

Abstract
A series of analogues of the dopamine D2 receptor antagonist L741,626 were synthesized and evaluated for binding and function at D2 family receptor subtypes. Several analogues showed comparable binding profiles to the parent ligand, however, in general, chemical modification served to reduce D2 binding affinity and selectivity.
Keywords: Dopamine D2 receptor antagonists
The neurotransmitter dopamine has been associated with fine movement coordination, cognition, emotion, affect, memory, and the regulation of prolactin secretion by the pituitary reward system.1 Alterations in dopaminergic function are not only involved in the pathogenesis of Parkinson’s disease2 and schizophrenia,3 but also occur as a consequence of acute and chronic abuse of pyschostimulants.4 Therefore, the D1-like (D1 and D5) and the D2-like (D2, D3, and D4) dopamine receptor families have been targets for the development of treatment medications for these disorders.5-8
The majority of antipsychotic medications are nonselective dopamine D2 receptor antagonists that frequently produce undesirable extrapyramidal side effects upon chronic exposure. As such, to date the discovery of highly selective dopamine D2 antagonists has been elusive in part because the therapeutic value of such agents has been perceived as minimal due to the association of this receptor exclusively with the unwanted side effects of nonselective D2 receptor antagonists. Furthermore the high degree of amino acid homology within the binding sites of the D2-like receptors provides a formidable challenge to discovering highly selective and potent D2 (or D3) antagonists.7, 9 Nevertheless, the discovery of D2 receptor selective antagonists and partial agonists would provide important pharmacological tools to determine the role of the D2-like receptor subtypes in 1) the mechanism of action of antipsychotic agents, 2) the etiology of drug-induced extrapyramidal side effects, and 3) the contribution of the D2 and D3 receptor subtypes in the CNS reward system. In addition, such D2 receptor selective compounds would also be a starting point for the development of radioligands.
A decade ago, a D2-selective antagonist, L741,626 (1) was reported amongst a series of D4-selective agents to bind with ∼40-fold higher affinity to D2 over D3 receptors and is currently being used as a D2-selective antagonist in animal models of drug abuse.10 During the course of our investigation, attempts to define the D2 receptor pharmacophore have led to the identification of D2-related SAR and several novel analogues (e.g. 2a and 2b) of L741,626 that show D2 selectivity over D3 and D4 receptors have been reported.11, 12 In this report, 2b demonstrated a >100-fold D3/D2 selectivity that appeared to be related to the addition of a 5-OCH3 onto the indolyl moiety of 2a. This study and many others describing D3-selective ligands demonstrate the importance and potential differences in pendant aryl ring substitution that may provide separation between D2 and D3 receptor recognition. Thus our approach to further define the D2 pharmacophore and design D2-selective and bioavailable ligands was to 1) examine the effects of various substituents on the pendant piperidinol-phenyl ring, 2) retain the sp3 piperidine C-4, as determined by others to retain D2 affinity but to investigate substitutions of the OH that are either isosteric or would not be prone to elimination in vivo, 3) examine heteroatom replacements in and substitutions on the indole ring and 4) combine some of these features for comparison with compounds recently reported.10, 11
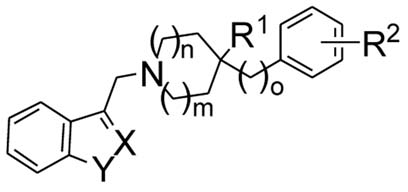
A general synthetic strategy for analogues 5-32 of L741,626 is depicted in Scheme 1. Phenyl-piperidines 3 were reacted with gramine (4a, X-Y = CH-NH), 2-methyl-gramine (4b, X-Y = CMe-NH), or (1H-indazol-3-ylmethyl)-dimethyl-amine (4c, X-Y = N-NH)13 in refluxing pyridine overnight to give products 5-32. 3-Chloromethyl-benzofuran14 was converted to analogues 33-35 in the presence of sodium bicarbonate in refluxing acetonitrile. Regioselective alkylation of the indole nitrogen (36, 37) was achieved by reaction of the dianion of L741,626 (generated by treatment with 2 equiv. sodium hydride) with the methyl iodide or hexyl bromide in THF. The 2-indolyl analog 38 of L741,626 was prepared from the corresponding amide of 2-indole-carboxylic acid by reduction with lithium aluminum hydride.
