were analyzed using a FACSCalibur system (BD Biosciences), counting a minimum of 10,000 cells. In addition to FACS analyses, apoptosis was assessed using standard morphological analysis of distinctive apoptotic morphology. Samples (100 μl) of cells were cytocentrifuged, fixed in methanol, stained with Reastain Quick-Diff (Reagena, Garridor, UK), and examined using oil immersion microscopy. Total cell counts were performed in duplicate by haemocytometer and light microscopy (using standard methodology) for each well upon removal of gently resuspended cells for FACS analysis and cytospin.
Western immunoblotting
Fresh human blood neutrophils (7.5×106), per condition, in 5 ml RPMI-1640 media [supplemented with 10% (v/v) heat-inactivated FBS, 1% (v/v) L-glutamine, and 1 nM sodium pyruvate] were exposed to LL-37 at the concentrations detailed or endotoxin-free water as a vehicle control and incubated at 37°C, 5% CO2, for 4 h. Cells were washed with ice-cold PBS containing 1 mM sodium orthovanadate (Sigma-Aldrich) and lysed with 150 μl 1% Triton X-100, 10 mM Tris, pH 7.5, 150 mM NaCl, 2 mM EDTA, 1 mM phenylmethylsulfonyl fluoride buffer containing Sigma protease inhibitor cocktail [104 mM 4-(2-aminoethyl) benzenesulfonyl fluoride, 0.08 mM aprotinin, 2 mM leupeptin, 4 mM bestatin, 1.5 mM pepstatin A, 1.4 mM E-64] and phosphatase inhibitor cocktail 2 (sodium orthovanadate, sodium molybdate, sodium tartrate, imidazole). The protein concentrations of lysates were determined using a bicinchoninic acid assay (Pierce, Rockford, IL, or Cramlington, UK). Equivalent lysate (15–40 μg) was resolved on a 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA), which were blocked for 1 h with 20 mM Tris, pH 7.4, 150 mM NaCl, and 0.1% Tween 20 (TBST) containing 5% skimmed milk powder (TBST/milk). Subsequently, the nitrocellulose membranes were incubated with anti-Mcl-1, anti-BID, anticaspase-3, or anti-cleaved caspase-3 antibodies at 1/1000 dilution in TBST/milk for 1 h at room temperature. Membranes were washed for 30 min in TBST and then incubated with a 1/5000 dilution of HRP-conjugated goat antimouse or antirabbit antibody (in TBST/milk) for 1 h. The membranes were washed for 30 min in TBST and developed with chemiluminescence peroxidase substrate (Sigma-Aldrich), according to the manufacturer's instructions. To quantify bands, the luminescence was detected with Versadoc (Bio-Rad). The blots were stripped using Restore™ Western blot stripping buffer (Pierce), according to the manufacturer's instructions, and reprobed with an anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody, and densitometry was performed to allow correction for protein loading.
Chemotaxis assay
Transwell® polyester-permeable supports (pore size 3.0 μm, diameter 6.5 mm; Corning Life Sciences, UK) were used for chemotaxis studies. Briefly, each chemoattractant sample (600 μl) was loaded into the lower well, and 100 μl PBS without Ca2+ and Mg2+ containing 1 × 105 fresh human blood neutrophils was added to the apical compartment of each transwell. The plates were incubated for 60 min at 37°C in 5% CO2. Chemoattractants tested were sterile PBS without Ca2+ and Mg2+ (negative control) and WKYMVm. For inhibition studies, neutrophils were pretreated with 10 μM WRW4 for 30 min at room temperature prior to use in the chemotaxis assay. Following incubation, the supernatants from the upper and lower compartments were removed, and a cytospin was performed. The upper surface of the polyester membranes was wiped with cotton buds to remove nonmigrated cells, and the cells on the underside of the membranes were fixed with methanol and stained with a Reastain Quick-Diff (Reagena). The polyester membranes with migrated cells were removed with a curved, serrated scalpel, and mounted “cell side up” on a microscope slide. The migrated cells were then counted using a Zeiss light microscope at 1000× magnification. For each membrane, five fields of view were counted, and results were expressed as a chemotactic index correct for migration observed in the negative control.
Epithelial cell culture
Clonetics™ normal human bronchial epithelial (NHBE) cells were purchased from Cambrex BioScience Ltd. (Wokingham, UK) and were cultured and maintained in bronchial epithelial cell growth media (BEGM; Cambrex BioScience Ltd.), strictly in accordance with the manufacturer's instructions. BEGM is a basal medium (Cambrex BioScience Ltd.) supplemented with bronchial epithelial cell SingleQuots® growth factors and supplements (Cambrex BioScience Ltd.) as a serum substitute optimized for growth, and appropriate differentiation of these primary cells was used according to the manufacturer's instructions.
In situ cell death detection terminal deoxynucleotidyl transferase mediated deoxyuridine triphosphate nick-end labeling (TUNEL) assay
Transwell® polyester-permeable supports (pore size 0.4 μm, diameter 6.5 mm; Corning Life Sciences) were equilibrated for 45 min in BEGM before addition of 100 μl media containing 3.1 × 105 NHBE cells/ml into the apical compartment, with 600 μl medium in the basal compartment. Cells were allowed to adhere overnight at 37°C, 5% CO2. The cells were exposed in duplicate to LL-37 at the concentrations described or endotoxin-free water as a vehicle control in fresh BEGM with the addition of 10% (v/v) heat-inactivated FBS in the apical and basolateral compartments of all samples and incubated for 18–20 h at 37°C, 5% CO2. For caspase inhibition studies, cells were exposed to 50 μM caspase inhibitor I (Z-VAD-FMK) for 4 h prior to LL-37 treatment. Cells were fixed in 10% neutral-buffered formalin (3.7% formaldehyde) for 10 min, washed once in PBS, permeabilized in ice-cold 0.1% Triton X-100/0.1% sodium citrate for 3 min, and then washed twice with PBS. An in situ cell death detection kit (Roche Applied Science) was used according to the manufacturer's instructions. The polyester membranes with cells were removed with a curved, serrated scalpel and mounted cell side up on a microscope slide in 50 μl Vectashield® Hardset™ [containing 4′,6-diamidino-2-phenylindole (DAPI)]. For each membrane, three random fields of view were counted, each containing more than 100 cells, using an Axiovert S100 fluorescent microscope and analyzed using OpenLAB 3.0 software. The level of apoptosis was expressed as the percentage TUNEL-positive cells per DAPI-positive nuclei in the field of view.
Detection of cytokines
Fresh human blood neutrophils were plated at 4 × 105 cells in 400 μl RPMI-1640 media [supplemented with 10% (v/v) heat-inactivated FBS, 1% (v/v) L-glutamine, 1 nM sodium pyruvate], respectively, in 48-well plates. Cells were then incubated in media for 20 h in the presence of P. aeruginosa H103 LPS, LL-37, cytokines (at the stated concentrations), or endotoxin-free water as a vehicle control, in at least triplicate. Supernatants were collected and stored at −20°C until use. The concentrations of TNF-α and IL-8 in the supernatants were measured using commercially prepared ELISA plates in accordance with the manufacturer's instructions (BioSource International, Camarillo, CA).
Statistical analysis
Student's t-tests were performed to determine statistical significance, and P ≤ 0.05 was considered significant. Values shown are expressed as mean ± sem.
RESULTS
LL-37 inhibits neutrophil apoptosis
To evaluate the effect of LL-37 exposure on spontaneous neutrophil apoptosis, fresh human peripheral blood neutrophils were isolated and incubated in culture over a range of LL-37 concentrations in the presence of 10% FBS. The level of apoptosis was assessed by FACS quantification of FITC-labeled annexin V and propidium iodide staining. Confirmation of apoptosis was obtained by morphological evaluation of cytospins, revealing the classical appearance of cell shrinkage and nuclear condensation in apoptotic cells (Fig. 1A). In untreated cells, spontaneous apoptosis was observed in 60.6% ± 3.6% (n=9) of cells at 20 h. Exposure to LL-37 was demonstrated to inhibit neutrophil apoptosis significantly at 20 h (Fig. 1B). This inhibition was dose-dependent, with significant inhibition (P=0.004) at LL-37 concentrations of 250 ng/ml or greater, and profound inhibition of apoptosis observed at 10 μg/ml (diminished by 86% of control levels; P=1×10−9) or above.
