

Abstract
Chronic respiratory infections in cystic fibrosis result from CFTR channel mutations but how these impair antibacterial defense is less clear. Airway host defense depends on lactoperoxidase (LPO) that requires thiocyanate (SCN−) to function and epithelia use CFTR to concentrate SCN− at the apical surface. To test whether CFTR mutations result in impaired LPO-mediated host defense, CF epithelial SCN− transport was measured. CF epithelia had significantly lower transport rates, did not accumulate SCN− in the apical compartment. The lower CF [SCN−] did not support LPO antibacterial activity. Modeling of airway LPO activity suggested that reduced transport impairs LPO-mediated defense and cannot be compensated by LPO or H2O2 upregulation.
Keywords: Lactoperoxidase, CFTR, Host Defense, thiocyanate, hydrogen peroxide, cystic fibrosis
Cystic fibrosis (CF) is one of the most common genetic disorders among Caucasians. The disease is characterized by recurrent respiratory infections caused primarily by Staphylococcus aureus and Pseudomonas aeruginosa [1]. Lifelong respiratory infections result in progressive loss of lung function and shortened life expectancy. Chronic infection implies failure of airway host defenses against bacterial colonization. However, the exact defects in CF host defense that are primarily responsible chronic infection in this disease remain unclear. The normal airway barrier to infection is complex and multifactorial. One possible view is that mucus delays growth of pathogens and attachment of bacteria and other particles to epithelia cells until cilia mechanically clear them. The complex composition of mucus must provide the proper viscoelastic properties to permit ciliary clearance but also must contain broad-spectrum antibiotic activities in order to be effective.
CFTR, the gene product responsible for CF, contains an anion channel that has been extensively characterized [2]. CFTR mutations result in loss of functional anion channel activity on the apical cell surface, although, it is not fully understood how a loss of anion channel activity completely explains defective antibacterial defenses. Several hypotheses have been proposed to relate these two phenomena and focus on: a) the effects of airway surface liquid (ASL) dehydration that leads to impaired ciliary and cough clearance of mucus [3]; b) the effects of elevated ASL salt concentration on the activity of antimicrobials in mucus [4–6]; or c) CFTR mediated defective bacterial-epithelial interactions [for reviews, see 7,8–11].
Current views of CF pathogenesis may be limited by incomplete knowledge regarding the array of mechanisms at work in airway defense. For example, the lactoperoxidase (LPO) system has recently been shown to be present in airways, and to be important for airway antibacterial activity and bacterial clearance in both humans and sheep [12,13]. The LPO system works to preserve sterility of secretions [14],[15] in several species including human. Similar to myeloperoxidase (MPO) and eosinophil peroxidase (EPO), LPO uses H2O2 to catalyze oxidation of the anion SCN− to the antibiotic OSCN− [16] via an oxidized enzyme intermediate called compound I (LPO-O).
Both SCN− and H2O2 are required for LPO to function in the airway lumen. Normal human airway epithelia transport SCN− from the basolateral to the apical surface and concentrate it in airway secretions [17]. This transport utilizes the sodium/iodide symporter at the basolateral surface to mediate uptake of SCN− or I− into the cells followed by release to the apical surface that is regulated by CFTR [17]. Normal human airway epithelia also actively produce H2O2 at the apical surface using Duox and release it into the apical surface liquid [18]. This airway H2O2 production is increased through stimulation of purinergic receptors [18] that also act to increase mucus secretion [19,20] and ciliary beating [21–23] all through intracellular Ca2+ increases. Thus, a complete antibacterial LPO system is present in the airway lumen and can be regulated in concert with other facets of the airway host defense.
Since the LPO system is effective against staphylococci [24], E. coli and pseudomonads [24–26] and since SCN− is a requisite substrate for LPO, defects in SCN− transport and resulting loss of LPO activity could contribute, at least partially, to the chronic respiratory infections seen in CF patients. Although MPO was previously thought to primarily use chloride as a substrate, it is now recognized that MPO also uses SCN− and that SCN− maybe the physiological substrate for MPO [27]. In addition, hypochlorite (OCl−), the product of MPO oxidation of chloride, reacts almost instantaneously with SCN− at physiological concentrations [28] to give OSCN− suggesting that SCN− may have a central role not only in LPO antibacterial activity but also MPO antibacterial activity.
The described experiments test the hypothesis that CF airway epithelia are defective in SCN− transport to the airway surface and that this defect may be reflected by impaired or altered peroxidase antibiotic activity in CF epithelial cell secretions.
Methods
Cell Culture and Thiocyanate Transport Experiments
Human airways were obtained following IRB approved protocols from CF patients at the time of transplant or from organ donors whose lungs were not used for transplant. Epithelial cells were isolated, grown and differentiated on either 6.5 mm or 24 mm collagen-coated T-clear membranes (Costar #3450) at an air-liquid interface as previously described [29,30]. All experiments comparing non-CF and CF cells were performed with cultures grown simultaneously and matched in passage number, the number of cells plated, and days in culture. All cultures had a resistivity ≥ 300 Ω.cm. Prior to initiating experiments, surfaces of ALI cultures were washed with PBS, returned to the air interface and incubated 18–24 h with 70 μM 14C-SCN− (50 μCi/mM) in the basolateral media. To start the experiments, apical surfaces of the cultures were rapidly washed three times with Dulbecco's PBS (50 μL for 6.5mm filters or 500 μL for 24 mm filters). Following the third wash, additional aliquots were placed on the apical surface for sequential 2 min incubations at 37° in humidified 5% CO2. Basolateral media were sampled after the last wash. 14C-SCN− in media and apical washes was determined by liquid scintillation counting. All of the collected 14C-SCN− was soluble following 10% TCA precipitation showing that radiolabel was not covalently attached to protein.
PKA stimulation was accomplished by adding 500 μM dibutyryl cAMP and 10 μM forskolin to the PBS washes. To inhibit CFTR during PKA stimulation, glibenclamide (500 μM) was added to washes containing dibutyryl cAMP and forskolin.