Scheme 1.

General synthesis of the derivatives 5-3218
Thirty-four novel ligands were evaluated in competition binding assays in HEK 293 cells transfected with D2L, D3 or D4 human dopamine receptors15 using [125I]IABN16 as the radiolabeled ligand. In addition, intrinsic activities for selected compounds were determined in a functional assay using stimulation (agonist) or inhibition of quinpirole stimulation (antagonist) of mitogenesis in human dopamine D2 or D3 receptors transfected into Chinese hamster ovary (CHO) cells (Table 1). All of the compounds evaluated were antagonists in these functional tests. The parent molecule L741,626 was prepared by literature methods10 (Ki (D2) = 11.2 nM) and displayed a D3/D2 and D4/D2 selectivity ratio of 15-fold and 136-fold, respectively. In the functional assay L741,626 was a potent antagonist (EC50 (D2) = 4.46 nM) with some D2 selectivity (EC50 (D3) = 90.4 nM).
Table 1.
Binding and functional data for D2 compounds.
 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compd. | m | n | o | X-Y | R1 | R2 | D2a | D3a | D4a | D3/D2 | D4/D2 | D2 functionb | D3 functionb |
| Ki [nM] ± SEM | EC50 [nM] ± SEM | ||||||||||||
| 5 | 1 | 1 | 0 | CH-NH | OH | 2′-Cl | 382 ±33 | 1860 ± 340 | 2820 ± 64 | 5 | 7 | 586 ± 210 | ND |
| 6 | 1 | 1 | 0 | CH-NH | OH | 2′,3′-Cl | 26.1 ± 2.0 | 273 ± 42 | 1550 ± 190 | 10 | 59 | 34.3 ± 6.8 | 232 ±7.7 |
| 7 | 1 | 1 | 0 | CH-NH | OH | 3′-Cl | 129 ± 22 | 450 ± 80 | 3540 ± 660 | 3 | 27 | 83.4 ± 10 | ND |
| 8 | 1 | 1 | 0 | CH-NH | OH | 3′,4′-Cl | 6.9 ± 1.0 | 111 ± 17 | 1100 ± 210 | 16 | 159 | 15.3 ± 0.8 | 41.5 ± 9.1 |
| 9 | 1 | 1 | 0 | CH-NH | OH | 3′-CF3 | 106 ± 15 | 570 ± 44 | 3750 ± 140 | 5 | 35 | 69.1 ± 23 | ND |
| 10 | 1 | 1 | 0 | CH-NH | OH | 4′-F | 127 ± 7.2 | 490 ± 74 | 6090 ± 580 | 4 | 48 | ND | ND |
| 11 | 1 | 1 | 0 | CH-NH | OH | 4′-OH | 5360 ± 1100 | 16800 ± 6500 | 1360 ± 270 | 3 | 0.3 | ND | ND |
| 12 | 1 | 1 | 0 | CH-NH | OH | 4′-OMe | 177 ± 61 | 2130 ± 300 | 679 ± 60 | 12 | 4 | ND | ND |
| 13 | 1 | 1 | 0 | CH-NH | OH | 4′-OHex | 159 ± 60 | 1950 ± 350 | 1490 ± 150 | 12 | 9 | 216 ± 1.7 | ND |
| 14 | 1 | 1 | 0 | CH-NH | OH | 4′-Ph | 98.4 ± 19 | 467 ± 38 | 129 ± 26 | 5 | 1 | ND | ND |
| 15 | 2 | 1 | 0 | CH-NH | OH | 4′-Cl | 201 ± 21 | 2100 ± 490 | 3870 ± 770 | 10 | 19 | 260 ± 91 | ND |
| 16 | 1 | 1 | 1 | CH-NH | OH | 4′-Cl | 2890 ± 560 | 5480 ± 2900 | 3930 ± 1200 | 2 | 1 | ND | ND |
| 17 | 2 | 0 | 0 | CH-NH | OH | 4′-Cl | 4265 ± 2280 | 4270 ± 400 | 2440 ± 1400 | 1 | 1 | ND | ND |
| 18 | 1 | 1 | 0 | CH-NH | Me | 4′-Cl | 992 ± 192 | 2100 ± 150 | 2750 ± 140 | 2 | 3 | 314 ± 4.2 | 347±7.4 |
| 19 | 1 | 1 | 0 | CH-NH | NH2 | 4′-Cl | 184 ± 40 | 382 ± 55 | 2293 ± 545 | 2 | 12 | ND | ND |
