

Abstract
The HIV-1 capsid (CA) protein plays an important role in virus assembly and infectivity. Previously we showed that Ala substitutions in the N-terminal residues Trp23 and Phe40 cause a severely defective phenotype. In searching for mutations at these positions that result in a non-lethal phenotype, we identified one candidate, W23F. Mutant virions contained aberrant cores, but unlike W23A, also displayed some infectivity in a single-round replication assay and delayed replication kinetics in MT-4 cells. Following long-term passage in MT-4 cells, two second-site mutations were isolated. In particular, the W23F/V26I mutation partially restored the wild-type phenotype, including production of particles with conical cores and wild-type replication kinetics in MT-4 cells. A structural model is proposed to explain the suppressor phenotype. These findings describe a novel occurrence, namely suppression of a mutation in a hydrophobic residue that is critical for maintaining the structural integrity of CA and proper core assembly.
Keywords: HIV-1 capsid protein, second-site suppressors, HIV-1 viral cores, dominant-negative inhibition, HIV-1 assembly, structural models, reverse transcriptase, transmission electron microscopy
Introduction
The Gag polyprotein of human immunodeficiency virus type-1 (HIV-1), also known as Pr55gag, is the only viral protein required for particle assembly (Freed, 1998; Vogt, 1997; Wills and Craven, 1991). During or shortly after budding, the viral protease cleaves Gag into the mature HIV-1 proteins, which include (from the N- to C-terminus): matrix; capsid (CA); nucleocapsid (NC); and p6 (Freed, 1998; Swanstrom and Wills, 1997; Vogt, 1997). Proteolytic processing of Gag induces dramatic structural rearrangements (virus “maturation”), which lead to formation of mature, infectious HIV-1 virions containing electron-dense, cone-shaped cores (Gross et al., 1998; Gross et al., 2000; von Schwedler et al., 1998; Wiegers et al., 1998).
The HIV-1 core has been likened to a fullerene cone (Benjamin et al., 2005; Briggs et al., 2006; Ganser et al., 1999; Li et al., 2000) and is assembled with hexameric arrays of N-terminal CA domains such that each hexamer is connected to six others by interactions with C-terminal CA dimers (Ganser et al., 1999; Ganser et al., 2003; Ganser-Pornillos et al., 2004; Huseby et al., 2005; Lanman et al., 2003; Lanman and Prevelige, 2005; Li et al., 2000; Mayo et al., 2003). Helices I and II line the inner holes of each hexameric ring and are important for holding the rings together (Ganser-Pornillos et al., 2004; Lanman et al., 2003; Li et al., 2000). Based on the N-terminal domain structure of murine leukemia virus (MuLV) CA, it has been suggested that helix III also participates in formation of the inner lining of the hexameric ring (Mortuza et al., 2004). Evidence from numerous studies supports the idea that proper assembly of conical cores is required for HIV-1 infectivity (Auerbach et al., 2006; Dorfman et al., 1994; Fitzon et al., 2000; Forshey et al., 2002; Reicin et al., 1996; Scholz et al., 2005; Tang et al., 2003a; Tang et al., 2001; von Schwedler et al., 1998; von Schwedler et al., 2003).
The mature HIV-1 CA protein consists of 231 residues, which are folded into two independent domains connected by a short linker (Freed, 1998): an N-terminal “core” domain (residues 1-145) (Gitti et al., 1996; Momany et al., 1996; Tang, Ndassa, and Summers, 2002) and a C-terminal “dimerization” domain (residues 151-231) (Gamble et al., 1997). In general, mutation of N-terminal residues does not interfere with particle production, but mutant virions often exhibit defects in reverse transcription and core assembly as well as loss of infectivity (Tang et al., 2003b and references therein; Auerbach et al., 2006; Rulli et al., 2006; Scholz et al., 2005).
In earlier work, we found that Ala substitutions at Trp23 (helix I) and Phe40 (helix II), members of a conserved group of N-terminal hydrophobic residues in HIV-1 CA (Momany et al., 1996), result in a postentry defect with the following phenotype: virions produced are noninfectious, have aberrant cores, and are unable to initiate reverse transcription in infected cells, despite the presence of a functional reverse transcriptase (RT) in virus particles (Tang et al., 2001). Additionally, mutant cores retain an excessive amount of CA and contain almost no RT (Tang et al., 2003b). In view of recent findings emphasizing the critical role of helices I and II for CA structure and our data demonstrating the specific importance of Trp23 and Phe40 for proper core assembly and overall folding of CA, we initiated a study to provide new information on the plasticity of CA, i.e., its ability to tolerate change in residues crucial for CA structure without total abrogation of biological activity. More specifically, we wanted to determine (i) whether Trp23 or Phe40 can be changed to other residues that would permit some retention of infectivity and (ii) if so, whether the virus can rescue the mutant phenotype by generating second-site suppressor mutations.
Here we report that a search for other substitutions at positions 23 and 40. which could fulfill the requirement for some viral infectivity, identified one such mutation, i.e., Trp23 to Phe. Thus, W23F exhibits a low level of infectivity in a single-round replication assay and delayed replication kinetics in MT-4 cells. Isolation of second-site suppressor mutations in CA is rare and might not occur in the case of a mutation that has a major effect on CA structure. Yet surprisingly, long-term passage of W23F in MT-4 cells led to isolation of two second-site mutations, one of which, W23F/V26I (helix I), partially restores the wild-type (WT) phenotype. A structural model that accommodates the spatial changes induced by the W23F and V26I mutations shows that hydrophobic interactions between Ile26 and Phe23 are possible and helps to explain the suppressor phenotype. The data illustrate an unusual example in which a residue so intimately involved in maintaining the structural integrity of CA can be mutated without a complete loss of functional activity and the mutation can be rescued, at least partially, by a second-site suppression mechanism.
Results
New mutations at positions 23 and 40 in the N-terminal domain of CA
In earlier studies, we showed that mutants bearing Ala substitutions for Trp23 and Phe40 (Fig. 1) have severe defects in virus replication, virion ultrastructure, and biochemical properties (Tang et al., 2001; Tang et al., 2003b). To further elucidate the role of these residues in maintaining CA function, we asked whether other substitutions besides Ala would result in a more moderate phenotype. A total of 13 new mutations at Trp23 (W23C, D, F, G, H, L, M, N, P, S, T, V, and Y), two aromatic substitution mutations at Phe40 (F40W and F40Y), and one double mutation, W23F/F40W, were generated. The culture supernatants from transfected HeLa cells were assayed for RT activity and virus production was found to be ~60 to 90% of the WT value (data not shown). However, when infectivity was measured in a single-cycle assay using LuSIV cells (Roos et al., 2000), only W23F displayed any infectivity, albeit at a very low level (2% of the WT value) (see below); noninfectious mutants had ~0.5% of WT infectivity (data not shown).
Fig. 1.
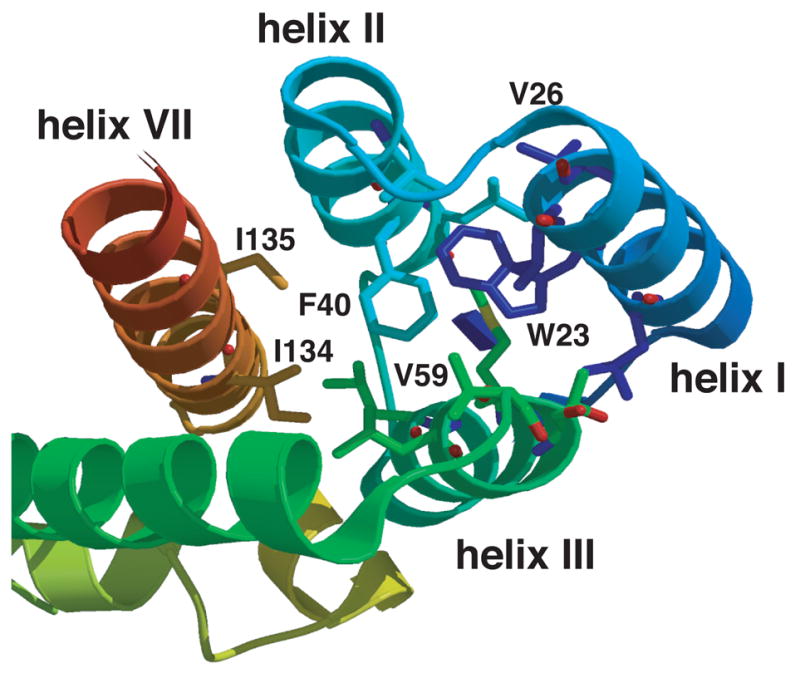
Ribbon diagram highlighting helices I, II, III, and VII in the N-terminal domain of HIV-1 CA. The ribbon diagram shows a top view of helices I, II, III, and VII in the N-terminal domain structure. The positions of two conserved hydrophobic residues Trp23 (helix I) and Phe40 (helix II) are shown. The positions of Val26 (helix I), Val59 (helix III), as well as I134 and I135 (helix VII) are also illustrated in the figure.