Assay of Antibacterial Activity
Antibacterial activity assays were performed essentially as described previously [12]. Pseudomonas aeruginosa (ATCC 27853) were grown in LB broth overnight, collected in the stationary phase, diluted to 5 – 6 x 104 / ml in LB broth with 15% glycerol and stored at −80ºC. At least 24h prior to the experiments, basolateral media of matched non-CF and CF ALI cultures (except controls) were supplemented with 100 μM SCN−. The apical surfaces of the cultures were washed with 0.25 ml PBS and washes were pooled and stored at −20ºC. SCN− dependent antibiotic activity was assayed using 560 μl of apical culture washes, adjusted to pH 5.7, and containing 2400–3600 P. aeruginosa. Since LPO was not secreted by the cells under these culture conditions, washes were supplemented with 1.1 μg/ml LPO and 10−5 M H2O2. Control experiments were performed in the absence of basolateral addition of SCN− or using only PBS never added to the cultures and adjusted to pH 5.7. Growth of bacteria was not affected by addition of 1.125 μg/ml LPO, 10−5 M H2O2, or 5 x 10−4 M SCN− singly or in pairs. However, addition of all three reconstituted a functional LPO system that was bactericidal. To show SCN− dependence, apical CF culture washes were also supplemented to 5 x 10−4 M SCN− in some experiments. Mixtures were sampled immediately and 4 h after incubation at room temperature and CFU were determined by plating on LB Agar. Antibacterial activity was expressed as a ratio of CFU after 4 h to the starting CFU in the sample to control for slight differences in the starting number of bacteria.
Computational Modeling of LPO Activity
Differential equations describing LPO enzymatic activity were reiteratively integrated over 0.1 sec intervals using IgorPro (WaveMetrics, Portland, OR). Kinetic constants (k1 and k4) were those published for human salivary lactoperoxidase [31] at pH 6.8 with the normal steady state assumptions. Initial values for reactants, products and LPO are shown in table 1. The used equations were:
Table 1.
Initial conditions for computations shown in Figure 5 were derived from estimates or measured amounts of enzyme and substrates [12], transport rates (preliminary data, above), and volumes [3]. Transport rates described in the text (shown in Figure 1) are recalculated to reflect the estimated ASL volume on 1 cm of epithelial surface. CF LPO was increased 2 fold over normal to accommodate the reduced ASL volume in CF.
| Normal | CF | units | |
|---|---|---|---|
| [H2O2] | 0 | 0 | M |
| [SCN−] | 4 x 10−4 | 1 x 10−4 | M |
| [LPO] | 7.2 x 10−8 | 14.4 x 10−8 | M |
| SCN− Transport | 1.04 x 10−6 | 0.263 x 10−6 | M· sec−1 |
| H2O2 production | 1 x 10−6 | 1 x 10−6 | M· sec−1 |
| ASL Volume (1 cm2) | 2.5 x 10−6 | 1.25 x 10−6 | 1 |
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
where k2 and k3 are the H2O2 production rate and SCN− transport rates respectively. The Igor Pro procedure code is available on request to the authors.
Results
Thiocyanate Transport
We have previously shown that normal human bronchial epithelial cells actively transport SCN− from the basolateral to apical surfaces and that the transport was regulated by CFTR as it was stimulated by cAMP analogs and by forskolin, and inhibited by apical glibenclamide, diphenylamine-2-carboxylic acid and 5-nitro-2- (3-phenylpropylamino)-benzoate [17]. Since a loss of SCN− in the airway lumen might result in a loss of LPO antibacterial activity and perhaps also affect neutrophil MPO antibacterial activity, we assessed whether SCN− transport was defective in CF bronchial epithelial cells compared to normal epithelia. Airway epithelial cells, obtained from CF lungs or non-CF lungs at the time of transplant or organ donation, were passaged twice (thereby de-differentiated) and then cultured at an air-liquid interface (ALI) for re-differentiation [29,30]. As expected CF cell cultures showed no change in short circuit current in Ussing chambers following stimulation with forskolin
To measure the transport of SCN− across the epithelium, 14C-SCN− was added to the basolateral media and, after overnight incubation to allow an approach to steady state, its rate of appearance in the apical surface liquid was monitored. Airway epithelial cell cultures from three separate CF patients were compared to non-CF control cultures from three individuals. CF cells showed 4 fold reduced SCN− transport rates when compared to normal (CF, 2.3 ± 0.5 nmol/h/cm2 vs. non-CF, 9.3 ± 1.7 nmol/h/cm2; n=18; 3 donors, p < 0.0002, one way ANOVA) (Fig. 1A). All three individuals had one ΔF508 allele, one had a second W1282X allele (nonsense mutation), while the second alleles in the other two patients were unidentified unknown. However, all were diagnosed with CF and underwent lung transplant. CFTR function as diagnosed by an abnormal sweat test. Transport rates for cultures from each CF individual were: ΔF508/W1282X, 1.69 ± 0.32 (n=8) and 2.41 ± 0.72 (n=5) as well as 3.65 ± 2.34 (n=3) for the other two. The differences in SCN− transport between non-CF and CF cultures were independent of the age of the cultures (repeated measurements from 2 – 8 week old cultures), suggesting that a possible difference in time to reach full differentiation between CF and non-CF cells was not responsible for these observations.
Figure 1. Thiocyanate transport in CF airway epithelium is defective.
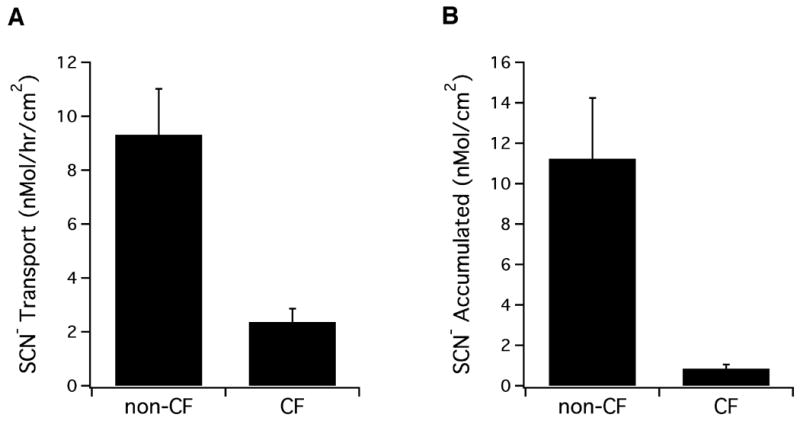
Panel A. 14C-SCN− (70 μM) was added to the basolateral media of non-CF and CF cultures for 18 – 24 h and stable unstimulated transport rates measured as described previously [17]. Non-CF cultures showed a 4-fold higher rate of transport using 18 from three non-CF and three CF individuals (n= 18, p < 0.0001). Panel B. 14C-SCN− (70 μM) was added to the basolateral media of cultures for 18 – 24 h. The 14C -SCN− accumulated on the apical surface was collected in a PBS wash and measured by liquid scintillation counting (n = 18 cultures each from three non-CF and three CF, p < 0.007).