Fig. 1.
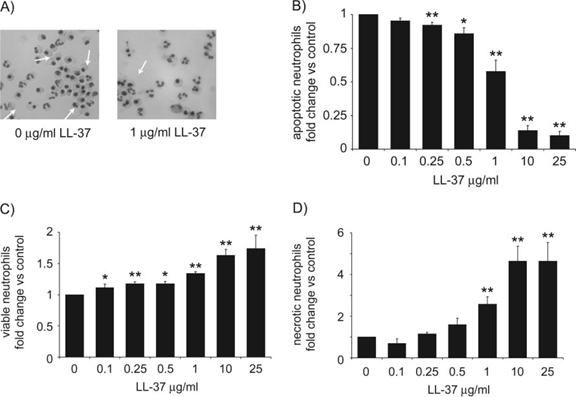
Inhibition of neutrophil apoptosis by LL-37. Neutrophils were incubated for 20 h over a range of LL-37 concentrations in the presence of 10% FBS. Modulation of spontaneous apoptosis was examined by FACS analysis and morphology. (A) Representative fields from cytospins. White arrows indicate examples of apoptotic neutrophils, 320× original magnification. (B–D) FACS analysis was used to determine the percentage of neutrophils, which were (B) apoptotic (FITC-annexin V-positive, propidium iodide-negative), (C) viable (FITC-annexin V-negative, propidium iodide-negative), and (D) necrotic (FITC-annexin V-positive, propidium iodide-positive). Figures represent the percentage of cells in LL-37-treated samples as a proportion of that observed in the vehicle alone-treated controls to correct for donor variation and indicate mean values ± sem, for n ≥ 6 for each condition, performed in duplicate, using seven different donors. Significance was assessed using absolute values in LL-37-treated samples compared with vehicle alone-treated controls by paired sample t-test analyses. *, P ≤ 0.05; **, P ≤ 0.01.
The consequence of LL-37-mediated inhibition of apoptosis in neutrophils at 20 h was evaluated by FACS analysis, assessing the number of annexin V-negative/propidium iodide-negative (viable) and annexin V-positive/propidium iodide-positive (necrotic) cells. In the absence of LL-37, 26.2% ± 2.6% (n=9) of cells were viable, and 11.6% ± 1.5% (n=9) of cells were necrotic at 20 h. Exposure to LL-37 resulted in dose-dependent increases in the proportions of viable (Fig. 1C) and necrotic (Fig. 1D) cells. The proportion of viable cells increased significantly after exposure to 100 ng/ml LL-37 (P=0.04) or greater and by up to 63% of control levels at 10 μg/ml. In contrast, the proportion of necrotic cells did not increase significantly at concentrations of LL-37 below 1 μg/ml but was greatly increased by exposure to higher levels of LL-37, by approximately fourfold of control levels at 10 μg/ml.
To confirm that exposure to LL-37 did not induce early neutrophil cytolysis, cells were also examined over a time course using FACS analyses and total cell counts. Negligible levels of apoptosis and necrosis were observed by FACS at 0 h (data not shown), 1 h (Fig. 2A), and 4 h (Fig. 2B), but inhibition of apoptosis by LL-37 was confirmed at 20 h, with resultant increases in the numbers of viable and necrotic cells observed (Fig. 2C), as described. In addition, total cell counts were performed by haemocytometer (Fig. 2D) to examine the possibility that cytolytic destruction could remove cells from FACS and cytological analyses. No significant loss of cells was observed at 0, 1, or 4 h after exposure to LL-37 at any concentration tested or at 20 h at ≤10 μg/ml LL-37. However, a significant decrease (P=0.02) in total cell number was observed only after 20 h exposure to quite high concentrations of LL-37 (25 μg/ml). Consequently, as a proportion of the initial cell population, FACS and cytospin-based analyses could have modestly underestimated the degree of LL-37-mediated inhibition of apoptosis and the increase in necrosis, induced by the most extreme conditions examined (20 h exposure to 25 μg/ml of LL-37).
Fig. 2.
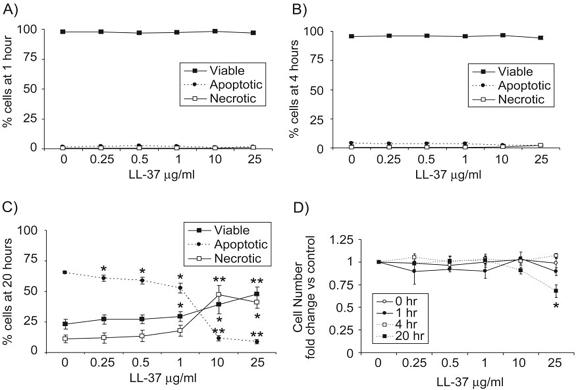
LL-37-mediated inhibition of neutrophil apoptosis occurs in the absence of cytolysis. Neutrophils were incubated over a range of LL-37 concentrations in the presence of 10% FBS for 0, 1, 4, or 20 h. (A–C) FACS analysis was used to determine the percentage of neutrophils that were apoptotic (FITC-annexin V-positive, propidium iodide-negative), viable (FITC-annexin V-negative, propidium iodide-negative), and necrotic (FITC-annexin V-positive, propidium iodide-positive) at (A) 1 h, (B) 4 h, and (C) 20 h. (D) Total cell counts were performed in duplicate using a haemocytometer and represented as a proportion of the number of cells in the vehicle-alone controls at each time-point. No significant difference was observed between control samples over the time course. Figures indicate mean values ± sem for n = 4 for each condition using four different donors. Significance was assessed using absolute values in LL-37-treated samples compared with vehicle alone-treated controls by paired sample t-test analyses. *, P ≤ 0.05; **, P ≤ 0.01.
LL-37 exposure modulates expression of Mcl-1 and inhibits cleavage of BID and procaspase-3 in neutrophils
To study the mechanisms by which LL-37 can modulate the apoptosis of neutrophils, the expression of critical apoptosis-regulating Bcl-2 family proteins and the cleavage of procaspase-3 were examined by Western immunoblot. Fresh human peripheral blood neutrophils were examined at 4 h after exposure to LL-37 in the presence of 10% FBS, a time-point chosen to precede any substantial apoptosis or necrosis with 94–96% cell viability observed, irrespective of exposure to LL-37 (Fig. 2B). Expression of the antiapoptotic protein Mcl-1 is of particular importance in neutrophils [29] and was found to be significantly higher in neutrophils after 4 h exposure to LL-37 (Fig. 3A) with dose-dependent regulation. Cleavage of the “BH3 domain only” Bcl-2 protein BID by caspase-8 generates a proapoptotic fragment [30, 31] and was demonstrated to be inhibited significantly in neutrophils exposed to LL-37 (Fig. 3B). In addition, significantly higher levels of the inactive procaspase-3 protein, with a corresponding, significant decrease in levels of active-cleaved caspase-3, were observed after exposure to LL-37 (Fig. 4, A and B), indicating inhibition of the activation pathways for the effector caspase-3 and correlating with LL-37-mediated inhibition of neutrophil apoptosis.
Fig. 3.
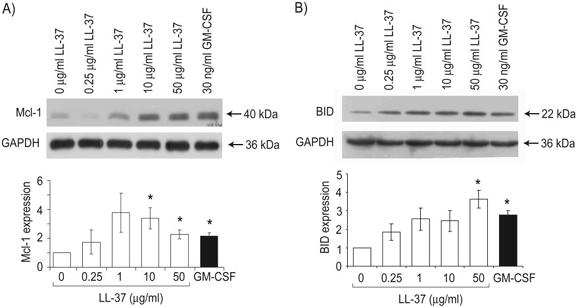
Modulation of neutrophil Mcl-1 expression and cleavage of BID in response to LL-37. Neutrophils were exposed to a concentration range of 0.25–50 μg/ml LL-37, 30 ng/ml GM-CSF as a positive control, or endotoxin-free water as a vehicle control for 4 h in the presence of 10% FBS. Whole cell protein lysates were prepared and analyzed by SDS-PAGE and Western immunoblotting. Immunoblots for expression of (A) Mcl-1 and (B) uncleaved BID are shown, each representative of n = 3 different donors, and expression of the housekeeping protein GAPDH was assessed as a loading control. Quantitative densitometry was performed, corrected for protein loading, expressed as a proportion of the vehicle alone-treated control sample, and displayed as mean values ± sem for n = 3 different donors. t-Test analyses were used to compare Mcl-1 or BID expression in LL-37- or GM-CSF-exposed samples with vehicle alone-treated controls. *, P ≤ 0.05.