The replication capacity of W23F was evaluated in long-term culture experiments by transfecting T-cell lines, i.e., H9, Jurkat, CEM (12D7), and MT-4 cells, with WT or W23F plasmid DNAs. As expected, WT virus replicated at high levels in all of these cell lines, with peak virus production reached within the first week posttransfection (Fig. 2A; data not shown). In contrast, W23F replicated only in MT-4 cells, but with significantly delayed replication kinetics and a small peak of virus production evident only on day 18 posttransfection (Fig. 2A; data not shown). This result could reflect the more permissive nature of MT-4 cells, which are infected with HTLV-1 and express an active Tax protein that interacts with HIV-1 Tat to upregulate transcription of HIV-1 genes (Cheng, Tarnok, and Parks, 1998; Zack et al., 1988).
Fig. 2.
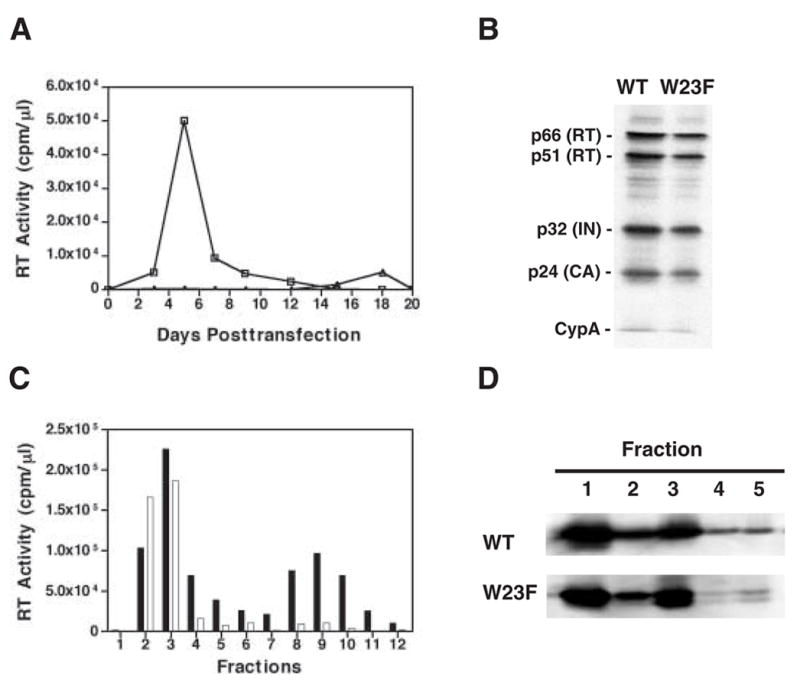
W23F phenotype. (A) Replication kinetics. MT-4 cells were transfected with WT and W23F plasmid DNAs. Cells were split 1:3 every 2 or 3 days. Virus production was monitored by assaying RT activity in the culture supernatants for each time point. Symbols: WT, open squares; W23F, open triangles; mock, open circles. (B) Incorporation of CypA into mature virus particles. HeLa cells were transfected with WT and W23F plasmid DNAs. Virion-associated proteins as well as CypA were detected by Western blot analysis, using anti-HIV RT and IN (Klutch et al., 1998) as well as anti-CA and anti-CypA (Tang et al., 2003b). The positions of individual viral proteins and CypA are indicated to the left of the gel. (C) Retention of HIV-1 RT in WT and W23F viral cores. Virions isolated from the supernatant fluids of transfected HeLa cells were treated with 0.3% NP-40 and were sedimented through 20% to 70% (wt/wt) linear sucrose gradients at 4°C for 16 h at 120,000 × g in a Beckman SW55Ti rotor (Tang et al., 2003b). Twelve fractions were collected from the top of the gradient. The bar graph shows RT activity in the fractions collected from WT (solid bars) or W23F (open bars) samples. (D) Retention of HIV-1 CA protein in viral cores. Detergent-treated virions were sedimented through sucrose step gradients centrifuged at 4°C for 60 min at 120,000 × g in a Beckman SW55Ti rotor (Tang et al., 2003b). Five fractions were collected from top of the gradients and were analyzed by Western blot using AIDS patient sera. HIV-1 CA protein bands are shown. Fractions 1 and 2 represent the soluble and detergent-soluble fractions, respectively. Fractions 3, 4, and 5 represent the detergent-resistant core fractions. The smaller band seen in W23F fractions 3 to 5 appears to be a CA degradation product.
W23F Phenotype
Since the replication data described above (Fig. 2A) suggested that W23F might be less defective than W23A, it was of interest to characterize the W23F phenotype in more detail. Interestingly, like W23A and F40A (Tang et al., 2003b), most of the new mutants were unable to incorporate normal levels of CypA (data not shown). However, as illustrated in Fig. 2B, the protein composition of W23F was essentially the same as that of WT and the ratio of CypA incorporated by W23F relative to the level of integrase was 97% of the WT value.
It was also of interest to determine the amounts of RT and CA retained in viral cores isolated by detergent-treatment of virions and subsequent sedimentation in sucrose density gradients (Tang et al., 2003b). As anticipated, the WT sample had two peaks of RT activity, one in the soluble fractions 2 to 4, and the other in the high-density core fractions 8 to 10 (Fig. 2C) (Tang et al., 2003b). In contrast, like the original mutants W23A and F40A (Tang et al., 2003b), W23F had only one peak of RT activity, which was located in the soluble fractions 2 to 4 (Fig. 2C). Quantification of the data indicated that less than 5% of total RT activity was retained in W23F cores, whereas ~28% of RT activity was detected in WT cores (also see Tang et al., 2003b).
To examine CA content in viral cores, detergent-treated virions were sedimented in a sucrose step gradient (Fig. 2D). Proteins associated with viral cores were found primarily in fraction 3, but some material was also detected in fractions 4 and 5 (Fig. 2D) (Tang et al., 2003b). The results of Western blot analysis showed that the distribution of CA protein in the sucrose fractions of W23F and WT samples was very similar. Densitometry scanning of the CA bands showed that W23F cores retained ~ 44% of total CA. This value is dramatically lower than the values reported for W23A and F40A (~70% of total CA) (Tang et al., 2003b), but is still somewhat higher than the WT value of ~30% shown here and in a previous report (Tang et al., 2003b) (see below).
The ultrastructure of W23F particles was investigated by transmission electron microscopy. The images showed that WT particles contained typical cone-shaped cores (Fig. 3A; Table 1), whereas the W23F virions had round, centric or acentric cores (defined in Table 1) and no conical cores (Fig. 3B; Table 1). Interestingly, the architecture of W23F resembles that of CA mutant D51A (Tang et al., 2001; von Schwedler et al., 1998), but is quite different from the abnormal ultrastructure seen with W23A (Tang et al., 2001). In addition to the lack of conical cores, the diameter of W23F virions (183 ± 40 nm) was significantly larger than that of WT (125 ± 13 nm) (compare Figs. 3A and 3B; Table 1). These findings are consistent with rate zonal sucrose density gradient sedimentation analysis (data not shown) and are in accord with the conclusion that the CA domain in Gag is a major determinant of virus size and morphology (Ako-Adjei, Johnson, and Vogt, 2005).
Fig. 3.