Exact determination of apical SCN− concentration was not possible in these experiments because the small apical surface volumes naturally present on ALI cultures prevented accurate sampling of CF cultures. However, comparison of the total amounts of SCN−, recovered in the initial 0.5 ml PBS wash of the apical surface after overnight incubations with 14C-SCN− in the basolateral media, showed 11-fold higher levels in non-CF compared to CF cultures (non-CF, 11.2 ± 3.0 nmol/cm2 vs. CF, 0.8 ± 0.2 nmol/cm2, n = 18, p < 0.0008, one way ANOVA; Fig. 1B). Even though CF cultures are reported to have about one half the amount of apical surface fluid when compared to non-CF [3], the difference in accumulated apical 14C-SCN− between CF and non-CF cultures would remain significant, even if this volume difference is taken into account (5.5 fold). Thus, it appears that non-CF epithelial layers accumulate SCN− to significantly higher levels than CF. On the occasions when apical surface liquid was visible on non-CF cultures that had been pre-incubated with basolateral 14C-SCN−, direct sampling (2–5 μl) allowed measurement of the [SCN−] and showed that it was concentrated 6 fold over the 0.07 mM in the basolateral media to 0.43 ± 0.13 mM, n = 3. This concentration of SCN−, measured in undiluted non-CF apical surface liquid, is close to levels measured in undiluted airway secretions collected from patients, where we measured 0.5 mM [12]. Liquid was never observed on the apical surface of CF cultures, as expected from a previous report [3] and therefore, direct measurement of [SCN−] in CF apical surface fluid could not be made. However, by estimating the values for CF cells using the difference to non-CF cells measured above (5.5 fold), the apical SCN− concentration appears likely to be close to that in the basolateral media (0.4 mM divided by 5.5 would equal 0.072 mM).
Since previous studies showed that CFTR played a role in SCN− transport from the serosal to mucosal compartments [17], we evaluated the possible contribution of CFTR anion channel activity to the SCN− transport observed in cultures. Non-CF cultures showed the expected increase in SCN− transport after stimulation of PKA by apical addition of dibutyryl cAMP (0.5 mM) and forskolin (10 μM), while CF cultures did not (Fig. 2, A–C). In non-CF airway epithelia, both the PKA-stimulated and baseline SCN− transport are completely and reversibly blocked by glibenclamide [17]. Previous studies showed that SCN− transport in normal airway epithelia was also sensitive to diphenylamine-2-carboxylic acid and 5-nitro-2- (3-phenylpropylamino)-benzoate but not affected by DIDS and DNDS [17]. Thus the lack of cAMP stimulated SCN− transport by CF epithelia is consistent with the idea that SCN− is delivered to the apical surface through CFTR.
Figure 2. PKA stimulation increases thiocyanate transport in non-CF but not in CF airway epithelia.
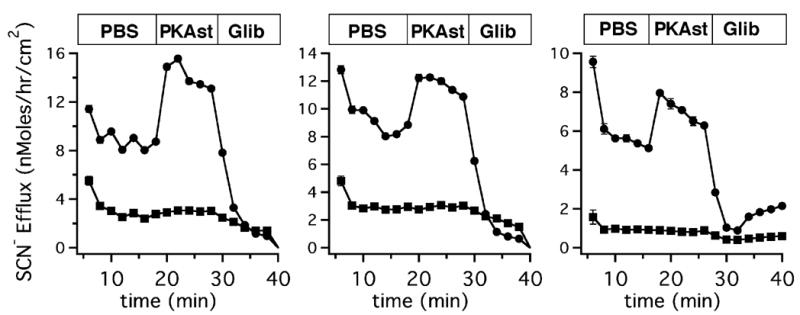
To initiate the experiments, apical surfaces of the cultures incubated overnight in 14C-SCN− were rapidly washed three times. Additional PBS aliquots were placed on the apical surface for sequential 2 min washes and 14C-SCN− was determined by liquid scintillation counting. cAMP stimulation of SCN− efflux was demonstrated by adding dibutyryl cAMP and forskolin to the PBS washes (18 min, indicated by the bar at top). The stimulated transport was inhibited by addition of glibenclamide (28 min) to washes containing dibutyryl cAMP and forskolin. SCN− flux is expressed in nMoles of SCN−/h normalized to 1 cm2 filter area. In the CF culture, SCN− transport was greatly diminished and not stimulated by cAMP. Bars at the top of graphs indicate the presence of PBS alone, dibutyryl cAMP and forskolin (PKAst), or dibutyryl cAMP, forskolin and glibenclamide (Glib). Panels A–C show single experiments comparing pairs of cultures obtained from three different CF patients and three different non-CF patients.
Thiocyanate Transport Defects and In Vitro Antibacterial Activity
To assess the effects of reduced SCN− transport on LPO-mediated antibacterial activity, the apical surfaces of cultures were washed with PBS and these washes were used in antibacterial assays previously developed to assess LPO activity in human tracheal secretions [12]. The day prior to washing, SCN− (100 μM) was added to the basolateral media of some cultures to allow transport to the apical surface. Since culture conditions did not result in LPO synthesis and secretion in these ALI cultures, washes were supplemented with exogenous bovine milk LPO and H2O2 (10−5 M). Supplementing washes from cells cultured in the absence of basolateral SCN−, with this concentration of H2O2 alone or in combination with LPO did not give rise to any antibacterial activity (Fig. 3, compare panel A and B). However, incubation of P. aeruginosa with LPO/H2O2 supplemented washes from non-CF cultures grown with basolateral SCN− resulted in LPO-dependent killing of bacteria (Fig. 3, panel C). In contrast, incubation of P. aeruginosa with LPO/H2O2 supplemented washes from CF cultures grown with basolateral SCN− had no detectable LPO-dependent killing activity. Addition of exogenous SCN− (0.5 mM) to LPO/H2O2 supplemented culture washes generated antibacterial activity in CF samples grown with basolateral SCN−, demonstrating that the lack of antibacterial activity was the result of defective SCN− transport by the CF epithelial cells (see below). Addition of the same amount of SCN− to the non-CF culture washes grown with basolateral SCN− increased the LPO-dependent activity, already present, due to the further increase in [SCN−]. Thus, the lack of SCN− transport by CF cells, compromised an in vitro LPO system reconstituted in apical washes of the ALI cultures [32]. Although small numbers of bacteria were used in the assays, substrates were not replenished and were limiting.