Fig. 4.
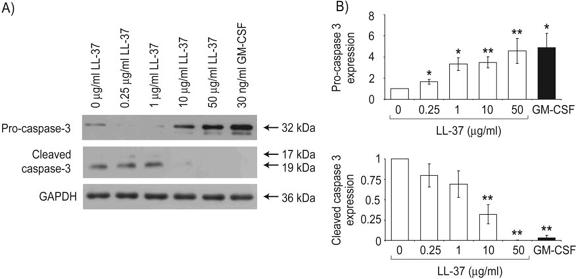
Modulation of procaspase-3 cleavage in LL-37-treated neutrophils, which were exposed to a concentration range of 0.25–50 μg/ml LL-37, 30 ng/ml GM-CSF as a positive control, or endotoxin-free water as a vehicle control for 4 h in the presence of 10% FBS. Whole cell protein lysates were prepared and analyzed by SDS-PAGE and Western immunoblotting. (A) Immunoblots for expression of inactive procaspase-3 and active-cleaved caspase-3 are shown, representative of n = 5 different donors, and expression of the housekeeping protein GAPDH was assessed as a loading control. (B) Quantitative densitometry was performed, corrected for protein loading, expressed as a proportion of the vehicle alone-treated control sample, and displayed as mean values ± sem for n = 5 different donors. t-Test analyses were used to compare procaspase-3 and cleaved caspase-3 expression in LL-37- or GM-CSF-exposed samples with vehicle alone-treated controls. *, P ≤ 0.05; **, P ≤ 0.01.
LL-37-induced inhibition of neutrophil apoptosis involves multiple signaling pathways
The immunomodulatory effects of LL-37 have been proposed to be dependent on signaling through a number of receptors, potentially relevant in this system, including the G-protein-coupled receptor FPRL-1 [19] and the purinergic receptor P2X7 [32]. Additional, as-yet-unidentified G-protein-coupled receptors and undetermined high- and low-affinity receptors have also been proposed [33-35]. To assess the possible significance of these receptors, human peripheral blood neutrophils were preincubated with PTX (to inhibit G-protein-coupled receptors), WRW4, or oxidized ATP (specific inhibitors of FPRL-1 [36] and P2X7 [32] receptors, respectively) before exposure to a concentration range of LL-37 in the presence of 10% FBS. The levels of spontaneous apoptosis were subsequently determined after 20 h incubation (Fig. 5A). No significant LL-37-mediated inhibition of neutrophil apoptosis occurred in the presence of oxidized ATP or PTX at concentrations of LL-37 up to 1 μg/ml, demonstrating effective blockade of LL-37 activity. In contrast, WRW4, at the optimal concentration for near-complete inhibition of the FPRL-1-specific agonist WKYMVm [36], did not impair the activity of LL-37, and significant LL-37-mediated inhibition of neutrophil apoptosis was observed at 250 ng/ml (P=0.009) and 1 μg/ml LL-37 (P=0.01) in the presence of WRW4. To further evaluate the role of FPRL-1 in this system, human peripheral blood neutrophils were incubated with the FPRL-1-specific agonist WKYMVm at 0.2 μM and 10 μM (approximately equimolar with 1 μg/ml and 50 μg/ml LL-37, respectively). No significant effects of this agonist were observed on the level of spontaneous apoptosis after 20 h incubation (Fig. 5B). To confirm the biological activity of our WKYMVm and WRW4 peptides, chemotaxis of fresh human neutrophils was studied. As described previously [37], significant chemotaxis of neutrophils was observed in response to 10 μM WKYMVm (P=0.001). This chemotaxis was inhibited significantly by preincubation with 10 μM WRW4 (P=0.02; Fig. 5C). These data suggest that P2X7 receptors and an undetermined G-protein-coupled receptor other than FPRL-1 are required for LL-37-mediated inhibition of neutrophil apoptosis.
Fig. 5.
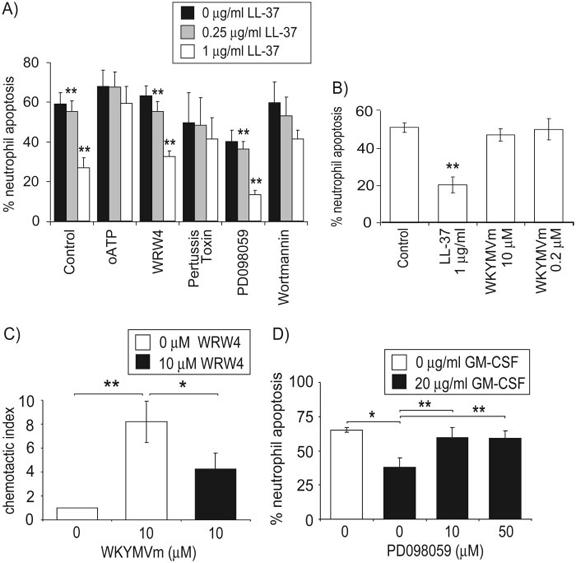
LL-37-induced inhibition of neutrophil apoptosis involves multiple signaling pathways. Neutrophil apoptosis over 20 h incubation was examined in duplicate by FACS analysis for FITC-annexin V-positive, propidium iodide-negative cells after (A) incubation with 0.25 μg/ml or 1 μg/ml LL-37 or endotoxin-free water as a vehicle control, added 30 min after 100 μM-oxidized ATP, 10 μM WRW4, 200 ng/ml PTX, 50 μM PD098059, 5 μM wortmannin, or a vehicle-alone control in the presence of 10% FBS. Results represent the percentage of apoptotic cells as mean ± sem for n ≥ 4 per condition from five different donors. Paired sample t-test analyses were used to compare LL-37-treated samples with controls under the same inhibitor conditions. **, P ≤ 0.01, or (B) incubation with 1 μg/ml LL-37, 0.2 μM or 10 μM WKYMVm, or a vehicle-alone control. Results represent the percentage of apoptotic cells as mean ± sem for n = 3 from three different donors. Paired sample t-test analyses were used to compare treated samples with controls. **, P ≤ 0.01. (C) Neutrophil chemotaxis was assessed in triplicate in response to 10 μM WKYMVm or vehicle-alone control after preincubation with 10 μM WRW4 or vehicle-alone control, and the chemotactic index was displayed as mean ± sem for n = 3 from three different donors. Paired sample t-test analyses were used to compare WKYMVm-treated samples with controls and WRW4-pretreated samples with WKYMVm alone. *, P ≤ 0.05; **, P ≤ 0.01. (D) Neutrophil apoptosis over 20 h incubation was examined in duplicate by FACS analysis after incubation with 20 μg/ml GM-CSF or endotoxin-free water as a vehicle control, added 30 min after 10 μM or 50 μM PD098059 or vehicle-alone control. Results represent the percentage of apoptotic cells as mean ± sem for n = 3 per condition from three different donors. Paired sample t-test analyses were used to compare GM-CSF-treated samples with controls and PD098059-pretreated samples with GM-CSF alone. *, P ≤ 0.05; **, P ≤ 0.01.