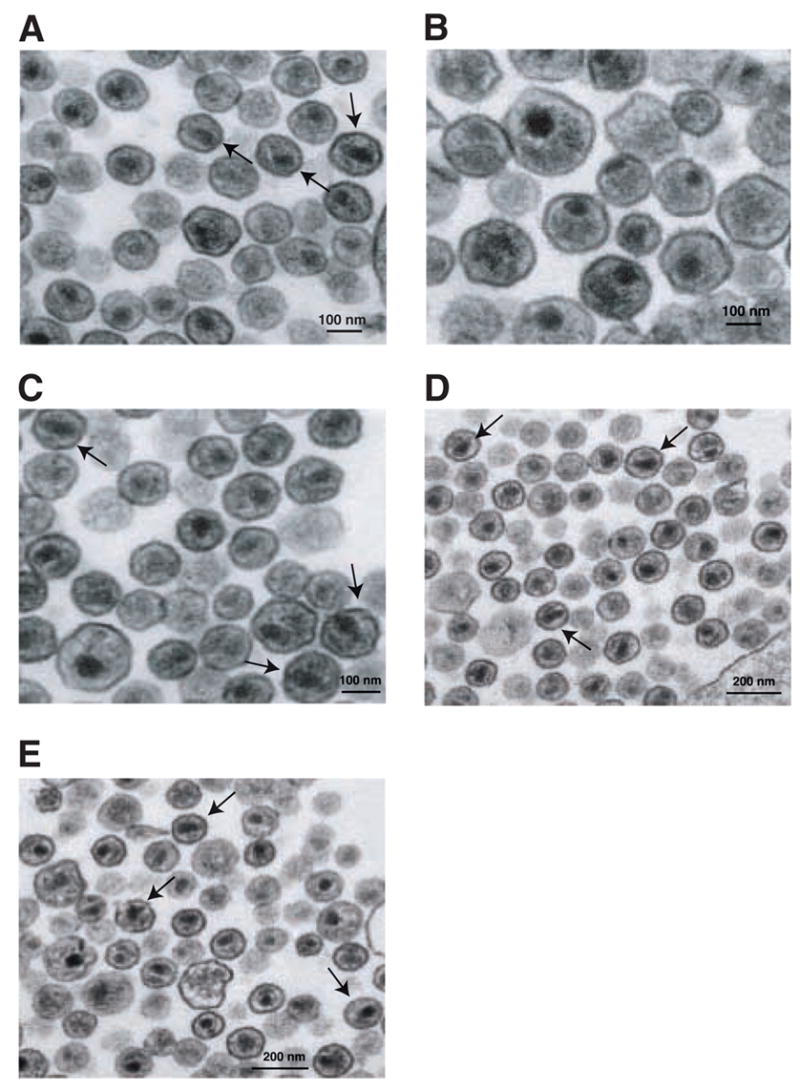
Transmission electron microscopy of WT and mutant virions produced in HeLa and MT-4 cells. HeLa cells: (A) HIV-1 WT; (B) W23F. MT-4 cells: (C) WT; (D) W23F/V26I; (E) W23F/V26I/R154K. The arrows point to representative particles in the field with a WT phenotype (i.e., having conical cones). The scale bars are 100 nm (A, B, and C) or 200 nm (D and E).
Table 1.
Electron microscopic analysis of HIV-1 WT and mutant particles in HeLa and MT-4 cells
| No. of particles whose cores are:
|
||||||
|---|---|---|---|---|---|---|
| Virus | Cell type | No. of particles | Particle size (nm) | Cone-shaped | Centrica | Acentricb |
| WT | HeLa | 105 | 125 ± 13 | 21 (20)c | 53 (50) | 31 (30) |
| W23F | HeLa | 82 | 183 ± 40 | 0 | 28 (34) | 66 (66) |
| WT | MT-4 | 113 | 116 ± 8.1 | 49 (43) | 51 (45) | 13 (12) |
| W23F/V26I | MT-4 | 118 | 115 ± 8.5 | 32 (27) | 36 (31) | 20 (17) |
| W23F/V26I/R154K | MT-4 | 79 | 121 ± 11 | 26 (33) | 39 (49) | 14 (18) |
Electron-dense cores that are centrally located in the particle.
Electron-dense round cores that are pushed up against or close to the viral membrane.
Numbers in parentheses are percentage of the total number of particles.
Taken together, the data demonstrate that with the exception of W23F, the new Trp 23 and Phe 40 mutations constructed in this study had a severe impact on maintaining proper CA structure and virus infectivity. In contrast, substitution of Phe at position 23 led to a somewhat less defective phenotype with respect to virion ultrastructure, incorporation of CypA, retention of CA in viral cores, and even viral infectivity. Only the RT content of cores remained as low in W23F as in the original W23A and F40A mutants.
W23F exhibits dominant-negative inhibition of WT infectivity
Since CA protein multimerizes (Campbell and Vogt, 1995; Ehrlich, Agresta, and Carter, 1992; Ganser et al., 2003; Ganser-Pornillos et al., 2004; Gross, Hohenburg, and Kräusslich, 1997; Lanman et al., 2003; Mayo et al., 2003; von Schwedler et al., 1998) when Gag molecules assemble, we investigated the possibility that W23F and W23A CA might affect the function of WT CA in trans and thereby inhibit WT infectivity (Fig. 4).
Fig. 4.
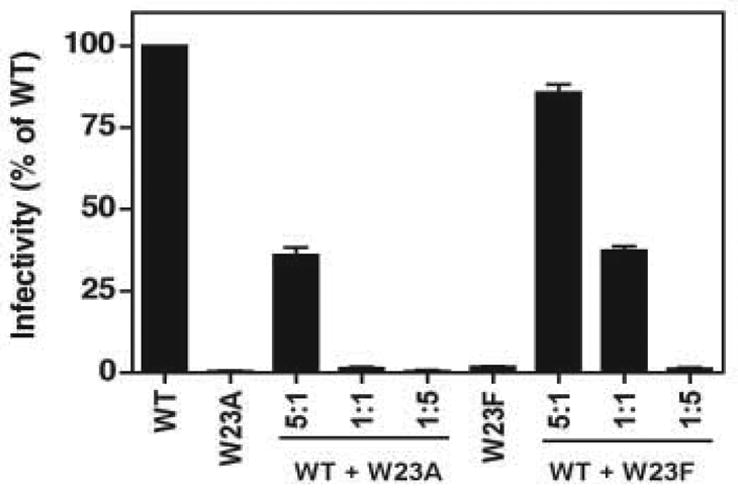
Dominant negative activity of W23A and W23F. HeLa cells were transfected with 5 μg of the following plasmid DNAs: WT alone, W23A or W23F alone, and 5:1, 1:1, and 1:5 mixtures of WT and either W23A or W23F. Virus stocks were harvested, normalized for RT activity, and used to infect LuSIV cells (Roos et al., 2000). The values in the bar graph represent infectivity as the percentage of the WT value, which was set at 100%. Here as well as in Figs. 5 and 6, error bars indicate the standard deviation from at least three independent experiments.
With a 5-fold excess of WT:W23A DNA, viral infectivity was reduced to 36% of the normal WT level, whereas with a 1:1 DNA ratio, the resulting virus had only 1.3% of WT infectivity. With a 1:5 ratio for WT:W23A, infectivity was further reduced to 0.5%. In contrast, W23F displayed a moderate inhibitory effect. At a ratio of 5:1, the resulting virus population retained significant infectivity (84% compared with WT alone) and with 1:1 ratio, infectivity was 37% of the WT level. Infectivity was reduced to 1.3% when there was a five-fold excess of W23F DNA.
These findings indicate that the transdominant inhibitory effect of W23A is ~5-fold greater than that of W23F, in accord with the greater severity of the observed W23A phenotype. Other HIV-1 (Furuta et al., 1997; Mammano et al., 1994; Scholz et al., 2005; Shimano et al., 1998; Trono, Feinberg, and Baltimore, 1989) and MuLV (Rulli et al., 2006) CA mutants, which are capable of dominant-negative interference, have also been described.
Second-site suppressors of W23F
The more moderate phenotype of W23F compared with that of W23A (Figs. 2 and 4) and the ability of W23F to replicate in MT-4 cells (Fig. 2A) encouraged us to try to isolate second-site suppressor mutations by prolonged passage of W23F in MT-4 cells. For the initial infection and for each subsequent passage, virions harvested at the peak of virus replication were used to infect fresh MT-4 cells. The experiment was terminated when the replication kinetics of the passaged virus were the same as the kinetics for the WT. In all, four passaging cycles were performed (data not shown).