Figure 3. Thiocyanate-mediated bacterial killing in ALI culture secretions.
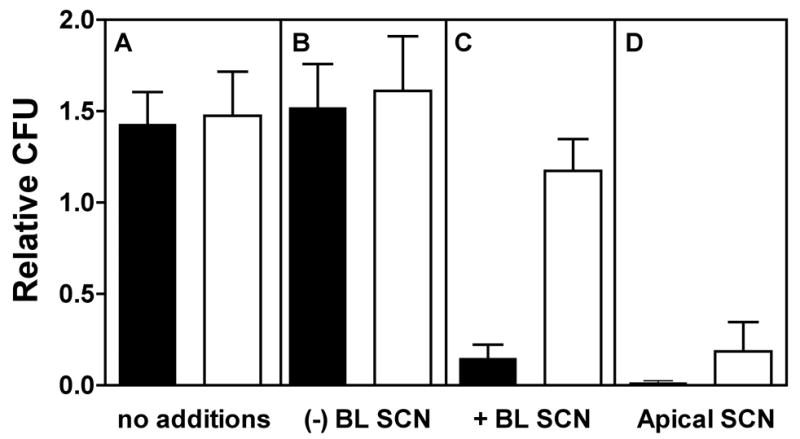
Non-CF (solid bars, n = 6 cultures on separate days) and CF (open bars, n = 4 cultures on separate days) ALI cultures were incubated in the presence or absence of SCN− in the basolateral media. Apical surfaces were washed with PBS and combined for assay of antibacterial activity as described in Methods. Assays were performed with no additions (panel A) or with LPO and H2O2 added to the washes (panel B). LPO and H2O2 did not increase the antibacterial activity of the washes in the absence of SCN−, however in the presence of basolateral SCN− (panel C), only non-CF culture washes showed antibacterial activity. Addition of SCN− to washes of CF cultures restored the antibacterial activity and increased the activity of non-CF culture washes (panel D).
Computational Modeling of LPO activity in Airway Surface Liquid (ASL)
Since LPO activity depends on adequate [SCN−], it is important to understand whether lower LPO activity due to lower [SCN−], could be up-regulated by compensatory changes in enzyme concentration or H2O2 production. To visualize the effects of decreased [SCN−] on LPO enzyme activity, we estimated LPO activity as a function of [SCN−] by calculating reaction rates based on kinetic constants and the velocity equation published by Pruitt et al. [31].
As expected, the lower SCN− concentrations found in apical CF culture washes resulted in lower enzymatic activity Fig 4A and B). Interestingly, LPO has increased activity with lower [SCN−] at lower pH that might be encountered during infection or with reduced bicarbonate transport seen in CF (Fig. 4A and B). Increased initial LPO activity due to lower initial pH resulted in more rapid depletion of SCN− and was not able to compensate for the loss of SCN− transport (not shown).
Figure 4. LPO activity varies with thiocyanate concentration and with pH.
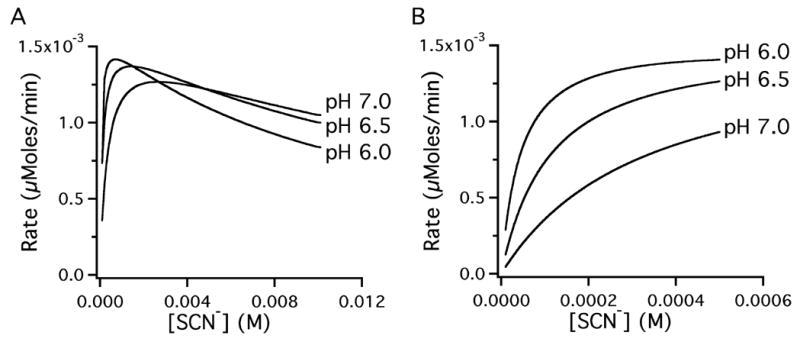
An arithmetic model of LPO activity using the kinetic constants published by Pruitt et al. [31] calculated the expected LPO activity with [SCN−] between 0.0001 M and 0.01 M (left panel) at pH 7.0, pH 6.5 and pH 6.0. The right panel shows an expanded view between 0 and 0.5 mM (0.4 mM being the concentration found in human airway secretions).
To approximate the effects of lower SCN− transport, differential equations describing changes in reactants were used to model LPO activity in ASL at pH 6.8. These computations allowed assessment of the possible effects of increasing [LPO] and [H2O2] on LPO activity in an otherwise normal airway except for a sharply reduced [SCN−] transport ass measured here in CF airway epithelial cultures. The model used LPO kinetic constants at pH 6.8 [31], the measured amounts of LPO in tracheal secretions [12], the measured rates of SCN− transport by non-CF [17] and CF airway epithelia (above), as well as estimates of ASL volume [3] per unit surface area. The concentration of H2O2 in ASL is not known. Therefore initial values of [H2O2] were set to zero and H2O2 production rates were set to balance the transport of SCN− and its consumption by LPO in normal airways. The CF model utilized an ASL volume equal to half that of the normal model [3] to account for a possible increase in [SCN−] due to a reduced volume. To account for equivalent synthesis and secretion of LPO to into this reduced volume of ASL in CF, initial [LPO] was 2-fold normal in the reduced transport (CF) model.
The simulation was carried out for 600 sec. At 300 sec, LPO was increased 10 fold over the initial concentration in both the CF and in the non-CF model to simulate possible compensatory increases in enzyme concentration. Figure 5 shows the calculated concentrations of the substrates and the rate. Figure 5A depicts [SCN−] changes in the normal and CF models. In the low SCN− transport CF model, SCN− is rapidly consumed resulting in decreased rate of product formation (Fig. 5C) and a corresponding increase in [H2O2] (Fig. 5B) as catalytic consumption depletes SCN− faster than it is replenished by diminished transport and thus, leading to decreased use of H2O2. To demonstrate that an increase of LPO could not offset the loss of SCN− transport, [LPO] was increased at 300 sec, and only resulted in a short transient increase in reaction velocity (Fig. 5C).