LL-37 has also been demonstrated to induce MAPK activation in a cell type-specific manner, through a PTX-insensitive pathway [22, 38], and to enhance the GM-CSF-dependent activation of these pathways in primary monocytes [38]. GM-CSF is a potent inhibitor of neutrophil apoptosis, with this effect mediated through the activation of ERK1/2 MAPK and PI-3K pathways [39]. Thus, to determine the significance of these pathways in this system, human peripheral blood neutrophils were preincubated with PD098059 (inhibitor of the ERK1/2 MAPK pathway via MAPK kinase) or wortmannin (PI-3K inhibitor) before exposure to a dose range of LL-37. The levels of spontaneous apoptosis were subsequently determined after 20 h incubation (Fig. 5A). The inhibition of the ERK1/2 MAPK pathway alone resulted in a degree of inhibition of neutrophil apoptosis but did not impair the effects of LL-37, and significant LL-37-mediated inhibition of neutrophil apoptosis was observed at 250 ng/ml (P=0.0005) and 1 μg/ml LL-37 (P=0.005) in the presence of PD098059. In contrast, although some inhibition of LL-37 was observed in the presence of wortmannin, concentrations of LL-37 up to 1 μg/ml were unable to inhibit neutrophil apoptosis significantly in the presence of this PI-3K blockade. To confirm the biological activity of our PD098059, GM-CSF-mediated inhibition of neutrophil apoptosis was demonstrated to be blocked significantly by preincubation with 10 μM or 50 μM PD098059 (P=0.002 and P=0.006, respectively; Fig. 5D), as described previously [39]. These data suggest that LL-37 may use the PI-3K but not the ERK1/2 MAPK pathway in the inhibition of neutrophil apoptosis.
LL-37 differentially modulates neutrophil cytokine responses to inflammatory stimuli
To evaluate the function of primary human neutrophils prevented from undergoing spontaneous apoptosis by exposure to LL-37, cytokine responses to inflammatory stimuli were examined. Neutrophils were incubated for 20 h with LPS, IL-1β, or TNF-α in the presence of 10 μg/ml LL-37 (the optimal concentration for the inhibition of apoptosis in the absence of cytolytic cell loss at this time-point) and 10% FBS, with subsequent determination of the cytokine responses by ELISA analysis of cell supernatants (Fig. 6, A and B). As expected, on the basis of the well-characterized, anti-endotoxic effects ascribed to this peptide in the responses of other cell types, LL-37 inhibited the release of IL-8 and TNF-α in response to LPS exposure. However, in contrast, LL-37 exposure of neutrophils enhanced significantly, but modestly, the release of IL-8 and TNF-α (P=0.04 and P=0.01, respectively) in response to stimulation with IL-1β and mediated a trend toward enhanced IL-8 production in response to TNF-α (with significant up-regulation in two out of four donors; P<0.01). These data demonstrate functional cytokine responses in LL-37-treated neutrophils and indicate that LL-37 modulates the cytokine responses of neutrophils to inflammatory signals in a stimulus-specific manner.
Fig. 6.
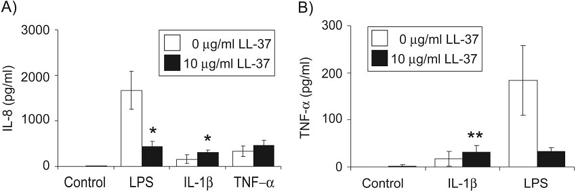
Modulation of neutrophil cytokine responses by LL-37. The IL-8 or TNF-α production by neutrophils was assessed by ELISA analysis of culture supernatants following incubation of cells for 20 h in the presence of (A) 100 ng/ml LPS, 10 ng/ml IL-1β, 100 ng/ml TNF-α, or vehicle-alone control, with 10 μg/ml LL-37 or vehicle-alone control in the presence of 10% FBS; or (B) 100 ng/ml LPS, 50 ng/ml IL-1β, or vehicle-alone control with 10 μg/ml LL-37 or vehicle-alone control in the presence of 10% FBS. Data represent means ± sem for n ≥ 3 replicates per condition from four different donors. Paired sample t-test analyses were used to compare LL-37-treated samples with controls under the same stimulatory conditions. *, P ≤ 0.05; **, P ≤ 0.01.
LL-37 induces apoptosis in primary airway epithelial cells
In contrast to the potent LL-37-mediated inhibition of apoptosis observed in neutrophils, we have recently described the capacity of LL-37 to mediate a dose-dependent induction of apoptosis in immortalized airway epithelial cells lines [40]. Incubation with 25 μg/ml LL-37 or higher concentrations induced a human serum-sensitive induction of TUNEL positivity and cleavage of caspase-3 (to the active form) in A549 human lung epithelial cells and 16HBE4o-human airway epithelial cells [40]. To confirm the significance of these observations in nonimmortalized cells, the effect of LL-37 on primary human bronchial epithelial cells cultured on semipermeable growth support membranes was evaluated in parallel with the freshly isolated human neutrophils described above, incubated for 20 h over a concentration range of LL-37 or vehicle-alone control, all in the presence of 10% FBS (Fig. 7A). Analysis demonstrated a significant LL-37-mediated increase in TUNEL-positive cells, suggestive of caspase-activated DNase activity during apoptosis upon exposure to 10 μg/ml LL-37 (P=0.003) and above, and up to 55% of cells were TUNEL-positive after exposure to 100 μg/ml LL-37. Preincubation with a caspase inhibitor blocked the proapoptotic effects of exposure to 25 μg/ml and 50 μg/ml LL-37. This was statistically significant at 50 μg/ml LL-37 (where a greater induction of apoptosis was observed; P=0.05) but despite being consistently inhibitory, failed to reach statistical significance at 25 μg/ml LL-37 (where variation in the lower absolute level of induction of apoptosis was observed; P=0.08). These data are indicative of caspase-dependent, programmed cell death in these LL-37-treated primary airway epithelial cells, although the possibility of an additional, contributory caspase-independent apoptosis, as described previously in a carcinoma cell line exposed to an LL-37 derivative [41], cannot be excluded.
Fig. 7.
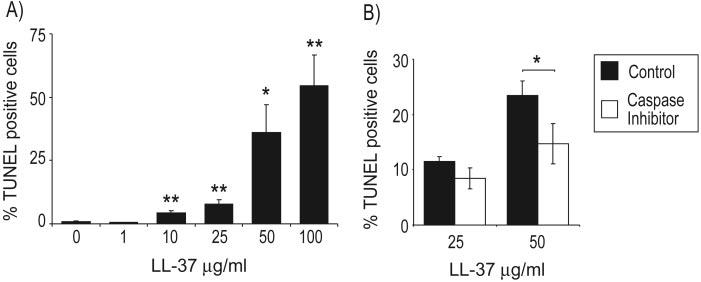
Induction of caspase-dependent cell death in primary bronchial epithelial cells by LL-37. Primary human bronchial epithelial cells were exposed to (A) a concentration range of 1–100 μg/ml LL-37 or endotoxin-free water as a vehicle control, all in the presence of 10% FBS for 20 h or (B) pretreated with 50 μM caspase inhibitor I (Z-VAD-FMK) for 4 h prior to the addition of 25 μg/ml or 50 μg/ml LL-37 in the presence of 10% FBS for 20 h. Cells were fixed, and apoptosis was assessed by TUNEL assay. Three random fields of view, each containing more than 100 cells, were counted for each sample, and the percentage of TUNEL-positive cells in the field of view was expressed as a percentage of the number of DAPI-positive nuclei. Data represent means ± sem for n = 6 (A) or n = 3 (B). t-Test analyses were used to compare LL-37-treated samples with controls. *, P ≤ 0.05; **, P ≤ 0.01.
DISCUSSION
Various cationic host-defense peptides [including LL-37 and the murine homologue cathelicidin-related antimicrobial peptide (mCRAMP)] have been demonstrated recently to have multiple immunomodulatory capabilities, potentially representing key mechanisms by which these peptides can enhance host clearance of infection in vivo. The full extent of these immunomodulatory functions and the precise mechanisms by which these peptides contribute to innate immunity remain undetermined in vivo and are of clear significance in the development of such peptides and their synthetic analogs as novel, antimicrobial therapeutics for multiple antibiotic-resistant infections.