| 20 | 1 | 1 | 0 | CH-NH | NHAc | 4′-Cl | 83.1 ± 47 | 193 ± 80 | 348 ± 39 | 2 | 4 | 24.8 ± 7.6 | ND |
| 21 | 1 | 1 | 0 | CH-NH | F | 4′-Cl | 120 ± 34 | 211 ± 67 | 365 ± 63 | 2 | 3 | ND | ND |
| 22 | 1 | 1 | 0 | CH-NH | CN | 4′-Cl | 199 ± 24 | 684 ± 180 | 2270 ± 350 | 3 | 11 | 225 ± 80 | ND |
| 23 | 1 | 1 | 0 | CH-NH | COOMe | 4′-Cl | 60.0 ± 9.5 | 544 ± 128 | 3930 ± 2500 | 9 | 66 | 76.7 ± 21 | ND |
| 24 | 1 | 1 | 0 | CH-NH | COOEt | 4′-Cl | 58.8 ± 20 | 724 ± 120 | 3520 ± 1320 | 12 | 60 | ND | ND |
| 25 | 1 | 1 | 0 | CH-NH | CH2NH2 | 4′-Cl | >10000 | >5000 | >20000 | ND | ND | ||
| 26 | 1 | 1 | 0 | CH-NH | CH2NHAc | 4′-Cl | 2000 ± 640 | 12500 ±1300 | 4450 ± 750 | 6 | 2 | ND | ND |
| 27 | 1 | 1 | 0 | CH-NH | CH2OH | 4′-Cl | 2370 ± 560 | 106 ±19 | 1940 ± 250 | 0.04 | 1 | ND | ND |
| 28 | 1 | 1 | 0 | CH-NH | OMe | 4′-Cl | 68.1 ± 22 | 262 ± 73 | 34.1 ± 6.0 | 4 | 1 | 34.7 ± 2.9 | 248 ± 84 |
| 29 | 1 | 1 | 0 | CH-NH | OAllyl | 4′-Cl | 165 ± 91 | 718 ± 35 | 1190 ± 360 | 4 | 7 | ND | ND |
| 30 | 1 | 1 | 0 | CMe-NH | OH | 4′-Cl | 15.4 ± 14 | 44.1 ± 11 | 2990 ± 180 | 3 | 194 | 2.13 ± 0.3 | 20.2 ± 4.2 |
| 31 | 1 | 1 | 0 | N-NH | OH | 3′,4′-Cl | 438 ± 46 | 156 ± 22 | 677 ± 148 | 0.4 | 2 | ND | ND |
| 32 | 1 | 1 | 0 | N-NH | OH | 4′-Cl | 548 ± 80 | 440 ± 88 | 2146 ± 616 | 1 | 4 | ND | ND |
| 33 | 1 | 1 | 0 | CH-O | OH | 3′,4′-Cl | 21.0 ± 4 | 28.0 ± 6 | 167 ± 35 | 1 | 8 | ND | ND |
| 34 | 1 | 1 | 0 | CH-O | OH | 4′-Cl | 21.0 ± 4 | 53.0 ± 12.5 | 404 ± 24 | 3 | 19 | ND | ND |
| 35 | 1 | 1 | 0 | CH-O | OH | 4′-SMe | 54.0 ±13 | 139 ± 7.9 | 153 ± 35 | 3 | 3 | ND | ND |
| 36 | 1 | 1 | 0 | CH-NMe | OH | 4′-Cl | 29.6 ± 6.2 | 238 ± 100 | 5140 ± 3330 | 8 | 174 | 20.0 ± 4.5 | 98.6 ± 29 |
| 37 | 1 | 1 | 0 | CH-NHex | OH | 4′-Cl | 188 ± 27 | 488 ± 280 | 8890 ± 5600 | 3 | 47 | 166.0 ± 35 | 661 ± 56 |
| 38 | 1 | 1 | 0 | n/ac | OH | 4′-Cl | >10000 | ND | >20000 | ND | ND | ||
| 2a11 | 1 | 1 | 0 | CH-NH | OH | 4′-SMe | 23.9 ± 5.5 | 638 ± 159 | 319 ± 58 | 27 | 13 | ND | ND |
| 2b11 | 1 | 1 | 0 | CH-NHd | OH | 4′-SMe | 5.5 ± 0.1 | 580 ± 92 | 567 ± 140 | 105 | 103 | ND | ND |
| 1, L741,62610 | 1 | 1 | 0 | CH-NH | OH | 4′-Cl | 11.2 ± 0.8 | 163 ± 32 | 1520 ± 280 | 15 | 136 | 4.46 ± 0.9 | 90.4 ± 15 |
Inhibtion of binding assay in HEK 293 cells transfected with either hD2L, hD3, or hD4 dopamine receptors, radioligand 125I-IABN.15, 16
Functional assays using inhibition of quinpirole stimulation in hD2 and hD3 receptors transfected into CHO cells. These data were obtained through the service of CTDP, Division of Treatment Research and Development, NIDA, using a contract (N01DA-1-8816) protocol. ND = not determined.