Figure 5. Defective thiocyanate transport predicts a loss of LPO antibacterial activity.
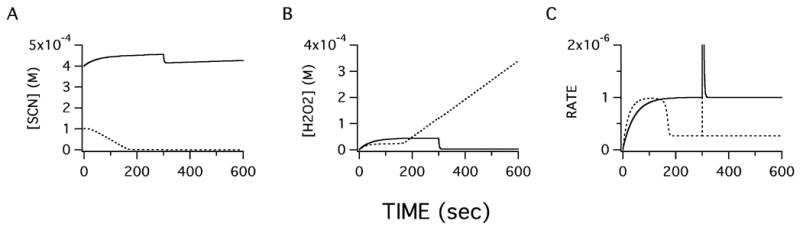
Computational modeling of LPO enzymatic activity in airway surface liquid shows predicted changes in response to altered conditions in CF airways, i.e. decreased volume and decreased SCN− transport rates. Initial conditions (shown in Table 1) were chosen based on published values, except [H2O2] that was arbitrarily set to zero. The H2O2 production rate was set to balance SCN− transport rates in normal cultures. Integration steps were 0.1 sec. Shown below are values of non-CF (solid line) and CF (dotted line) conditions. Changes in [SCN−], (panel A) and [H2O2], (panel B) include consumption by LPO catalysis and appearance due to production or transport. Differentiation of OSCN− product concentration with respect to time is shown in panel C. After 300 sec, LPO concentration was reset to a level 10 fold higher than the CF initial [LPO] to mimic possible upregulation of the enzyme concentration in response to lower SCN− transport. Despite a higher initial CF [LPO] to accommodate decreased CF ASL volume, computation showed that SCN− could not be replaced fast enough given the transport defect in CF. An increase in [LPO] (300 sec) was unable to restore LPO reaction rate in CF due to faster consumption of SCN−. The model predicts a loss of antibacterial activity in CF airways that cannot be compensated by increased LPO or H2O2.
Similar upregulation of H2O2 production did not restore LPO activity in the CF model, however, increased H2O2 production did stimulate LPO activity in the normal model until [SCN−] fell due to inability of SCN− transport to sustain the increased catalysis (not shown). Interestingly, increasing SCN− transport rate under normal conditions made little difference in LPO activity as the enzyme is nearly saturated with substrate at normal transport rates. Computation using LPO rate constants at pH 5.6 that could be encountered locally during infection accelerated the loss of SCN− in the low transport CF model. Thus, this computational model supports the idea that the reduced [SCN−] predicted by the above transport studies cannot be compensated by upregulation of LPO or H2O2 or changes in pH. The model also predicted that decreased SCN− transport may cause CF ASL to be deficient in SCN− rendering the LPO system unable to generate OSCN−, therefore impairing the antibacterial properties of mucus secretions.
Discussion
The experiments presented here showed that CF airway epithelia, cultured at the ALI, were defective in transporting SCN− from the basolateral to the apical compartment. The absence of PKA stimulated SCN− transport in CF cultures is consistent with a role for CFTR in SCN− transport suggested by our previous studies [17]. The measured defect in SCN− transport by CF ALI cultures was reflected in a reduction of SCN− accumulation on the apical surface and this reduction resulted in a loss of reconstituted LPO antibacterial activity that relied on substrate transported by the cultured epithelia. Finally, a computational model of LPO activity supported the concept that the effect of the measured decrease in SCN− transport on LPO enzymatic activity could not be compensated by up-regulating either LPO or H2O2 and thus predicted a loss of LPO-mediated host defense in CF airways. If the measured difference in apical SCN− between CF and non-CF airway epithelial cells is reflected in vivo, it is likely to be functionally significant since OSCN− production by LPO at the relevant SCN− concentrations are significantly different [33] and bacterial growth in vitro is inversely proportional to the [OSCN−] [34]. Although certain changes in CF epithelial phenotype in culture have been noted by others, these changes were seen in primary cultures and disappeared after 6–11 days in culture [35]. Our re-differentiated cultures (passaged once) have been in culture for much longer and are not expected to exhibit these changes noted in primary cultures.
Exact determination of SCN− concentration in normal and CF airway secretions is not currently possible. Thiocyanate content in normal secretions is near the limit of detection of colorimetric assays [12] and dilutions associated with bronchoalveolar lavage will require development of more sensitive methods. Our model predicted complete depletion of SCN− in CF secretions and the presence of large amounts of MPO due to chronic infection of CF airways virtually assures no SCN− to be measurable in secretions. Thus, accurate comparison of SCN− levels in CF and normal airways will require development of a highly sensitive assay method and will be best done in patients early in life prior to establishment of chronic infection and neutrophilic inflammation.
In lieu of direct measurement of ASL [SCN−], the computational model presented here predicted that decrease SCN− transport of the magnitude measured results in complete depletion of this LPO substrate and that upregulation of LPO enzyme activity cannot compensate for lower SCN− transport measured in the in vitro system. The model also suggested that H2O2 normally produced by airway epithelia [18,36,37] and consumed by LPO [38] will increase if SCN− transport is not able to provide sufficient substrate and might perhaps lead to increased oxidative damage in the CF airway. Thus the model predicted that changes in the LPO and H2O2 concentrations in CF ASL cannot offset the measured loss of SCN− transport.
It is important to note that the computational model used empirically determined rate constants for purified human LPO from saliva and thus the model does not take into account the presence of other components normally found in normal and CF airways. The model therefore cannot accurately predict the effects of other potential antibacterial peroxidase substrates such as nitrite. Nor does the model take into account the formation of LPO compound II that can generate SCN radicals and is now thought to be an important part of the normal peroxidase catalytic cycle [39]. The model also does not address MPO activities and cannot predict a complete loss of peroxidase mediated antibacterial activity but instead primarily serves to predict that upregulation of LPO expression cannot compensate for the observed reduction in SCN− transport.