The microbicidal activity of LL-37 is acutely sensitive to serum proteins and largely inhibitable under physiologically relevant, ionic conditions [42], although other recently proposed cleavage forms of hCAP-18 may have more effective microbicidal activities [43, 44]. However, various immunomodulatory properties have been demonstrated in physiological ionic environments, including leukocyte chemotaxis, stimulation of epithelial cell IL-8 production, post-translational modification of IL-1β, modulation of macrophage transcription, angiogenesis, enhanced wound-healing, modulation of dendritic cell differentiation and function, and adjuvant properties [12, 14, 19-22, 32, 34, 35, 45-48]. Demonstrating the relative contribution of direct microbicidal activity versus antimicrobial immunomodulatory functions in vivo remains challenging. Studies of mCRAMP indicate that despite high minimum inhibitory concentration values (even against a susceptible mutant bacteria), direct microbicidal activity may contribute to host defense in vivo in specific systems, perhaps at higher concentrations under favorable conditions in a protected niche or functioning synergistically [49, 50]. However, we have recently described in vivo protection against infection in animal models using a synthetic CHDP derivative with no direct in vitro antimicrobial activity [42]. Thus, the multiple immunomodulatory properties described for LL-37 in vitro and in vivo seem likely to be fundamental to the importance of this peptide to the innate immune system.
The precise mechanisms underlying the immunomodulatory effects of LL-37 are in many cases unknown. A number of identified and undefined receptors have been described for LL-37, including FPRL-1 and other G-protein-coupled receptors, epidermal growth factor receptor (EGFR), responsive via metalloproteinase-mediated cleavage of membrane-anchored EGFR ligands, and P2X7 receptors acting via caspase-1 [19, 22, 32-35, 38]. However, the relative significance of these receptors remains unclear, and traditional, ligand-binding receptor mechanisms may not fully determine peptide activities [44]. In addition, MAPK activation is involved in G-protein-independent LL-37 stimulation of monocytes [38], and endocytic peptide uptake in airway epithelial cells in vitro is required for LL-37-induced IL-8 expression [33, 42].
We demonstrate that in addition to the immunomodulatory functions of LL-37 described previously, this CHDP is capable of modulating apoptotic pathways in primary human innate-immune effector cells. The capacity of LL-37 to act as a potent inhibitor of spontaneous neutrophil apoptosis was demonstrated to involve signaling via P2X7 receptors and G-protein-coupled receptors other than FPRL-1. These data confirm and complement some of the observations made in a recent related paper [51]. However, in contrast to our observations, Nagaoka et al. [51] observed a role for FPRL-1 in LL-37-mediated inhibition of neutrophil apoptosis. The reasons for this difference remain to be determined but may relate to alternate sources of FPRL-1 antagonistic and agonistic peptides (subjected to functional validation by reproducing well-characterized activities of these peptides in other systems in our study), different peptide solvents, or technical differences in neutrophil purification, which might result in differential background levels of contaminant cell types with FPRL-1-mediated responses to LL-37, which could influence the response of neutrophils in these assays. Nevertheless, the observation that a combination of receptor types could be involved in LL-37 signaling was also the conclusion of a recent study examining the effects of LL-37 on keratinocyte functions [44]. That study, interestingly, also demonstrated similar responses when using D-LL-37 (composed of amino acids in the synthetic D form), arguing against a highly structure-specific interaction between LL-37 and cell surface receptors. Thus, we cannot exclude a complexity exceeding the use of P2X7 receptors and G-protein-coupled receptors in LL-37-mediated inhibition of neutrophil apoptosis. Regardless, our data suggest that this process involves subsequent downstream activation of PI-3K but not the ERK1/2 MAPK pathway. The latter observation is in keeping with the recent demonstration that in contrast to its effects in monocytes and epithelial cells [22, 38], LL-37 does not stimulate ERK1/2 phosphorylation in neutrophils [20].
We also demonstrate that LL-37 mediates antiapoptotic effects in neutrophils via an inhibitory effect on the activation of the effector caspase-3 and by altering the balance of Bcl-2 family proteins. It is interesting to note that these effects, observed after 4 h of incubation with LL-37, preceded substantial membrane translocation of phosphatidylserine (PS), assessed by FACS analyses of FITC annexin V/propidium iodide-stained cells. Although translocation of PS was not observed in substantial numbers of neutrophils at 4 h, compared with later time-points, there was an increase in FITC-annexin V-positive/propidium iodide-negative cells from ∼1% at 1 h to ∼4% at 4 h in control cells (Fig. 2, A and B). After this time-point, apoptosis determined by translocation of PS increased rapidly, and ∼15% of control neutrophils were FITC-annexin V-positive/propidium iodide-negative at 6 h (data not shown), demonstrating the temporal sequence of events in apoptosis of these cells. These data support previous studies indicating that the translocation of PS is a caspase-3-dependent event downstream of caspase activation [52] and that LL-37-mediated inhibition of caspase-3 cleavage precedes effects on translocation of PS in neutrophils undergoing spontaneous apoptosis.
LL-37 modulated expression of the anti-inflammatory protein Mcl-1, preventing the loss of expression associated with spontaneous apoptosis. This Bcl-2 family protein has a short half-life, providing rapid response to environmental stimuli and playing a critical role in promoting neutrophil survival by inhibiting mitochondrial damage and cytochrome c release [29, 53]. LL-37 also inhibited cleavage of the BH3 domain-only Bcl-2 protein BID, therefore decreasing generation of the proapoptotic p15 BID cleavage fragment, believed to translocate to the mitochondria and trigger release of cytochrome c during the induction of apoptosis [30, 31]. Cleavage of BID by caspase-8 is critical for death receptor-induced apoptosis but also plays an important role in spontaneous neutrophil apoptosis, even in the absence of death receptor stimulation [54]. In addition to the inhibition of cleavage of the key effector caspase-3, these observations suggest that LL-37 exerts its antiapoptotic effect on neutrophils by acting upstream of mitochondria on intrinsic and traditionally extrinsic pathways for the induction of programmed cell death.
The consequences for LL-37-mediated inhibition of neutrophil apoptosis in vitro were demonstrated to be dependent on the concentration of LL-37. After 20 h exposure to lower concentrations of LL-37, significant increases were observed in the proportion of viable cells at the expense of apoptotic cells. However, at higher concentrations of LL-37, despite further increases in the proportion of viable cells, the more dramatic decreases in apoptotic cells were accompanied by an increase in neutrophil necrosis. This switch in cellular fate, evident at approximately 10 μg/ml, could represent a transition to inflammatory concentrations of LL-37 in vivo [4]. The neutrophil necrosis observed is likely a consequence of the inhibition of apoptotic pathways, rather than secondary necrosis of apoptotic cells, given the potent inhibition of apoptotic pathways observed after 4 h exposure to LL-37, before any substantial, detectable cell death, and bears similarities to the recently reported effects of LPS in inhibiting apoptosis but inducing necrosis in neutrophils [55]. In contrast, the antiapoptotic factor GM-CSF acts primarily as a potent neutrophil-survival factor [56]. Thus, although LL-37-mediated inhibition of apoptosis led to a dose-dependent increase in neutrophil survival, higher concentrations of peptide also promoted an additional switch from apoptotic to necrotic cell death.
The in vivo consequences of LL-37-mediated inhibition of neutrophil apoptosis remain to be determined. Low-level, acute increases in LL-37 may primarily enhance neutrophil survival, promoting the clearance of infection. However, although necrosis induced by higher concentrations of LL-37 could be beneficial to the host by amplifying the acute inflammatory response (as proposed previously for LPS-induced necrosis [55]), the uncontrolled release of neutrophil contents in chronic inflammation would be expected to result in host damage and impair the resolution of inflammation. In addition to enhancing neutrophil survival, LL-37 has previously been demonstrated to be chemotactic for neutrophils in vitro, directly [19] and indirectly, by inducing IL-8 production by epithelial cells [12], and the murine homologue CRAMP has been shown to induce neutrophil recruitment in vivo in a mouse model [21]. Further, LL-37 release by recruited neutrophils would therefore amplify these responses and the effects of LL-37 on neutrophil apoptosis.