1-((1H-indol-2-yl)methyl)-4-(4′-chlorophenyl)piperidin-4-ol.
1-((5-Methoxy-1H-indol-3-yl)methyl)-4-(4′-(methylthio)phenyl)piperidin-4-ol
In the aryl ring substituted analogues (5-14) the chloro-substituent and its position on the phenyl-piperidinol moiety was pivotal fo r high affinity binding at the dopamine D2 receptor and for selectivities over the D3 and D4 receptors. Only the 3′,4′-dichloro-substituted derivative 8 demonstrated the same binding and selectivity profile as the parent compound L741,626, however in the functional assay 8 (EC50 (D2) = 15.3 nM) was 3-fold less potent at D2 than the parent molecule. Variation of the substituent of the phenyl-piperidinol moiety (9-14) resulted in decreased dopamine D2 receptor binding affinities. A 4′-OH group (11, Ki (D2) = 5360 nM) was particularly poorly tolerated, whereas the 4′-OCH3 analogue 12 (Ki (D2) = 177 nM) regained some affinity at the dopamine D2 receptor. Interestingly, the 4′-SCH3 derivative 2a had a 7-fold higher affinity at the dopamine D2 receptor than the 4′-OCH3 analog 12, indicating that a more polarizable 3rd row element may be required to achieve high dopamine D2 receptor binding affinity.
Retention of the C-4 piperidine sp3 carbon was previously reported to be preferred by the D2 receptor12 and replacement of the 4-piperidinol function with other groups was explored (15-29). In general, none of these analogues showed higher affinity for D2 than the parent compound. Introduction of a methylene spacer between the C-4 and OH (27) or other functional groups (25, 26) resulted in very low D2 affinities (< 2 μM). Interestingly, the derivative 27 showed higher affinity for the dopamine D3 than D2 receptor, D2/D3 = 22 and D4/D3 = 18. Despite lower D2 affinities than L741,626, both the 4-carboxylic methyl ester (23) and its ethyl analogue (24) demonstrated similar D3/D2 selectivity. Isosteric replacement of the 4-OH group with a 4-methyl group (18), 4-amino group (19) or a 4-fluoro group (21) resulted in a loss of affinity at the dopamine D2 receptor up to 88-fold. All of these derivatives were nonselective over the dopamine D3 receptor, only the 4-amino derivative 19 was to some extent selective over D4 (D4/D2 = 12). The 4-amino acetyl analogue 20 (Ki (D2) = 83.1 nM) displayed a slightly higher binding affinity at the dopamine D2 receptor than the 4-amino derivative 19 (Ki (D2) = 184 nM) but the D4/D2 selectivity was 3-fold lower.
Haloperidol and its homopiperidine analogue have been reported to be equipotent at the dopamine D2 receptor,17 however t he corresponding azepine analogue of L741,626, 15 showed an 18-fold lower binding affinity at the dopamine D2 receptor and reduced D4/D2 selectivity (7-fold). In addition, the replacement of the 4′-chloro-phenyl ring of L741,626 by a 4′-chloro-benzyl group (16) was not tolerated (Ki (D2) = 2800 nM). The 3-phenyl-piperidol analogue 17 was inactive at dopamine D2-like receptor subtypes.