Although most studies of MPO-mediated bacterial killing have concentrated on its use of Cl−, SCN− has recently been shown to be the preferred substrate of MPO [27,40] and normally OCl− immediately reacts with SCN− to produce OSCN− [28]. The presence of high levels of MPO in CF airways, that also consumes SCN− [e.g. 27], virtually ensures the full consumption of SCN− in the presence of an impaired transport system and suggests an increased use of chloride as a substrate and generation of OCl−. Since OCl− is believed to very rapidly react with SCN− in physiological conditions, increased production of OCl− that might also contribute to increased oxidative damage in the airway. Although hypochlorite itself is a potent antibacterial, CF airway secretions are reported to have high levels of taurine (> 3 mM) [41] that reacts extremely rapidly with hypochlorite and thus is expected to buffer the antibacterial effects [42,43]. Nevertheless mono and dichlorotaurine are also antibacterial, although much less so than HOCl [44,45]. The mechanisms by with chlorotaurines are antibacterial are not known. Clearly, a full understanding of the effects of SCN− transport changes in CF airways will require more investigation including studies of the phenotypic changes in bacteria that occur after chronic colonization. Thus, impaired SCN− transport to the airway lumen could potentially cause significant alteration of both the epithelial-derived LPO system as well as the neutrophil-derived MPO antibacterial activity.
The SCN− permeability and conductance of the CFTR anion channel is known [e.g. 46,47,48] and suggested that a SCN− transport deficiency might be relevant to increased infection in CF. Our previous studies on SCN− transport by airway epithelia support a role for CFTR in SCN− transport [17] and the decreased SCN− transport and lack of PKA stimulation in the CF cultures shown here are also consistent with a role for CFTR. We have not ruled out the possibility that NaI symporter in the basolateral membrane is also reduced in CF although such a reduction is not expected to be relevant in the absence of functional CFTR.
Since LPO has antimicrobial activity against a broad spectrum of pathogens including respiratory pathogens relevant in CF [e.g. 12], a CFTR dependent defect in LPO-mediated host defense might be relevant to cystic fibrosis. In our studies shown here, and in previous studies [12], small numbers of bacteria were used in an in vitro system that was not replenished with either SCN− or H2O2 during the assay. In vivo, continuous production of H2O2 [18,36,49] and SCN− transport in the presence of LPO are expected to continuously produce larger quantities of OSCN− for antibacterial host defense that are expected to prevent colonization of the airway but may not be able to eradicate an established infection.
There may be several reasons that the role of LPO in airway host defense, and a possible loss of its activity due to the anion transport defect in CF, have not previously attracted attention. First, studies that focus on antibacterial activity of airway secretions do not typically detect LPO activity since LPO requires H2O2 as a substrate and H2O2 is rapidly consumed and not present in secretions after collection and storage [38]. Thus, to detect LPO activity requires replenishing secretions with the estimated low physiological [H2O2] something not generally added in studies of antibacterial activity in airway secretions [e.g. 50]. These levels of H2O2, added to in vitro peroxidase antibacterial systems, are well below the levels needed for killing of bacteria by H2O2 alone [e.g. 12]. Second, broad screens of antibacterial substances have often focused on components with molecular masses below that of LPO [e.g. 51]. Finally LPO has an acidic pH optimum (~ pH 5.5) and shows reduced activity above pH 7.0. Thus, LPO’s characteristics and substrate needs may have slowed appreciation of its role in airway host defense.
Chronic infection in CF appears to arise from multiple defects in airway host defense. For example, inadequate ASL leads to dehydrated mucus, reduced mucociliary clearance [3], and decreased neutrophil migration into mucus [52]. Since LPO appears to be constitutively present in normal airway secretions, loss of its activity due to a SCN− transport defect may be an additional defect in CF airway host defense. Thus the data presented here support further studies of SCN− levels in CF airways.
Acknowledgments
We thank Dr. Adam Wanner for his thoughtful advice and strong support throughout this work. This work also acknowledges the memory of James R. Haft M.D. who inspired us by his lifelong determination and his dedication to medicine before his death from cystic fibrosis. This work was supported in part by the grants from NHLBI (GEC and MS) and The Cystic Fibrosis Foundation (SHR). Correspondence and requests for materials should be addressed to GEC. (Email: gconner@miami.edu)
Abbreviations
- ALI
air-liquid interface
- ASL
airway surface liquid
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane regulator
- LPO
lactoperoxidase
- MPO
myeloperoxidase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Boat TF, Boucher RC. In: Textbook of Respiratory Medicine. Murray JF, Nadel JA, editors. Vol. 2. W.B. Saunders Co; Philadelphia: 1994. pp. 1418–1450. [Google Scholar]
- 2.Sheppard DN, Welsh MJ. Structure and function of the CFTR chloride channel. Physiol Rev. 1999;79:S23–45. doi: 10.1152/physrev.1999.79.1.S23. [DOI] [PubMed] [Google Scholar]
- 3.Matsui H, Grubb BR, Tarran R, Randell SH, Gatzy JT, Davis CW, Boucher RC. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell. 1998;95:1005–1015. doi: 10.1016/s0092-8674(00)81724-9. [DOI] [PubMed] [Google Scholar]