We demonstrate that neutrophils undergoing LL-37-mediated inhibition of apoptosis continued to respond to inflammatory stimuli, indicating continued functional competence. Furthermore, we show that in contrast to the inhibition of LPS-induced cytokine production (as observed previously in vivo [12, 15] and in other cell types in vitro [12, 45, 57]), LL-37 failed to inhibit and indeed, slightly enhanced the cytokine response of neutrophils to IL-1β or TNF-α. These data confirm our recent observations, suggesting that LL-37 is not anti-inflammatory per se [45] but might differentially modulate responses to endogenous host inflammatory signals as compared with exogenous pathogen-derived molecular patterns. It is interesting that LL-37 has also previously been shown to promote IL-1β processing and release by LPS-primed monocytes [32]. We have demonstrated that LL-37 has direct effects on the proinflammatory and anti-inflammatory properties of mammalian cells [12] and inhibited LPS-induced, proinflammatory cytokine expression, in part by acting directly on the Toll-like receptor to nuclear factor-κB pathway [45]. However, the potent effects of LL-37 on LPS-induced responses also likely reflect the ability of LL-37 to bind LPS [58], inhibit LPS-binding protein interaction [59], and effectively neutralize the LPS. The mechanisms responsible for the modest but significant increases in cytokine production by LL-37-treated neutrophils following exposure to IL-1β or TNF-α remain undetermined but could simply reflect the increased numbers of viable neutrophils in the LL-37-treated population as a consequence of LL-37-mediated inhibition of apoptosis.
In contrast to the LL-37-mediated inhibition of apoptosis observed in neutrophils, we demonstrate that LL-37 induced caspase-dependent cell death in primary airway epithelial cells. These data are compatible with our recent observation of LL-37-induced apoptosis in immortalized epithelial cell lines [40] but demonstrate an increased sensitivity of these primary cells to lower doses of LL-37. In addition, these data contrast with the effects demonstrated in neutrophils, suggesting the induction of apoptosis in primary airway epithelial cells in response to concentrations of LL-37, which mediated inhibition of apoptosis in neutrophils at the same time-point and under identical serum conditions. The underlying mechanisms that result in these strikingly divergent consequences for LL-37-mediated modulation of apoptotic pathways in different human primary innate-immune effector cells remain unclear. However, it is interesting to note that although our data implicate P2X7 receptors in LL-37-mediated inhibition of apoptosis in neutrophils, the induction of apoptosis has been described in human cervical epithelial cells following activation of these receptors with ATP, mediated predominantly by the caspase-9 (instrinsic mitochondrial) pathway [60]. Indeed P2X7 receptor activation has recently been proposed to be capable of inducing a state of initially reversible “pseudoapoptosis” in the human embryonic kidney 293 epithelial cell line, which can proceed to apoptotic and/or necrotic cell death dependent on the length of receptor activation [61]. This raises the possibility of a common receptor use in different cell types but resulting in the initiation of contrasting downstream signaling effects in epithelial cells as compared with neutrophils. Future assessment and comparison of these processes are expected to be instructive in dissecting the mechanisms underlying these contrasting immunomodulatory activities of LL-37.
In addition, the consequences of LL-37-induced epithelial cell apoptosis in the context of an infectious insult remain to be determined. This response could be detrimental to the host if excessive cell death results in a breach of epithelial integrity. However, apoptosis of infected epithelial cells has been proposed as an innate defense mechanism for clearance of P. aeruginosa by airway epithelium [62, 63] and a defense against Escherichia coli infection in the bladder epithelium [64]. In this context, LL-37-induced apoptosis could represent an innate host defense response, complemented by an LL-37-induced enhancement of wound-healing for resolution of inflammation [14, 46, 47]
Thus, raised levels of LL-37 in an acute inflammatory scenario could contribute to innate host defenses by mediating recruitment and enhanced survival of neutrophils, apoptosis of infected epithelial cells, and the modulation of innate immune cell responses to endogenous inflammatory signals to enhance the resolution of an acute, infectious insult. A full knowledge of the in vivo significance of these LL-37-mediated immunomodulatory activities and their applicability to other CHDP is essential in developing the potential of CHDP as future immunomodulatory, antimicrobial therapeutics.
ACKNOWLEDGMENTS
This work was supported by the Wellcome Trust, the Norman Salvesen Emphysema Research Trust, the Miss A. M. Urquhart's Charitable Trust, the Canadian Cystic Fibrosis Foundation, and the Pathogenomics of Innate Immunity grant provided through Genome British Columbia. T. S. W. and A. J. S. are funded by the Jules Thorn Charitable Trust, D. M. E. B. is supported by a Canadian Institutes for Health Research fellowship, R. E. W. H. holds a Canada Research Chair, and D. J. D. is a Wellcome Trust Career Development Fellow (078265). The authors thank Adriano Rossi, Ian Dransfield, Simon Hart, Trevor Walker, and John Savill for helpful advice and discussions; Kanwaldeep Dhaliwal, Jelena Pistolic, Jie Yu, and Linda Rehaume for advice and technical assistance; and Lesley Farrell, Kallirroi Kefala, Tara Sheldrake, Sarah McMeekin, and George Song-Zhao for technical assistance.
REFERENCES
- 1.Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415:389–395. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- 2.Ganz T, Metcalf JA, Gallin JI, Boxer LA, Lehrer RI. Microbicidal/cytotoxic proteins of neutrophils are deficient in two disorders: Chediak-Higashi syndrome and “specific” granule deficiency. J Clin Invest. 1988;82:552–556. doi: 10.1172/JCI113631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Putsep K, Carlsson G, Boman HG, Andersson M. Deficiency of antibacterial peptides in patients with morbus Kostmann: an observation study. Lancet. 2002;360:1144–1149. doi: 10.1016/S0140-6736(02)11201-3. [DOI] [PubMed] [Google Scholar]
- 4.Schaller-Bals S, Schulze A, Bals R. Increased levels of antimicrobial peptides in tracheal aspirates of newborn infants during infection. Am J Respir Crit Care Med. 2002;165:992–995. doi: 10.1164/ajrccm.165.7.200110-020. [DOI] [PubMed] [Google Scholar]
- 5.Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, Gallo RL, Leung DY. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med. 2002;347:1151–1160. doi: 10.1056/NEJMoa021481. [DOI] [PubMed] [Google Scholar]
- 6.Nizet V, Ohtake T, Lauth X, Trowbridge J, Rudisill J, Dorschner RA, Pestonjamasp V, Piraino J, Huttner K, Gallo RL. Innate antimicrobial peptide protects the skin from invasive bacterial infection. Nature. 2001;414:454–457. doi: 10.1038/35106587. [DOI] [PubMed] [Google Scholar]
- 7.Wilson CL, Ouellette AJ, Satchell DP, Ayabe T, Lopez-Boado YS, Stratman JL, Hultgren SJ, Matrisian LM, Parks WC. Regulation of intestinal α-defensin activation by the metalloproteinase matrilysin in innate host defense. Science. 1999;286:113–117. doi: 10.1126/science.286.5437.113. [DOI] [PubMed] [Google Scholar]