The results of sequencing DNA samples collected at the time of peak virus replication showed that viral DNA from each passage retained the original W23F mutation. However, passage 3 DNA contained an additional mutation at nucleotide (nt) 1261 (G→A, codon change of GTA to ATA) that resulted in an Ile for Val26 substitution (V26I); passage 4 DNA contained a third mutation at nt 1646 (G→A, codon change of AGA to AAA) that led to substitution of Arg for Lys at position 154 (R154K) (data not shown). Val26 is located in helix I of the N-terminal domain of HIV-1 CA and is close to Trp23 (Fig. 1), whereas Lys154 is found at the beginning of the MHR in the C-terminal domain. The emergence of the V26I and V26I/R154K mutations in virions derived from long-term passage of W23F in MT-4 cells suggests that the conversion from delayed replication kinetics to WT kinetics is the consequence of these additional mutations.
Defects in W23F are partially repaired by mutations V26I or V26I/R154K
To further evaluate whether the two mutations, V26I and V26I/R154K, were responsible for the improved replication capacity of the W23F-derived virus, double (W23F/V26I) and triple (W23F/V26I/R154K) mutants were constructed in a pNL4-3 background (Adachi et al., 1986). Unlike the parental clone W23F (Fig. 2A), the replication kinetics of W23F/V26I and W23F/V26I/R154K in MT-4 cells exhibited a pattern close to that of WT (delay of only two days) (Fig. 5A). Furthermore, the infectivity of the double and triple mutants in the single-cycle LuSIV assay showed a modest increase from the value for W23F (2% of WT infectivity) to 12% and 16% of the WT level (virus produced in HeLa cells) and 14% and 17% (virus produced in MT-4 cells), respectively (Fig. 5B). Similar results were obtained when mutant virions isolated from transfected MT-4 cells were tested in another single-cycle assay that uses TZM-bl cells (data not shown). Thus, the second-site suppressor mutations appeared to be capable of partial rescue of the replication defect imposed by the W23F mutation.
Fig. 5.
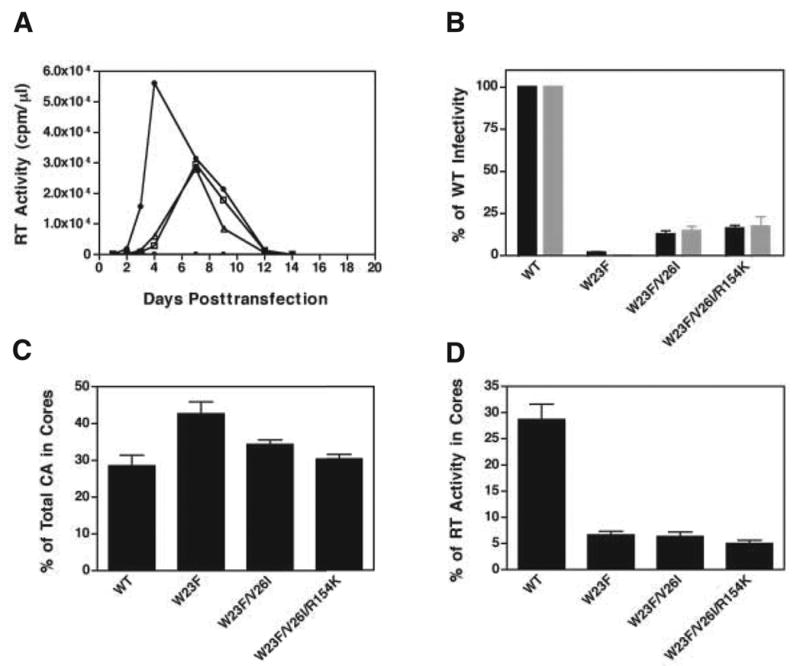
Phenotypes of second-site suppressor mutants. (A) WT proviral DNA and proviral DNAs bearing the indicated double or triple mutations were used to transfect MT-4 cells in a long-term culture experiment. RT activity in the culture supernatants was assayed to monitor the replication of WT (closed circles), W23F/V26I (open squares), and W23F/V26I/R154K (open triangles). A mock-infected control (no DNA) was also included (open circles). (B) Virus particles obtained from the supernatant fluids of transfected HeLa cells (black bars) and MT-4 cells (gray bars) were normalized and were used to infect LuSIV cells (Roos et al., 2000). The results are reported as percentage of WT infectivity, which was set at 100%. (C) and (D) Assays for detection of HIV-1 CA and RT retention in viral cores were performed as described previously (Tang et al., 2003b). The amounts of CA in the detergent-resistant core fractions were quantified and plotted as the percentage of total CA protein recovered from the sucrose step gradient (C). RT activity in the core fractions isolated from 20% to 70% (wt/wt) linear sucrose density gradients was quantified and plotted as the percentage of total RT activity (D). The values for WT and W23F in panels (C) and (D) were calculated from data in Fig. 2.
We next sought to determine whether the suppressor mutations could reverse the W23F core defects in RT and CA content observed with particles produced in HeLa cells. As may be seen in Fig. 5C, the amount of CA in the cores of these mutants was ~35% of total CA protein, which is close to the WT value of ~30% and lower than the value for W23F (~44%). Surprisingly, the amount of RT retained in W23F/V26I and W23F/V26I/R154K cores was approximately the same as in W23F cores, i.e., around 5% of total RT activity (Fig. 5D). This does not appear to be the result of a defect in viral protein expression, encapsidation, and processing since both the double and triple mutants have protein profiles similar to that of WT (data not shown). Taken together, these results demonstrate that the phenotypes of the double and triple mutants are very similar and represent a significant improvement in some, but not all aspects of the W23F defect.
Phenotypic analysis of CA mutations generated in the pNL4-3 WT clone
To examine the effects of the individual mutations that appeared after long-term passage of W23F and in particular, to determine whether both the W26I and R154K mutations are required for suppression, we constructed a series of single or double mutations (Fig. 6). The single mutants V26I and R154K as well as the double mutant V26I/R154K showed WT or nearly WT replication kinetics in MT-4 cells, i.e., a replication peak appeared 4 to 7 days posttransfection (Fig. 6A). Moreover, the infectivity of these mutants measured in the single-cycle LuSIV assay was between 82- to- 120% of the WT level (Fig. 6B). These results demonstrate that V26I, R154K, and the double mutant V26I/R154K behave like WT, presumably because these mutations do not affect CA function. However, the W23F/R154K mutant failed to replicate in MT-4 cells (Fig. 6A) and displayed only ~2% of WT infectivity (Fig. 6B). This finding taken together with the very slight (albeit consistent) improvement in infectivity (Fig. 5B), CA content (Fig. 5C), and the percentage of conical cores in the virus population (Table 1) seen with the W23F/V26I/R154K mutant (compared with W23F/V26I) suggests that the R154K mutation had only a minimal effect on the second-site suppressor phenotype.
Fig. 6.
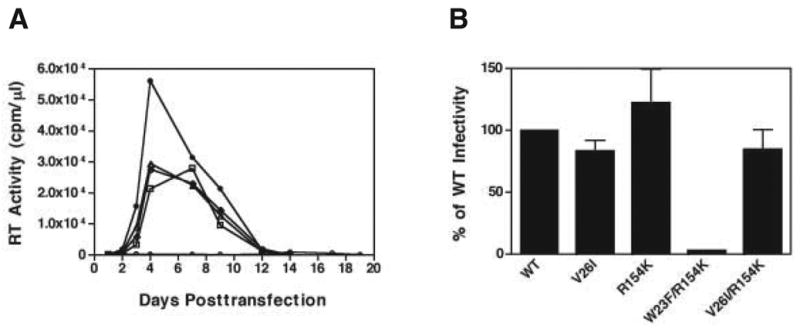
Phenotypes of single or double CA mutants. (A) The replication kinetics of WT and the following CA mutants in MT-4 cells are shown: WT (closed circles), V26I (open squares), R154K (open triangles), W23F/R154K (open circles) and V26I/R154K (open diamonds). (B) Virus infectivity was plotted as the percentage of WT infectivity, which was set at 100%. Note that the data of Fig. 5A and 6A were obtained from the same experiment; thus, the WT curves in both figures are identical.