Substitution or heteroatom replacement in the indole moiety was also explored (30-38). A methyl group in the 2-position of the indole moiety (30) was well tolerated at the dopamine D2 receptor (Ki (D2) = 15.4 nM, EC50 (D2) = 2.13 nM), however this modification resulted in a 5-fold reduction of D3/D2 selectivity, while D4/D2 selectivity was unaffected. In contrast, moving the piperidinol moiety to the 2-position of the indole was not tolerated. Binding affinities of > 10,000 nM were determined at both D2 and D4 dopamine receptor subtypes for the 1H-indol-2-yl analogue 38, and no further testing was performed. Both the indazole derivatives 31 and 32 had low binding affinities at D2 receptors (Ki (D2) = 438 and 548 nM, respectively). Notably the 3′,4′-dichloro analogue 31 was nonselective over the dopamine D2-like subtypes. The benzofurans 33-35 showed moderate binding affinity at the dopamine D2 receptor but the D3/D2 and D4/D2 selectivity was low (< 20-fold) even when substitution on the piperidinol phenyl ring was made optimal for D2 (33 and 35 compared to 8 and 2a, 2b, respectively).
In summary, we have used the D2 selective antagonist L741,626 as a lead compound to further elucidate D2 receptor SAR in order to identify novel D2 selective agents that can be used as research tools to more precisely define the physiological role of the dopamine D2 receptor in psychiatric and neurodegenerative disorders. However, the high degree of amino acid homology between the D2 and D3 dopamine receptor subtypes has thus far precluded our discovery of compounds that bind with a high degree of selectivity at the D2 receptor. Nevertheless, clues to D2/D3 separability continue to be revealed and some of these compounds may still prove useful for in vivo investigation as they have comparable profiles to L741,626.
Figure 1.

Acknowledgement
The research reported herein was supported by funds from the NIDA Intramural Research Program and 1 DA 13584. P.G. was supported by a National Institutes of Health (NIH) Visiting Fellowship. S.L.J.H. was supported by a NIH summer IRTA fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Physiol. Rev. 1998;78:189. doi: 10.1152/physrev.1998.78.1.189. [DOI] [PubMed] [Google Scholar]
- 2.Lotharius J, Brundin P. Nature Rev. Neurosci. 2002;3:932. doi: 10.1038/nrn983. [DOI] [PubMed] [Google Scholar]
- 3.Kapur S, Mamo D. Prog. Neuropsychopharm. Bio. Psych. 2003;27:1081. doi: 10.1016/j.pnpbp.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 4.Volkow ND, Fowler JS, Wang GJ, Swanson JM. Mol. Psychiatry. 2004;9:557. doi: 10.1038/sj.mp.4001507. [DOI] [PubMed] [Google Scholar]
- 5.Platt DM, Rowlett JK, Spealman RD. Psychopharmacology. 2002;163:265. doi: 10.1007/s00213-002-1137-8. [DOI] [PubMed] [Google Scholar]
- 6.Seeman P. Expert Opinion On Therapeutic Targets. 2006;10:515. doi: 10.1517/14728222.10.4.515. [DOI] [PubMed] [Google Scholar]
- 7.Newman AH, Grundt P, Nader MA. J. Med. Chem. 2005;48:3663. doi: 10.1021/jm040190e. [DOI] [PubMed] [Google Scholar]
- 8.Zhang A, Kan Y, Li FY. Expert Opinion On Therapeutic Patents. 2006;16:587. doi: 10.1517/13543776.16.8.1183. [DOI] [PubMed] [Google Scholar]