- 8.Salzman NH, Ghosh D, Huttner KM, Paterson Y, Bevins CL. Protection against enteric salmonellosis in transgenic mice expressing a human intestinal defensin. Nature. 2003;422:522–526. doi: 10.1038/nature01520. [DOI] [PubMed] [Google Scholar]
- 9.Iimura M, Gallo RL, Hase K, Miyamoto Y, Eckmann L, Kagnoff MF. Cathelicidin mediates innate intestinal defense against colonization with epithelial adherent bacterial pathogens. J Immunol. 2005;174:4901–4907. doi: 10.4049/jimmunol.174.8.4901. [DOI] [PubMed] [Google Scholar]
- 10.Bals R, Weiner DJ, Moscioni AD, Meegalla RL, Wilson JM. Augmentation of innate host defense by expression of a cathelicidin antimicrobial peptide. Infect Immun. 1999;67:6084–6089. doi: 10.1128/iai.67.11.6084-6089.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bowdish DM, Davidson DJ, Hancock RE. A re-evaluation of the role of host defense peptides in mammalian immunity. Curr Protein Pept Sci. 2005;6:35–51. doi: 10.2174/1389203053027494. [DOI] [PubMed] [Google Scholar]
- 12.Scott MG, Davidson DJ, Gold MR, Bowdish D, Hancock RE. The human antimicrobial peptide LL-37 is a multifunctional modulator of innate immune responses. J Immunol. 2002;169:3883–3891. doi: 10.4049/jimmunol.169.7.3883. [DOI] [PubMed] [Google Scholar]
- 13.Welling MM, Hiemstra PS, van den Barselaar MT, Paulusma-Annema A, Nibbering PH, Pauwels EK, Calame W. Antibacterial activity of human neutrophil defensins in experimental infections in mice is accompanied by increased leukocyte accumulation. J Clin Invest. 1998;102:1583–1590. doi: 10.1172/JCI3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koczulla R, Von Degenfeld G, Kupatt C, Krotz F, Zahler S, Gloe T, Issbrucker K, Unterberger P, Zaiou M, Lebherz C, et al. An angiogenic role for the human peptide antibiotic LL-37/hCAP-18. J Clin Invest. 2003;111:1665–1672. doi: 10.1172/JCI17545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fukumoto K, Nagaoka I, Yamataka A, Kobayashi H, Yanai T, Kato Y, Miyano T. Effect of antibacterial cathelicidin peptide CAP18/LL-37 on sepsis in neonatal rats. Pediatr Surg Int. 2005;21:20–24. doi: 10.1007/s00383-004-1256-x. [DOI] [PubMed] [Google Scholar]
- 16.Finlay BB, Hancock RE. Can innate immunity be enhanced to treat microbial infections? Nat Rev Microbiol. 2004;2:497–504. doi: 10.1038/nrmicro908. [DOI] [PubMed] [Google Scholar]
- 17.Zanetti M. Cathelicidins, multifunctional peptides of the innate immunity. J Leukoc Biol. 2004;75:39–48. doi: 10.1189/jlb.0403147. [DOI] [PubMed] [Google Scholar]
- 18.Chen CI, Schaller-Bals S, Paul KP, Wahn U, Bals R. β-Defensins and LL-37 in bronchoalveolar lavage fluid of patients with cystic fibrosis. J Cyst Fibros. 2004;3:45–50. doi: 10.1016/j.jcf.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 19.De Yang, Chen Q, Schmidt AP, Anderson GM, Wang JM, Wooters J, Oppenheim JJ, Chertov O. LL-37, the neutrophil granule- and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (FPRL1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells. J Exp Med. 2000;192:1069–1074. doi: 10.1084/jem.192.7.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tjabringa GS, Ninaber DK, Drijfhout JW, Rabe KF, Hiemstra PS. Human cathelicidin LL-37 is a chemoattractant for eosinophils and neutrophils that acts via formyl-peptide receptors. Int Arch Allergy Immunol. 2006;140:103–112. doi: 10.1159/000092305. [DOI] [PubMed] [Google Scholar]
- 21.Kurosaka K, Chen Q, Yarovinsky F, Oppenheim JJ, Yang D. Mouse cathelin-related antimicrobial peptide chemoattracts leukocytes using formyl peptide receptor-like 1/mouse formyl peptide receptor-like 2 as the receptor and acts as an immune adjuvant. J Immunol. 2005;174:6257–6265. doi: 10.4049/jimmunol.174.10.6257. [DOI] [PubMed] [Google Scholar]
- 22.Tjabringa GS, Aarbiou J, Ninaber DK, Drijfhout JW, Sorensen OE, Borregaard N, Rabe KF, Hiemstra PS. The antimicrobial peptide LL-37 activates innate immunity at the airway epithelial surface by transactivation of the epidermal growth factor receptor. J Immunol. 2003;171:6690–6696. doi: 10.4049/jimmunol.171.12.6690. [DOI] [PubMed] [Google Scholar]
- 23.Simon HU. Neutrophil apoptosis pathways and their modifications in inflammation. Immunol Rev. 2003;193:101–110. doi: 10.1034/j.1600-065x.2003.00038.x. [DOI] [PubMed] [Google Scholar]
- 24.Borregaard N, Cowland JB. Granules of the human neutrophilic polymorphonuclear leukocyte. Blood. 1997;89:3503–3521. [PubMed] [Google Scholar]
- 25.Darveau RP, Hancock RE. Procedure for isolation of bacterial lipopolysaccharides from both smooth and rough Pseudomonas aeruginosa and Salmonella typhimurium strains. J Bacteriol. 1983;155:831–838. doi: 10.1128/jb.155.2.831-838.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gough M, Hancock RE, Kelly NM. Antiendotoxin activity of cationic peptide antimicrobial agents. Infect Immun. 1996;64:4922–4927. doi: 10.1128/iai.64.12.4922-4927.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dransfield I, Buckle AM, Savill JS, McDowall A, Haslett C, Hogg N. Neutrophil apoptosis is associated with a reduction in CD16 (Fc γ RIII) expression. J Immunol. 1994;153:1254–1263. [PubMed] [Google Scholar]
- 28.Boyum A, Lovhaug D, Tresland L, Nordlie EM. Separation of leucocytes: improved cell purity by fine adjustments of gradient medium density and osmolality. Scand J Immunol. 1991;34:697–712. doi: 10.1111/j.1365-3083.1991.tb01594.x. [DOI] [PubMed] [Google Scholar]
- 29.Moulding DA, Akgul C, Derouet M, White MR, Edwards SW. BCL-2 family expression in human neutrophils during delayed and accelerated apoptosis. J Leukoc Biol. 2001;70:783–792. [PubMed] [Google Scholar]
- 30.Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- 31.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 32.Elssner A, Duncan M, Gavrilin M, Wewers MD. A novel P2X7 receptor activator, the human cathelicidin-derived peptide LL37, induces IL-1 β processing and release. J Immunol. 2004;172:4987–4994. doi: 10.4049/jimmunol.172.8.4987. [DOI] [PubMed] [Google Scholar]
- 33.Lau YE, Rozek A, Scott MG, Goosney DL, Davidson DJ, Hancock RE. Interaction and cellular localization of the human host defense peptide LL-37 with lung epithelial cells. Infect Immun. 2005;73:583–591. doi: 10.1128/IAI.73.1.583-591.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davidson DJ, Currie AJ, Reid GS, Bowdish DM, MacDonald KL, Ma RC, Hancock RE, Speert DP. The cationic antimicrobial peptide LL-37 modulates dendritic cell differentiation and dendritic cell-induced T cell polarization. J Immunol. 2004;172:1146–1156. doi: 10.4049/jimmunol.172.2.1146. [DOI] [PubMed] [Google Scholar]
- 35.Niyonsaba F, Iwabuchi K, Someya A, Hirata M, Matsuda H, Ogawa H, Nagaoka I. A cathelicidin family of human antibacterial peptide LL-37 induces mast cell chemotaxis. Immunology. 2002;106:20–26. doi: 10.1046/j.1365-2567.2002.01398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bae YS, Lee HY, Jo EJ, Kim JI, Kang HK, Ye RD, Kwak JY, Ryu SH. Identification of peptides that antagonize formyl peptide receptor-like 1-mediated signaling. J Immunol. 2004;173:607–614. doi: 10.4049/jimmunol.173.1.607. [DOI] [PubMed] [Google Scholar]
- 37.Dahlgren C, Christophe T, Boulay F, Madianos PN, Rabiet MJ, Karlsson A. The synthetic chemoattractant Trp-Lys-Tyr-Met-Val-DMet activates neutrophils preferentially through the lipoxin A(4) receptor. Blood. 2000;95:1810–1818. [PubMed] [Google Scholar]