Ultrastructure of W23F/V26I and W23F/V26I/R154K virions produced in MT-4 cells
Since the double and triple mutants exhibit close to WT replication kinetics in MT-4 cells (Fig. 5A), it was also of interest to determine whether these mutants exhibit WT virus morphology. MT-4 cells transfected with either WT or the double and triple mutants were fixed and examined by transmission electron microscopy (Fig. 3). Interestingly, many of the particles in each mutant sample (Figs. 3D and 3E) resembled WT virions (Fig. 3C) and had conical cores (note positions of arrows), which is consistent with the replication data. Table 1 quantifies the distribution of different core structures (cone-shaped, centric, or acentric) in the WT and mutant samples. The percentage of cone-shaped cores in the double and triple mutant samples was 27% and 33%, respectively, compared to 43% for the WT. (The mutant values were therefore 63% and 76%, respectively, of the WT value). The particle sizes of the two mutants (double mutant, 115 ± 8.5 nm; triple mutant, 121 ± 11 nm) were essentially the same as the size of WT virions (116 ± 8.1 nm) (Table 1). Thus, these findings demonstrate that the second-site suppressor mutations largely restored proper assembly of virion cores and maturation of virus particles in MT-4 cells.
Discussion
The goal of this study was to determine whether HIV-1 can adapt to changes in residues that are important for maintaining CA structure without losing all biological activity. Our approach was to make mutant constructs that might retain the ability to replicate and thereby present an opportunity to isolate second-site suppressors. Out of 13 mutations in Trp23, two in Phe 40 (F40W, F40Y), and a double mutation (W23F/F40Y), we found only one substitution, W23F, that could satisfy this requirement. This observation emphasizes the critical contribution of Trp23 and Phe40 to the local structure of helices I and II, respectively. Note the proximity of these residues in the view shown in Fig. 1 and the central location of Phe in the hydrophobic core (Fig. 1).
The data clearly indicate a strict requirement for an aromatic residue at position 23 and it is interesting that even Tyr cannot substitute for Trp23 (or Phe40), presumably because the OH group of Tyr makes it too hydrophilic. The finding that Trp cannot replace Phe40 may reflect a steric requirement for a less bulky residue at position 40. These results are consistent with the crucial role of residues in helices I and II in the assembly of the HIV-1 capsid (Ganser et al., 2003; Ganser-Pornillos et al., 2004; Lanman et al., 2003; Li et al., 2000). In this regard, it is also of interest to note that deuterium exchange studies have identified a CA peptide consisting of residues 23 to 40, which appears to be involved in N-terminal homotypic domain interactions in the in vitro assembled CA protein as well as in immature and mature HIV-1 virus-like particles (Lanman et al., 2004; Lanman and Prevelige, 2005).
Isolation of second-site suppressors is a powerful genetic technique to obtain additional insights into the structural and functional effects of particular mutations. However, there are only a few reports of second-site suppressors of CA mutations (see below), presumably because most mutations in CA are not well tolerated (Auerbach et al., 2003; von Schwedler et al., 2003) and the possibilities for suppressing these mutations may be limited. Until now, second-site suppressors have been described for CA mutations in (i) the HIV-1 CypA binding loop (Aberham, Weber, and Phares, 1996); (ii) an 11-residue segment (Guo et al., 2005) at the C-terminus of HIV-1 CA that is required for virus replication (Liang et al., 2002; Liang et al., 2003; Melamed et al., 2004); and (iii) the RSV MHR (Bowzard, Wills, and Craven, 2001).
In the present study, despite the low probability of success, we were able to isolate two second-site suppressor mutants, W23F/V26I and W23F/V26I/R154K, after prolonged passage in MT-4 cells. However, it appears that only the V26I substitution contributes significantly to suppression of the W23F mutation (Fig. 6), although the additional R154K mutation might have a subtle effect (Fig. 5 and Table 1). The suppression event was specific for MT-4 cells, since W23F could not replicate even with delayed kinetics in other cell lines (see above). Transmission electron microscopy showed that many of the suppressor mutant particles isolated from MT-4 cells have the appearance of WT virus and contain conical cores (Figs. 3C, 3D, 3E; Table 1). Although the infectivity of these mutants (produced in either HeLa or MT-4 cells) was higher than that of W23F, it was still much lower than the WT value (Fig. 5B). The reduced infectivity in CEM174 cells, which are used in the LuSIV assay (Roos et al., 2000), might be related to the fact that similar cells (CEM (12D7)) do not support long-term replication of any of our mutants (data not shown).
The original W23A and F40A mutants (Tang et al., 2003b) and the HIV-1 His mutant, H84A (Scholz et al., 2005), were found to have abnormally high amounts of CA in their cores. In the case of W23F, the CA content is somewhat higher than normal, but the amount of CA in the cores of the two second-site suppressor mutants is essentially like that of WT, i.e., ~30% core-associated CA protein (Fig. 5C) (Tang et al., 2003b). (A WT value of ~17% has also been reported (Forshey and Aiken, 2003).) These data are in accord with recent physical determinations indicating that of the ~5000 copies of Gag in the immature virion, only 1000-1500 copies of CA are associated with the core of the mature particle (Briggs et al., 2003; Briggs et al., 2004; Lanman et al., 2004; Vogt and Simon, 1999).
Another striking feature of W23A and F40A cores is a reduction in RT content from the WT value of ~30% of total virion RT (Fig. 5D) (Khan et al., 2001; Tang et al., 2003b) to ~5% (Tang et al., 2003b). Similar findings have been reported for the H84A mutant (Scholz et al., 2005) and an RSV MHR mutant L171I (Cairns and Craven, 2001). Surprisingly, the WT level of core-associated RT was not restored in either W23F or in the suppressor mutants (Fig. 5D) and this might account for the low level of infectivity observed in the LuSIV assay (Fig. 5B). More detailed studies of the structure and organization of the ribonucleoprotein complex in HIV-1 cores will be needed to fully understand the relationship between RT levels in cores, RT function, and the ability to generate infectious particles.
Finally, we ask: “How can we explain why V26I partially rescues the W23F mutation?” This question is especially relevant since Val26 is partly exposed on the surface of helix 1, whereas Trp23 is buried within the hydrophobic core (Fig. 1) (Momany et al., 1996; Tang, Ndassa, and Summers, 2002). Interestingly, the NMR structure of the N-terminal domain of CA (Tang, Ndassa, and Summers, 2002) indicates that Val26 and Trp23 are actually close to each other. Specifically, among the family of models in the structure file (1GWP.pdb), the smallest separation between the side chain heavy atoms of Trp23 and Val26 ranges from 3.8 to 4.7 Å, with a mean of 4.2 Å. Based on this information, a model of the W23F/V26I structure was constructed such that the additional non-polar methylene group in Ile (CH-CH2-CH3), i.e., compared with Val (CH-CH3), contacts the Phe side chain and at least partially fills the cavity created by the W23F mutation (Fig. 7) (S. R. Durell, personal communication).
Fig. 7.
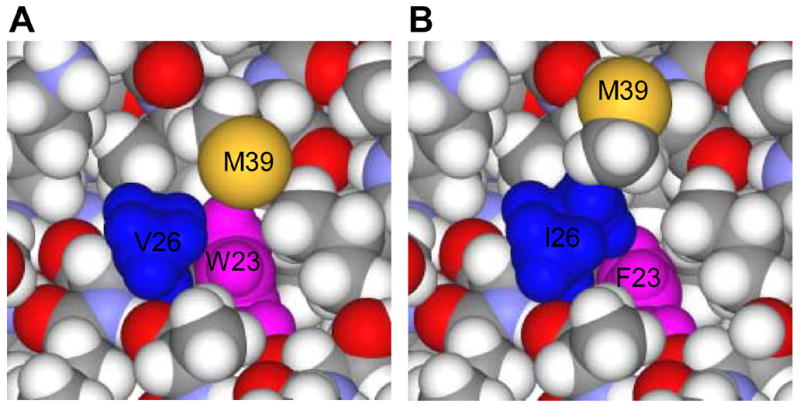
Structural models of WT CA and the W23F/V26I mutant illustrating contacts between the side chains of Trp23 and Val26 or the substituted Phe23 and Ile26 residues. (A) WT. Model number 10 in the 1GWP.pdb structure file of the N-terminal CA domain (Tang, Ndassa, and Summers, 2002) demonstrates the closest approach of the Trp23 (blue) and Val26 (magenta) side chains. Note that for clarity, the terminal methyl group of Met39 (yellow), which “hangs” over Trp23 is not shown. (B) W23F/V26I mutant. A possible model for the double mutant was constructed by mutating the Trp23 and Val26 residues and then slightly moving the Ile26 and Met39 side chains. Phe23 is shown in blue and Ile26 in magenta. This figure was kindly provided by Stewart R. Durell (NCI, NIH).