- 4.Smith JJ, Travis SM, Greenberg EP, Welsh MJ. Cystic fibrosis airway epithelia fail to kill bacteria because of abnormal airway surface fluid. Cell. 1996;85:229–236. doi: 10.1016/s0092-8674(00)81099-5. [DOI] [PubMed] [Google Scholar]
- 5.Zabner J, Smith JJ, Karp PH, Widdicombe JH, Welsh MJ. Loss of CFTR chloride channels alters salt absorption by cystic fibrosis airway epithelia in vitro. Mol Cell. 1998;2:397–403. doi: 10.1016/s1097-2765(00)80284-1. [DOI] [PubMed] [Google Scholar]
- 6.Goldman MJ, Anderson GM, Stolzenberg ED, Kari UP, Zasloff M, Wilson JM. Human beta-defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in cystic fibrosis. Cell. 1997;88:553–560. doi: 10.1016/s0092-8674(00)81895-4. [DOI] [PubMed] [Google Scholar]
- 7.Wine JJ. The genesis of cystic fibrosis lung disease. J Clin Invest. 1999;103:309–312. doi: 10.1172/JCI6222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Travis SM, Singh PK, Welsh MJ. Antimicrobial peptides and proteins in the innate defense of the airway surface. Curr Opin Immunol. 2001;13:89. doi: 10.1016/s0952-7915(00)00187-4. [DOI] [PubMed] [Google Scholar]
- 9.Bals R, Weiner DJ, Wilson JM. The innate immune system in cystic fibrosis lung disease. J Clin Invest. 1999;103:303–307. doi: 10.1172/JCI6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guggino WB. Cystic fibrosis and the salt controversy. Cell. 1999;96:607–610. doi: 10.1016/s0092-8674(00)80570-x. [DOI] [PubMed] [Google Scholar]
- 11.Goldberg JB, Pier GB. The role of the CFTR in susceptibility to Pseudomonas aeruginosa infections in cystic fibrosis. Trends Microbiol. 2000;8:514–520. doi: 10.1016/s0966-842x(00)01872-2. [DOI] [PubMed] [Google Scholar]
- 12.Wijkstrom-Frei C, El-Chemaly S, Ali-Rachedi R, Gerson C, Cobas MA, Forteza R, Salathe M, Conner GE. Lactoperoxidase and Human Airway Host Defense. Am J Respir Cell Mol Biol. 2003;29:206–212. doi: 10.1165/rcmb.2002-0152OC. [DOI] [PubMed] [Google Scholar]
- 13.Gerson C, et al. The lactoperoxidase system functions in bacterial clearance of airways. Am J Respir Cell Mol Biol. 2000;22:665–671. doi: 10.1165/ajrcmb.22.6.3980. [DOI] [PubMed] [Google Scholar]
- 14.Reiter N, Perraudin JP. In: Peroxidases in Chemistry and Biology. Everse J, Everse KE, Grisham MB, editors. I. CRC Press; Boca Raton FL: 1991. pp. 143–180. [Google Scholar]
- 15.Pruitt KM. The salivary peroxidase system: thermodynamic, kinetic and antibacterial properties. J Oral Pathol. 1987;16:417–420. doi: 10.1111/j.1600-0714.1987.tb02078.x. [DOI] [PubMed] [Google Scholar]
- 16.Thomas EL, Bozeman PM, Learn DB. In: Peroxidases in Chemistry and Biology. Everse J, Everse KE, Grisham MB, editors. CRC Press; Boca Raton FL: 1991. pp. 123–142. [Google Scholar]
- 17.Fragoso MA, Fernandez V, Forteza R, Randell SH, Salathe M, Conner GE. Transcellular thiocyanate transport by human airway epithelia. J Physiol. 2004;561:183–194. doi: 10.1113/jphysiol.2004.071548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Forteza R, Salathe M, Miot F, Forteza R, Conner GE. Regulated hydrogen peroxide production by Duox in human airway epithelial cells. Am J Respir Cell Mol Biol. 2005;32:462–469. doi: 10.1165/rcmb.2004-0302OC. [DOI] [PubMed] [Google Scholar]
- 19.Davis CW, Dowell ML, Lethem M, Van Scott M. Goblet cell degranulation in isolated canine tracheal epithelium: response to exogenous ATP, ADP, and adenosine. Am J Physiol. 1992;262:C1313–1323. doi: 10.1152/ajpcell.1992.262.5.C1313. [DOI] [PubMed] [Google Scholar]
- 20.Kim KC, Lee BC. P2 purinoceptor regulation of mucin release by airway goblet cells in primary culture. Br J Pharmacol. 1991;103:1053–1056. doi: 10.1111/j.1476-5381.1991.tb12299.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saano V, Virta P, Joki S, Nuutinen J, Karttunen P, Silvasti M. ATP induces respiratory ciliostimulation in rat and guinea pig in vitro and in vivo. Rhinology. 1992;30:33–40. [PubMed] [Google Scholar]
- 22.Wong LB, Yeates DB. Luminal purinergic regulatory mechanisms of tracheal ciliary beat frequency. Am J Respir Cell Mol Biol. 1992;7:447–454. doi: 10.1165/ajrcmb/7.4.447. [DOI] [PubMed] [Google Scholar]
- 23.Villalon M, Hinds TR, Verdugo P. Stimulus-response coupling in mammalian ciliated cells. Demonstration of two mechanisms of control for cytosolic [Ca2+] Biophys J. 1989;56:1255–1258. doi: 10.1016/S0006-3495(89)82772-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johansen C, Falholt P, Gram L. Enzymatic removal and disinfection of bacterial biofilms. Appl Environ Microbiol. 1997;63:3724–3728. doi: 10.1128/aem.63.9.3724-3728.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bjorck L, Rosen C, Marshall V, Reiter B. Antibacterial activity of the lactoperoxidase system in milk against pseudomonads and other gram-negative bacteria. Appl Microbiol. 1975;30:199–204. doi: 10.1128/am.30.2.199-204.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reiter B, Marshall VM, Bjorck L, Rosen CG. Nonspecific bactericidal activity of the lactoperoxidases-thiocyanate-hydrogen peroxide system of milk against Escherichia coli and some gram-negative pathogens. Infect Immun. 1976;13:800–807. doi: 10.1128/iai.13.3.800-807.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Dalen CJ, Whitehouse MW, Winterbourn CC, Kettle AJ. Thiocyanate and chloride as competing substrates for myeloperoxidase. Biochem J. 1997;327:487–492. doi: 10.1042/bj3270487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ashby MT, Carlson AC, Scott MJ. Redox buffering of hypochlorous acid by thiocyanate in physiologic fluids. J Am Chem Soc. 2004;126:15976–15977. doi: 10.1021/ja0438361. [DOI] [PubMed] [Google Scholar]