- 38.Bowdish DM, Davidson DJ, Speert DP, Hancock RE. The human cationic peptide LL-37 induces activation of the extracellular signal-regulated kinase and p38 kinase pathways in primary human monocytes. J Immunol. 2004;172:3758–3765. doi: 10.4049/jimmunol.172.6.3758. [DOI] [PubMed] [Google Scholar]
- 39.Klein JB, Rane MJ, Scherzer JA, Coxon PY, Kettritz R, Mathiesen JM, Buridi A, McLeish KR. Granulocyte-macrophage colony-stimulating factor delays neutrophil constitutive apoptosis through phosphoinositide 3-kinase and extracellular signal-regulated kinase pathways. J Immunol. 2000;164:4286–4291. doi: 10.4049/jimmunol.164.8.4286. [DOI] [PubMed] [Google Scholar]
- 40.Lau YE, Bowdish DM, Cosseau CC, Hancock RE, Davidson DJ. Apoptosis of airway epithelial cells: human serum sensitive induction by the cathelicidin LL-37. Am J Respir Cell Mol Biol. 2006;34:399–409. doi: 10.1165/rcmb.2005-0170OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okumura K, Itoh A, Isogai E, Hirose K, Hosokawa Y, Abiko Y, Shibata T, Hirata M, Isogai H. C-terminal domain of human CAP18 antimicrobial peptide induces apoptosis in oral squamous cell carcinoma SAS-H1 cells. Cancer Lett. 2004;212:185–194. doi: 10.1016/j.canlet.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 42.Bowdish DM, Davidson DJ, Lau YE, Lee K, Scott MG, Hancock RE. Impact of LL-37 on anti-infective immunity. J Leukoc Biol. 2005;77:451–459. doi: 10.1189/jlb.0704380. [DOI] [PubMed] [Google Scholar]
- 43.Murakami M, Lopez-Garcia B, Braff M, Dorschner RA, Gallo RL. Postsecretory processing generates multiple cathelicidins for enhanced topical antimicrobial defense. J Immunol. 2004;172:3070–3077. doi: 10.4049/jimmunol.172.5.3070. [DOI] [PubMed] [Google Scholar]
- 44.Braff MH, Hawkins MA, Nardo AD, Lopez-Garcia B, Howell MD, Wong C, Lin K, Streib JE, Dorschner R, Leung DY, Gallo RL. Structure-function relationships among human cathelicidin peptides: dissociation of antimicrobial properties from host immunostimulatory activities. J Immunol. 2005;174:4271–4278. doi: 10.4049/jimmunol.174.7.4271. [DOI] [PubMed] [Google Scholar]
- 45.Mookherjee N, Brown KL, Bowdish DM, Doria S, Falsafi R, Hokamp K, Roche FM, Mu R, Doho GH, Pistolic J, Powers JP, Bryan J, Brinkman FS, Hancock RE. Modulation of the TLR-mediated inflammatory response by the endogenous human host defense peptide LL-37. J Immunol. 2006;176:2455–2464. doi: 10.4049/jimmunol.176.4.2455. [DOI] [PubMed] [Google Scholar]
- 46.Heilborn JD, Nilsson MF, Kratz G, Weber G, Sorensen O, Borregaard N, Stahle-Backdahl M. The cathelicidin anti-microbial peptide LL-37 is involved in re-epithelialization of human skin wounds and is lacking in chronic ulcer epithelium. J Invest Dermatol. 2003;120:379–389. doi: 10.1046/j.1523-1747.2003.12069.x. [DOI] [PubMed] [Google Scholar]
- 47.Shaykhiev R, Beisswenger C, Kaendler K, Senske J, Puechner A, Damm T, Behr J, Bals R. The human endogenous antibiotic LL-37 stimulates airway epithelial cell proliferation and wound closure. Am J Physiol Lung Cell Mol Physiol. 2005;289:L842–L848. doi: 10.1152/ajplung.00286.2004. [DOI] [PubMed] [Google Scholar]
- 48.An LL, Yang YH, Ma XT, Lin YM, Li G, Song YH, Wu KF. LL-37 enhances adaptive antitumor immune response in a murine model when genetically fused with M-CSFR(J6–1) DNA vaccine. Leuk Res. 2005;29:535–543. doi: 10.1016/j.leukres.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 49.Braff MH, Zaiou M, Fierer J, Nizet V, Gallo RL. Keratinocyte production of cathelicidin provides direct activity against bacterial skin pathogens. Infect Immun. 2005;73:6771–6781. doi: 10.1128/IAI.73.10.6771-6781.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee PH, Ohtake T, Zaiou M, Murakami M, Rudisill JA, Lin KH, Gallo RL. Expression of an additional cathelicidin antimicrobial peptide protects against bacterial skin infection. Proc Natl Acad Sci USA. 2005;102:3750–3755. doi: 10.1073/pnas.0500268102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nagaoka I, Tamura H, Hirata M. An antimicrobial cathelicidin peptide, human CAP18/LL-37, suppresses neutrophil apoptosis via the activation of formyl-peptide receptor-like 1 and P2X7. J Immunol. 2006;176:3044–3052. doi: 10.4049/jimmunol.176.5.3044. [DOI] [PubMed] [Google Scholar]
- 52.Yu A, Byers DM, Ridgway ND, McMaster CR, Cook HW. Preferential externalization of newly synthesized phosphatidylserine in apoptotic U937 cells is dependent on caspase-mediated pathways. Biochim Biophys Acta. 2000;1487:296–308. doi: 10.1016/s1388-1981(00)00100-1. [DOI] [PubMed] [Google Scholar]
- 53.Michels J, Johnson PW, Packham G. Mcl-1. Int J Biochem Cell Biol. 2005;37:267–271. doi: 10.1016/j.biocel.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 54.Daigle I, Simon HU. Critical role for caspases 3 and 8 in neutrophil but not eosinophil apoptosis. Int Arch Allergy Immunol. 2001;126:147–156. doi: 10.1159/000049506. [DOI] [PubMed] [Google Scholar]
- 55.Turina M, Miller FN, McHugh PP, Cheadle WG, Polk HC., Jr. Endotoxin inhibits apoptosis but induces primary necrosis in neutrophils. Inflammation. 2005;29:55–63. doi: 10.1007/s10753-006-8970-6. [DOI] [PubMed] [Google Scholar]
- 56.Brach MA, deVos S, Gruss HJ, Herrmann F. Prolongation of survival of human polymorphonuclear neutrophils by granulocyte-macrophage colony-stimulating factor is caused by inhibition of programmed cell death. Blood. 1992;80:2920–2924. [PubMed] [Google Scholar]
- 57.Bowdish DM, Davidson DJ, Scott MG, Hancock RE. Immunomodulatory activities of small host defense peptides. Antimicrob Agents Chemother. 2005;49:1727–1732. doi: 10.1128/AAC.49.5.1727-1732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Turner J, Cho Y, Dinh NN, Waring AJ, Lehrer RI. Activities of LL-37, a cathelin-associated antimicrobial peptide of human neutrophils. Antimicrob Agents Chemother. 1998;42:2206–2214. doi: 10.1128/aac.42.9.2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scott MG, Vreugdenhil AC, Buurman WA, Hancock RE, Gold MR. Cutting edge: cationic antimicrobial peptides block the binding of lipopolysaccharide (LPS) to LPS binding protein. J Immunol. 2000;164:549–553. doi: 10.4049/jimmunol.164.2.549. [DOI] [PubMed] [Google Scholar]
- 60.Wang Q, Wang L, Feng YH, Li X, Zeng R, Gorodeski GI. P2X7 receptor-mediated apoptosis of human cervical epithelial cells. Am J Physiol Cell Physiol. 2004;287:C1349–C1358. doi: 10.1152/ajpcell.00256.2004. [DOI] [PubMed] [Google Scholar]
- 61.Mackenzie AB, Young MT, Adinolfi E, Surprenant A. Pseudoapoptosis induced by brief activation of ATP-gated P2X7 receptors. J Biol Chem. 2005;280:33968–33976. doi: 10.1074/jbc.M502705200. [DOI] [PubMed] [Google Scholar]
- 62.Grassme H, Kirschnek S, Riethmueller J, Riehle A, von Kurthy G, Lang F, Weller M, Gulbins E. CD95/CD95 ligand interactions on epithelial cells in host defense to Pseudomonas aeruginosa. Science. 2000;290:527–530. doi: 10.1126/science.290.5491.527. [DOI] [PubMed] [Google Scholar]
- 63.Pier GB, Grout M, Zaidi TS, Olsen JC, Johnson LG, Yankaskas JR, Goldberg JB. Role of mutant CFTR in hypersusceptibility of cystic fibrosis patients to lung infections. Science. 1996;271:64–67. doi: 10.1126/science.271.5245.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mulvey MA, Lopez-Boado YS, Wilson CL, Roth R, Parks WC, Heuser J, Hultgren SJ. Induction and evasion of host defenses by type 1-piliated uropathogenic Escherichia coli. Science. 1998;282:1494–1497. doi: 10.1126/science.282.5393.1494. [DOI] [PubMed] [Google Scholar]