In conclusion, we have shown that it is possible to make a substitution in a residue critical for maintaining HIV-1 CA structure and function, i.e., W23F, which results in virions with a low level of infectivity in a single-round replication assay and moderate loss of the WT phenotype. Since the N-terminal CA structure is defined by stringent parameters and only rarely accommodates even small perturbations at certain residues, it is especially surprising that a second-site suppressor of the W23F mutation could be isolated. These findings are novel and demonstrate that despite the limits imposed on assembly of proper CA structure, HIV-1 is able to partially adapt to even severe structural disruptions.
Materials and methods
Cell culture, transfection, and infection
HeLa cells were maintained as previously described (Tang et al., 2001) and were transfected by using the Trans IT transfection reagent (Mirus, Madison, WI), according to the manufacturer’s instructions. T-cell lines (H9, MT-4, Jurkat, CEM (12D7), and CEM174) were maintained as described (Murakami and Freed, 2000) and were transfected by using the DEAE-dextran procedure detailed by Freed and Martin (Freed and Martin, 1994). For infection of T-cell lines, virus was obtained from the culture fluids of HeLa or MT-4 cells transfected with pNL4-3 (Adachi et al., 1986) or with variants having mutations in the CA coding region. RT activity was detected by exogenous RT assay (Freed, Englund, and Martin, 1995).
Single-cycle infectivity assays
To measure viral infectivity, we used a single-cycle assay (Roos et al., 2000). Equal amounts of viral particles (106 cpm of RT activity under the conditions of our RT assay) were used to infect LuSIV cells, which are CEM174 cells stably transfected with the firefly luciferase reporter gene under the control of the simian immunodeficiency virus (SIV) long terminal repeat. Some single-cycle assays were also performed with TZM-bl cells, a HeLa cell line, which expresses large amounts of CD4 and CCR5 and has integrated copies of luciferase and β-galactosidase genes under the control of the HIV-1 promoter (Derdeyn et al., 2000; Platt et al., 1998; Wei et al., 2002). LuSIV cells (originally from Drs. J. W. Roos and J. E. Clements) and TZM-bl cells (originally from Dr. John Kappes, Dr. Xiaoyun Wu, and Tranzyme, Inc.) were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH.
Plasmids and site-specific mutagenesis
The cloning strategy used for introducing site-specific mutations into the CA coding region in pNL4-3 followed the procedures described by Tang et al. (Tang, Collier, and Elliott, 1999). Detailed information about the primers and cloning procedures used for the construction of mutations at residues Trp23 and Phe40 of HIV-1 CA, as well as the single mutations V26I and R154K are available upon request. The sequences of all CA mutants were confirmed by sequencing performed by the Biopolymer Lab (University of Maryland, College Park, MD). Plasmid DNAs were prepared with a QIAFilter Maxi Kit (Qiagen) or by Lofstrand.
Analysis and construction of revertant clones
In the long-term replication experiments, genomic DNA (template for PCR) was extracted from infected MT-4 cells collected at the virus replication peak, using a QIAamp DNA Mini Kit (Qiagen). A 1.3-kb fragment of viral DNA representing almost the entire HIV-1 gag region, i.e., between the BssHII and ApaI sites of pNL4-3 (nt 701-2018) (GenBank accession number M19921) was amplified with PCR SuperMix (Invitrogen). The primer sets used were as follows: JL392 (forward primer, 5′-GCT TGC TGA AGC GCG CAC GGC, nt 701 to 721); and JL569 (reverse primer, 5′-TCC TAG GGG CCC TGC AAT TTT TGG, nt 2018 to 1994). PCR products were purified using the QIAQuick PCR Purification Kit (Qiagen) and sequenced.
To construct the revertant clones, PCR products containing the double mutation W23F/V26I or the triple mutation W23F/V26I/R154K, were digested with BssHII and ApaI. The resulting fragments were then inserted into the pNL4-3 vector cut with the same enzymes. For construction of the double mutants W23F/R154K and V26I/R154K, W23F, V26I, and R154K plasmid DNAs were digested with BssHII and SpeI. Ligation of fragments containing the W23F and R154K mutations, respectively, produced CA mutant W23F/R154K, whereas ligation of fragments containing V26I and R154K, respectively, yielded CA mutant V26I/R154K.
Western blot analysis of WT and mutant viral proteins
The Western-Star chemiluminescence detection system (TROPIX Inc., Bedford, MA) was used for the analysis. The densities of the protein bands were quantified with a laser densitometer by using a model PDS1-P90 instrument from Molecular Dynamics Inc., Sunnyvale, CA. AIDS patient sera were obtained from the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH. The identity of individual viral protein bands was confirmed with specific antisera. Akira Ono, Department of Microbiology and Immunology, University of Michigan Medical School, Ann Arbor, MI (formerly at the HIV Drug Resistance Program, NCI at Frederick, Frederick, MD) kindly provided rabbit anti-HIV CA antibody. Rabbit anti-CypA was a generous gift from David Ott, SAIC Frederick, Inc., NCI at Frederick, Frederick, MD. Rabbit anti-HIV IN and RT antibodies have been described in a previous report (Klutch et al., 1998).
Transmission electron microscopy
Preparation of HeLa and MT-4 cells transfected with WT and mutant virions for examination by electron microscopy was performed as described previously (Freed and Martin, 1994; Tang et al., 2001). Images shown in Fig. 3 illustrate representative particles seen in at least eight to ten thin sections per sample.
Acknowledgments
We thank David Ott and Akira Ono for their generous gifts of anti-CypA and anti-CA sera, respectively, and Zhonglin Yang for generating the ribbon diagram used to illustrate N-terminal residues in the hydrophobic core of CA. We are also indebted to Stewart R. Durell for valuable insights into the structure of the N-terminal CA domain and for providing Fig. 7. This research was supported by the Intramural Research Program of the NIH (NICHD and NCI, Center for Cancer Research) and in part with federal funds from the NCI, NIH, under contract N01-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aberham C, Weber S, Phares W. Spontaneous mutations in the human immunodeficiency virus type 1 gag gene that affect viral replication in the presence of cyclosporins. J Virol. 1996;70:3536–3544. doi: 10.1128/jvi.70.6.3536-3544.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol. 1986;59:284–291. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ako-Adjei D, Johnson MC, Vogt VM. The retroviral capsid domain dictates virion size, morphology, and coassembly of Gag into virus-like particles. J Virol. 2005;79:13463–13472. doi: 10.1128/JVI.79.21.13463-13472.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach MR, Shu C, Kaplan A, Singh IR. Functional characterization of a portion of the Moloney murine leukemia virus gag gene by genetic footprinting. Proc Natl Acad Sci USA. 2003;100:11678–11683. doi: 10.1073/pnas.2034020100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach MR, Brown KR, Kaplan A, de Las Nueces D, Singh IR. A small loop in the capsid protein of Moloney murine leukemia virus controls assembly of spherical cores. J Virol. 2006;80:2884–2893. doi: 10.1128/JVI.80.6.2884-2893.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin J, Ganser-Pornillos BK, Tivol WF, Sundquist WI, Jensen GJ. Three-dimentional structure of HIV-1 virus-like particles by electron cryotomography. J Mol Biol. 2005;346:577–588. doi: 10.1016/j.jmb.2004.11.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowzard JB, Wills JW, Craven RC. Second-site suppressors of Rous sarcoma virus CA mutations: evidence for interdomain interactions. J Virol. 2001;75:6850–6856. doi: 10.1128/JVI.75.15.6850-6856.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs JAG, Wilk T, Welker R, Kräusslich H-G, Fuller SD. Structural organization of authentic, mature HIV-1 virions and cores. EMBO J. 2003;22:1707–1715. doi: 10.1093/emboj/cdg143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs JAG, Simon MN, Gross I, Kräusslich H-G, Fuller SD, Vogt VM, Johnson MC. The stoichiometry of Gag protein in HIV-1. Nat Struct Mol Biol. 2004;11:672–675. doi: 10.1038/nsmb785. [DOI] [PubMed] [Google Scholar]