- 9.Luedtke RR, Mach RH. Curr. Pharm. Design. 2003;9:643. doi: 10.2174/1381612033391199. [DOI] [PubMed] [Google Scholar]
- 10.Kulagowski JJ, Broughton HB, Curtis NR, Mawer IM, Ridgill MP, Baker R, Emms F, Freedman SB, Marwood R, Patel S, Ragan CI, Leeson PD. J. Med. Chem. 1996;39:1941. doi: 10.1021/jm9600712. [DOI] [PubMed] [Google Scholar]
- 11.Vangveravong S, McElveen E, Taylor M, Xu JB, Tu Z, Luedtke RR, Mach RH. Bioorg. Med. Chem. 2006;14:815. doi: 10.1016/j.bmc.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 12.Sikazwe DMN, Li SM, Mardenborough L, Cody V, Roth BL, Ablordeppey SY. Bioorg. Med. Chem. Lett. 2004;14:5739. doi: 10.1016/j.bmcl.2004.09.046. [DOI] [PubMed] [Google Scholar]
- 13.Snyder HR, Thompson CB, Hinman RL. J. Am. Chem. Soc. 1952;74:2009. [Google Scholar]
- 14.Shafiee A, Mohamadpour M. J. Heterocycl. Chem. 1978;15:481. [Google Scholar]
- 15.Grundt P, Carlson EE, Cao J, Bennett CJ, McElveen E, Taylor M, Luedtke RR, Newman AH. J. Med. Chem. 2005;48:839. doi: 10.1021/jm049465g. [DOI] [PubMed] [Google Scholar]
- 16.Luedtke RR, Freeman RA, Boundy VA, Martin MV, Huang YS, Mach RH. Synapse. 2000;38:438. doi: 10.1002/1098-2396(20001215)38:4<438::AID-SYN9>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 17.Lyles-Eggleston M, Altundas R, Xia J, Sikazwe DMN, Fan P, Yang Q, Li S, Zhang W, Zhu X, Schmidt AW, Vanase-Frawley M, Shrihkande A, Villalobos A, Borne RF, Ablordeppey SY. J. Med. Chem. 2004;47:497. doi: 10.1021/jm0301033. [DOI] [PubMed] [Google Scholar]
- 18.Notes: The 4-substituted piperdines required for compounds 18-29 were synthesized in adaptation of literature methods:Harriman GCB, Shao JX, Luly JR. Tetrahedron Lett. 2000;41:8853.Bernstein P, Cacciola J, Dedinas R, Shen L, Warwick P. WO 2004022539 Chem. Abstr. 2004;140:270744.Ghosh S, Patane MA, Carson KG, Chi I-CS, Ye Q, Elder AM, Jenkins TJ. WO 2003037271 Chem. Abstr. 2003;138:368921.Luly JR, Nakasato Y, Ohshima E, Harriman GCB, Carson KG, Ghosh S, Elder AM, Mattia KM. US 2005070549 Chem. Abstr. 2005;142:355256.; All final compounds were characterized by NMR. Microanalysis data were determined to ± 0.4 % of the calculated values. Representative Spectral Data: 30: 1H NMR (400 MHz, CDCl3): δ 1.67 (d, J 12.1, 2H), 2.06 (td, J 12.9, 4.1, 2H), 2.35 (s, 1H), 2.39 (s, 3H), 2.45 (td, J 10.6, 1.8, 2H), 2.85 (d, J 11.3, 2H), 3.71 (s, 2H), 7.07-7.19 (m, 2H), 7.24-7.30 (m, 3H), 7.40 (d, J 8.6, 2H), 7.67 (d, J 6.6, 1H), 7.90 (s, 1H). 32: 1H NMR (400 MHz, CD3OD): δ 1.66 (d, J 12.1, 2H), 2.05 (m, 2H), 2.66 (m, 2H), 2.85 (d, J 11.3, 2H), 3.97 (s, 2H), 7.12 (m, 1H), 7.24 (d, J 8.6, 2H), 7.34 (m, 1H), 7.41 (d, J 8.6, 2H), 7.47 (d, J 8.2, 1H), 7.86 (d, J 7.6, 1H). 34: 1H NMR (400 MHz, CDCl3): δ 1.68 (d, J 14.1, 2H), 1.82 (s, br., 1H), 2.07 (td, J 12.9, 4.5, 2H), 2.48 (td, J 12.1, 2H), 2.83 (d, 11.0, 2H), 3.69 (d, 0.8, 2H), 7.22-7.31 (m, 4H), 7.41 (d, J 9.0, 2H), 7.55 (s, 1H). 36: 1H NMR (400 MHz, CDCl3): δ 1.71 (d, J 12.8, 2H), 2.18 (t, J 11.4, 2H), 2.56 (t, J 11.8, 2H), 2.93 (d, J 10.8, 2H), 3.78 (s, 3H), 3.84 (s, 3H), 7.10 (s, 1H), 7.14 (m, 1H), 7.25 (m, 1H), 7.29 (d, J 8.8, 2H), 7.33 (m, 1H), 7.43 (d, J 8.4, 2H), 7.72 (d, J 8.0, 1H)