- 29.Nlend MC, Bookman RJ, Conner GE, Salathe M. Regulator of G-protein signaling protein 2 modulates purinergic calcium and ciliary beat frequency responses in airway epithelia. Am J Respir Cell Mol Biol. 2002;27:436–445. doi: 10.1165/rcmb.2002-0012OC. [DOI] [PubMed] [Google Scholar]
- 30.Fulcher ML, Gabriel S, Burns KA, Yankaskas JR, Randell SH. Well-differentiated human airway epithelial cell cultures. Methods Mol Med. 2005;107:183–206. doi: 10.1385/1-59259-861-7:183. [DOI] [PubMed] [Google Scholar]
- 31.Pruitt KM, Mansson-Rahemtulla B, Baldone DC, Rahemtulla F. Steady-state kinetics of thiocyanate oxidation catalyzed by human salivary peroxidase. Biochemistry. 1988;27:240–245. doi: 10.1021/bi00401a036. [DOI] [PubMed] [Google Scholar]
- 32.Mansson-Rahemtulla B, Rahemtulla F, Baldone DC, Pruitt KM, Hjerpe A. Purification and characterization of human salivary peroxidase. Biochemistry. 1988;27:233–239. doi: 10.1021/bi00401a035. [DOI] [PubMed] [Google Scholar]
- 33.Pruitt KM, Mansson-Rahemtulla B, Tenovuo J. Detection of the hypothiocyanite (OSCN-) ion in human parotid saliva and the effect of pH on OSCN- generation in the salivary peroxidase antimicrobial system. Arch Oral Biol. 1983;28:517–525. doi: 10.1016/0003-9969(83)90184-x. [DOI] [PubMed] [Google Scholar]
- 34.Tenovuo J, Makinen KK, Sievers G. Antibacterial effect of lactoperoxidase and myeloperoxidase against Bacillus cereus. Antimicrob Agents Chemother. 1985;27:96–101. doi: 10.1128/aac.27.1.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ribeiro CM, Paradiso AM, Schwab U, Perez-Vilar J, Jones L, O'Neal W, Boucher RC. Chronic airway infection/inflammation induces a Ca2+i-dependent hyperinflammatory response in human cystic fibrosis airway epithelia. J Biol Chem. 2005;280:17798–17806. doi: 10.1074/jbc.M410618200. [DOI] [PubMed] [Google Scholar]
- 36.Geiszt M, Witta J, Baffi J, Lekstrom K, Leto TL. Dual oxidases represent novel hydrogen peroxide sources supporting mucosal surface host defense. Faseb J. 2003;17:1502–1504. doi: 10.1096/fj.02-1104fje. [DOI] [PubMed] [Google Scholar]
- 37.Schwarzer C, Machen TE, Illek B, Fischer H. NADPH oxidase-dependent acid production in airway epithelial cells. J Biol Chem. 2004;279:36454–36461. doi: 10.1074/jbc.M404983200. [DOI] [PubMed] [Google Scholar]
- 38.El-Chemaly S, Salathe M, Baier S, Conner GE, Forteza R. Hydrogen peroxide-scavenging properties of normal human airway secretions. Am J Respir Crit Care Med. 2003;167:425–430. doi: 10.1164/rccm.200206-531OC. [DOI] [PubMed] [Google Scholar]
- 39.Tahboub YR, Galijasevic S, Diamond MP, Abu-Soud HM. Thiocyanate modulates the catalytic activity of mammalian peroxidases. J Biol Chem. 2005;280:26129–26136. doi: 10.1074/jbc.M503027200. [DOI] [PubMed] [Google Scholar]
- 40.Furtmuller PG, Burner U, Obinger C. Reaction of myeloperoxidase compound I with chloride, bromide, iodide, and thiocyanate. Biochemistry. 1998;37:17923–17930. doi: 10.1021/bi9818772. [DOI] [PubMed] [Google Scholar]
- 41.Cantin AM. Taurine modulation of hypochlorous acid-induced lung epithelial cell injury in vitro. Role of anion transport. J Clin Invest. 1994;93:606–614. doi: 10.1172/JCI117013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thomas EL, Grisham MB, Jefferson MM. Preparation and characterization of chloramines. Methods Enzymol. 1986;132:569–585. doi: 10.1016/s0076-6879(86)32042-1. [DOI] [PubMed] [Google Scholar]
- 43.Thomas EL, Grisham MB, Jefferson MM. Cytotoxicity of chloramines. Methods Enzymol. 1986;132:585–593. doi: 10.1016/s0076-6879(86)32043-3. [DOI] [PubMed] [Google Scholar]
- 44.Grisham MB, Jefferson MM, Melton DF, Thomas EL. Chlorination of endogenous amines by isolated neutrophils. Ammonia-dependent bactericidal, cytotoxic, and cytolytic activities of the chloramines. J Biol Chem. 1984;259:10404–10413. [PubMed] [Google Scholar]
- 45.Thomas EL. Myeloperoxidase, hydrogen peroxide, chloride antimicrobial system: nitrogen-chlorine derivatives of bacterial components in bactericidal action against Escherichia coli. Infect Immun. 1979;23:522–531. doi: 10.1128/iai.23.2.522-531.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Illek B, Tam AW, Fischer H, Machen TE. Anion selectivity of apical membrane conductance of Calu 3 human airway epithelium. Pflugers Archiv - European Journal of Physiology. 1999;437:812–822. doi: 10.1007/s004240050850. [DOI] [PubMed] [Google Scholar]
- 47.Hipper A, Mall M, Greger R, Kunzelmann K. Mutations in the putative pore-forming domain of CFTR do not change anion selectivity of the cAMP activated Cl- conductance. FEBS Letters. 1995;374:312–316. doi: 10.1016/0014-5793(95)01132-x. [DOI] [PubMed] [Google Scholar]
- 48.Tabcharani JA, Rommens JM, Hou YX, Chang XB, Tsui LC, Riordan JR, Hanrahan JW. Multi-ion pore behaviour in the CFTR chloride channel [see comments] Nature. 1993;366:79–82. doi: 10.1038/366079a0. [DOI] [PubMed] [Google Scholar]
- 49.Ali-Rachedi R, Gelin M, Forteza R, Conner GE. Hydrogen Peroxide Production in Airway Epithelial Cells Is Catalyzed by Gp91phox Homologs Highly Similar to Duox 1. Am J Respir Crit Care Med. 2003;167:A284. [Google Scholar]
- 50.Dajani R, et al. Lysozyme secretion by submucosal glands protects the airway from bacterial infection. Am J Respir Cell Mol Biol. 2005;32:548–552. doi: 10.1165/rcmb.2005-0059OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cole AM, Dewan P, Ganz T. Innate antimicrobial activity of nasal secretions. Infect Immun. 1999;67:3267–3275. doi: 10.1128/iai.67.7.3267-3275.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Matsui H, Verghese MW, Kesimer M, Schwab UE, Randell SH, Sheehan JK, Grubb BR, Boucher RC. Reduced three-dimensional motility in dehydrated airway mucus prevents neutrophil capture and killing bacteria on airway epithelial surfaces. J Immunol. 2005;175:1090–1099. doi: 10.4049/jimmunol.175.2.1090. [DOI] [PubMed] [Google Scholar]