- Briggs JAG, Grünewald K, Glass B, Förster F, Kräusslich HG, Fuller SD. The mechanism of HIV-1 core assembly: Insights from three-dimensional reconstructions of authentic virions. Structure. 2006;14:15–20. doi: 10.1016/j.str.2005.09.010. [DOI] [PubMed] [Google Scholar]
- Cairns TM, Craven RC. Viral DNA synthesis defects in assembly-competent Rous sarcoma virus CA mutants. J Virol. 2001;75:242–250. doi: 10.1128/JVI.75.1.242-250.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell S, Vogt VM. Self-assembly in vitro of purified CA-NC proteins from Rous sarcoma virus and human immunodeficiency virus type 1. J Virol. 1995;69:6487–6497. doi: 10.1128/jvi.69.10.6487-6497.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Tarnok J, Parks WP. Human immunodeficiency virus type 1 genome activation induced by human T-cell leukemia virus type 1 Tax protein is through cooperation of NF-κB and Tat. J Virol. 1998;72:6911–6916. doi: 10.1128/jvi.72.8.6911-6916.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derdeyn CA, Decker JM, Sfakianos JN, Wu X, O’Brien WA, Ratner L, Kappes JC, Shaw GM, Hunter E. Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J Virol. 2000;74:8358–8367. doi: 10.1128/jvi.74.18.8358-8367.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorfman T, Bukovsky A, Öhagen Å, Höglund S, Göttlinger HG. Functional domains of the capsid protein of human immunodeficiency virus type 1. J Virol. 1994;68:8180–8187. doi: 10.1128/jvi.68.12.8180-8187.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich LS, Agresta BE, Carter CA. Assembly of recombinant human immunodeficiency virus type 1 capsid protein in vitro. J Virol. 1992;66:4874–4883. doi: 10.1128/jvi.66.8.4874-4883.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzon T, Leschonsky B, Bieler K, Paulus C, Schröder J, Wolf H, Wagner R. Proline residues in the HIV-1 NH2-terminal capsid domain: structure determinants for proper core assembly and subsequent steps of early replication. Virology. 2000;268:294–307. doi: 10.1006/viro.1999.0178. [DOI] [PubMed] [Google Scholar]
- Forshey BM, von Schwedler U, Sundquist WI, Aiken C. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J Virol. 2002;76:5667–5677. doi: 10.1128/JVI.76.11.5667-5677.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forshey BM, Aiken C. Disassembly of human immunodeficiency virus type 1 cores in vitro reveals association of Nef with the subviral ribonucleoprotein complex. J Virol. 2003;77:4409–4414. doi: 10.1128/JVI.77.7.4409-4414.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed EO, Martin MA. Evidence for a functional interaction between the V1/V2 and C4 domains of human immunodeficiency virus type 1 envelope glycoprotein gp120. J Virol. 1994;68:2503–2512. doi: 10.1128/jvi.68.4.2503-2512.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed EO, Englund G, Martin MA. Role of the basic domain of human immunodeficiency virus type 1 matrix in macrophage infection. J Virol. 1995;69:3949–3954. doi: 10.1128/jvi.69.6.3949-3954.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed EO. HIV-1 Gag proteins: diverse functions in the virus life cycle. Virology. 1998;251:1–15. doi: 10.1006/viro.1998.9398. [DOI] [PubMed] [Google Scholar]
- Furuta RA, Shimano R, Ogasawara T, Inubushi R, Amano K, Akari H, Hatanaka M, Kawamura M, Adachi A. HIV-1 capsid mutants inhibit the replication of wild-type virus at both early and late infection phases. FEBS Letters. 1997;415:231–234. doi: 10.1016/s0014-5793(97)01132-0. [DOI] [PubMed] [Google Scholar]
- Gamble TR, Yoo S, Vajdos FF, von Schwedler UK, Worthylake DK, Wang H, McCutcheon JP, Sundquist WI, Hill CP. Structure of the carboxyl-terminal dimerization domain of the HIV-1 capsid protein. Science. 1997;278:849–853. doi: 10.1126/science.278.5339.849. [DOI] [PubMed] [Google Scholar]
- Ganser BK, Li S, Klishko VY, Finch JT, Sundquist WI. Assembly and analysis of conical models for the HIV-1 core. Science. 1999;283:80–83. doi: 10.1126/science.283.5398.80. [DOI] [PubMed] [Google Scholar]
- Ganser BK, Cheng A, Sundquist WI, Yeager M. Three-dimentional structure of the M-MuLV CA protein on a lipid monolayer: a general model for retroviral capsid assembly. EMBO J. 2003;22:2886–2892. doi: 10.1093/emboj/cdg276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganser-Pornillos BK, von Schwedler UK, Stray KM, Aiken C, Sundquist WI. Assembly properties of the human immunodeficiency virus type 1 CA protein. J Virol. 2004;78:2545–2552. doi: 10.1128/JVI.78.5.2545-2552.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitti RK, Lee BM, Walker J, Summers MF, Yoo S, Sundquist WI. Structure of the amino-terminal core domain of the HIV-1 capsid protein. Science. 1996;273:231–235. doi: 10.1126/science.273.5272.231. [DOI] [PubMed] [Google Scholar]
- Gross I, Hohenburg H, Kräusslich H-G. In vitro assembly properties of purified bacterially expressed capsid proteins of human immunodeficiency virus. Eur J Biochem. 1997;249:592–600. doi: 10.1111/j.1432-1033.1997.t01-1-00592.x. [DOI] [PubMed] [Google Scholar]
- Gross I, Hohenberg H, Huckhagel C, Kräusslich H-G. N-terminal extension of human immunodeficiency virus capsid protein converts the in vitro assembly phenotype from tubular to spherical particles. J Virol. 1998;72:4798–4810. doi: 10.1128/jvi.72.6.4798-4810.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross I, Hohenberg H, Wilk T, Wiegers K, Grättinger M, Müller B, Fuller S, Kräusslich H-G. A conformational switch controlling HIV-1 morphogenesis. EMBO J. 2000;19:103–113. doi: 10.1093/emboj/19.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Roy BB, Hu J, Roldan A, Wainberg MA, Liang C. The R362A mutation at the C-terminus of CA inhibits packaging of human immunodeficiency virus type 1 RNA. Virology. 2005;343:190–200. doi: 10.1016/j.virol.2005.08.031. [DOI] [PubMed] [Google Scholar]
- Huseby D, Barklis RL, Alfadhli A, Barklis E. Assembly of human immunodeficiency virus precursor Gag proteins. J Biol Chem. 2005;280:17664–17670. doi: 10.1074/jbc.M412325200. [DOI] [PubMed] [Google Scholar]
- Khan MA, Aberham C, Kao S, Akari H, Gorelick R, Bour S, Strebel K. Human immunodeficiency virus type 1 Vif protein is packaged into the nucleoprotein complex through an interaction with viral genomic RNA. J Virol. 2001;75:7252–7265. doi: 10.1128/JVI.75.16.7252-7265.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klutch M, Woerner AM, Marcus-Sekura CJ, Levin JG. Generation of HIV-1/HIV-2 cross-reactive peptide antisera by small sequence changes in HIV-1 reverse transcriptase and integrase immunizing peptides. J Biomed Sci. 1998;5:192–202. doi: 10.1007/BF02253469. [DOI] [PubMed] [Google Scholar]
- Lanman J, Lam TT, Barnes S, Sakalian M, Emmett MR, Marshall AG, Prevelige PE., Jr Identification of novel interactions in HIV-1 capsid protein assembly by high-resolution mass spectrometry. J Mol Biol. 2003;325:759–772. doi: 10.1016/s0022-2836(02)01245-7. [DOI] [PubMed] [Google Scholar]
- Lanman J, Lam TT, Emmett MR, Marshall AG, Sakalian M, Prevelige PE., Jr Key interactions in HIV-1 maturation identified by hydrogen-deuterium exchange. Nat Struct Mol Biol. 2004;11:676–677. doi: 10.1038/nsmb790. [DOI] [PubMed] [Google Scholar]
- Lanman J, Prevelige PE., Jr Kinetic and mass spectrometry-based investigation of human immunodeficiency virus type 1 assembly and maturation. Adv Virus Res. 2005;64:285–309. doi: 10.1016/S0065-3527(05)64009-2. [DOI] [PubMed] [Google Scholar]
- Li S, Hill CP, Sundquist WI, Finch JT. Image reconstructions of helical assemblies of the HIV-1 CA protein. Nature. 2000;407:409–413. doi: 10.1038/35030177. [DOI] [PubMed] [Google Scholar]
- Liang C, Hu J, Russell RS, Roldan A, Kleiman L, Wainberg MA. Characterization of a putative α-helix across the capsid-SP1 boundary that is critical for the multimerization of human immunodeficiency virus type 1 gag. J Virol. 2002;76:11729–11737. doi: 10.1128/JVI.76.22.11729-11737.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C, Hu J, Whitney JB, Kleiman L, Wainberg MA. A structurally disordered region at the C terminus of capsid plays essential roles in multimerization and membrane binding of the Gag protein of human immunodeficiency virus type 1. J Virol. 2003;77:1772–1783. doi: 10.1128/JVI.77.3.1772-1783.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammano F, Öhagen Å, Höglund S, Göttlinger HG. Role of the major homology region of human immunodeficiency virus type 1 in virion morphogenesis. J Virol. 1994;68:4927–4936. doi: 10.1128/jvi.68.8.4927-4936.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayo K, Huseby D, McDermott J, Arvidson B, Finlay L, Barklis E. Retrovirus capsid protein assembly arrangments. J Mol Biol. 2003;325:225–237. doi: 10.1016/s0022-2836(02)01176-2. [DOI] [PubMed] [Google Scholar]
- Melamed D, Mark-Danieli M, Kenan-Eichler M, Kraus O, Castiel A, Laham N, Pupko T, Glaser F, Ben-Tal N, Bacharach E. The conserved carboxy terminus of the capsid domain of human immunodeficiency virus type 1 Gag protein is important for virion assembly and release. J Virol. 2004;78:9675–9688. doi: 10.1128/JVI.78.18.9675-9688.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momany C, Kovari LC, Prongay AJ, Keller W, Gitti RK, Lee BM, Gorbalenya AE, Tong L, McClure J, Ehrlich LS, Summers MF, Carter C, Rossmann MG. Crystal structure of dimeric HIV-1 capsid protein. Nat Struct Biol. 1996;3:763–770. doi: 10.1038/nsb0996-763. [DOI] [PubMed] [Google Scholar]
- Mortuza GB, Haire LF, Stevens A, Smerdon SJ, Stoye JP, Taylor IA. High-resolution structure of a retroviral capsid hexameric amino-terminal domain. Nature. 2004;431:481–485. doi: 10.1038/nature02915. [DOI] [PubMed] [Google Scholar]
- Murakami T, Freed EO. The long cytopasmic tail of gp41 is required in a cell type-dependent manner for HIV-1 envelope glycoprotein incorporation into virions. Proc Natl Acad Sci USA. 2000;97:343–348. doi: 10.1073/pnas.97.1.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt EJ, Wehrly K, Duhmann SE, Chesebro B, Kabat D. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J Virol. 1998;72:2855–2864. doi: 10.1128/jvi.72.4.2855-2864.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reicin AS, Ohagen A, Yin L, Hoglund S, Goff SP. The role of Gag in human immunodeficiency virus type 1 virion morphogenesis and early steps of the viral life cycle. J Virol. 1996;70:8645–8652. doi: 10.1128/jvi.70.12.8645-8652.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos JW, Maughan MF, Liao Z, Hildreth JEK, Clements JE. LuSIV cells: a reporter cell line for the detection and quantitation of a single cycle of HIV and SIV replication. Virology. 2000;273:307–315. doi: 10.1006/viro.2000.0431. [DOI] [PubMed] [Google Scholar]
- Rulli SJ, Jr, Muriaux D, Nagashima K, Mirro J, Oshima M, Baumann JG, Rein A. Mutant murine leukemia virus Gag proteins lacking proline at the N-terminus of the capsid domain block infectivity in virions containing wild-type Gag. Virology. 2006;347:364–371. doi: 10.1016/j.virol.2005.12.012. [DOI] [PubMed] [Google Scholar]
- Scholz I, Arvidson B, Huseby D, Barklis E. Virus particle core defects caused by mutations in the human immunodeficiency virus capsid N-terminal domain. J Virol. 2005;79:1470–1479. doi: 10.1128/JVI.79.3.1470-1479.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimano R, Iida S, Fukumori T, Yamamoto Y, Kawamura M, Furuta RA, Adachi A. Inhibition of HIV replication by capsid mutant C6b. Biochem Biophy Res Comm. 1998;242:313–316. doi: 10.1006/bbrc.1997.7963. [DOI] [PubMed] [Google Scholar]
- Swanstrom R, Wills JW. Synthesis, assembly, and processing of viral proteins. In: Coffin JM, Hughes SH, Varmus HE, editors. Retroviruses. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, N. Y.: 1997. pp. 263–334. [PubMed] [Google Scholar]
- Tang C, Ndassa Y, Summers MF. Structure of the N-terminal 283-residue fragment of the immature HIV-1 Gag polyprotein. Nat Struct Biol. 2002;9:537–543. doi: 10.1038/nsb806. [DOI] [PubMed] [Google Scholar]
- Tang C, Loeliger E, Kinde I, Kyere S, Mayo K, Barklis E, Sun Y, Huang M, Summers MF. Antiviral inhibition of the HIV-1 capsid protein. J Mol Biol. 2003a;327:1013–1020. doi: 10.1016/s0022-2836(03)00289-4. [DOI] [PubMed] [Google Scholar]
- Tang S, Collier AJ, Elliott RM. Alterations to both the primary and predicted secondary structure of stem-loop IIIc of the hepatitis C virus 1b 5′ untranslated region (5′ UTR) lead to mutants severely defective in translation which cannot be complemented in trans by the wild-type 5′UTR sequence. J Virol. 1999;73:2359–2364. doi: 10.1128/jvi.73.3.2359-2364.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S, Murakami T, Agresta BE, Campbell S, Freed EO, Levin JG. Human immunodeficiency virus type 1 N-terminal capsid mutants that exhibit aberrant core morphology and are blocked in initiation of reverse transcription in infected cells. J Virol. 2001;75:9357–9366. doi: 10.1128/JVI.75.19.9357-9366.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S, Murakami T, Cheng N, Steven AC, Freed EO, Levin JG. Human immunodeficiency virus type 1 N-terminal capsid mutants containing cores with abnormally high levels of capsid protein and virtually no reverse transcriptase. J Virol. 2003b;77:12592–12602. doi: 10.1128/JVI.77.23.12592-12602.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trono D, Feinberg MB, Baltimore D. HIV-1 Gag mutants can dominantly interfere with the replication of the wild-type virus. Cell. 1989;59:113–120. doi: 10.1016/0092-8674(89)90874-x. [DOI] [PubMed] [Google Scholar]
- Vogt VM. Retroviral virions and genomes. In: Coffin JM, Hughes SH, Varmus HE, editors. Retroviruses. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, N. Y.: 1997. pp. 27–69. [PubMed] [Google Scholar]
- Vogt VM, Simon MN. Mass determination of Rous sarcoma virus virions by scanning transmission electron microscopy. J Virol. 1999;73:7050–7055. doi: 10.1128/jvi.73.8.7050-7055.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Schwedler U, Stemmler TL, Klishko VY, Li S, Albertine KH, Davis DR, Sundquist WI. Proteolytic refolding of the HIV-1 capsid protein amino-terminus facilitates viral core assembly. EMBO J. 1998;17:1555–1568. doi: 10.1093/emboj/17.6.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Schwedler UK, Stray KM, Garrus JE, Sundquist WI. Functional surfaces of the human immunodeciency virus type 1 capsid protein. J Virol. 2003;77:5439–5450. doi: 10.1128/JVI.77.9.5439-5450.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, Kappes JC. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob Agents Chemother. 2002;46:1896–1905. doi: 10.1128/AAC.46.6.1896-1905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegers K, Rutter G, Kottler H, Tessmer U, Hohenberg H, Kräusslich H-G. Sequential steps in human immunodeficiency virus particle maturation revealed by alterations of individual Gag polyprotein cleavage sites. J Virol. 1998;72:2846–2854. doi: 10.1128/jvi.72.4.2846-2854.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills JW, Craven RC. Form, function, and use of retroviral Gag proteins. AIDS. 1991;5:639–654. doi: 10.1097/00002030-199106000-00002. [DOI] [PubMed] [Google Scholar]
- Zack JA, Cann AJ, Lugo JP, Chen IS. HIV-1 production from infected peripheral blood T cells after HTLV-1 induced mitogenic stimulation. Science. 1988;240:1026–1029. doi: 10.1126/science.2835813. [DOI] [PubMed] [Google Scholar]