

Abstract
Late-onset GM2-gangliosidosis (GM2) is composed of two related, autosomal recessive, neurodegenerative diseases, both resulting from deficiency of lysosomal, heterodimeric β-hexosaminidase A (Hex A, αβ). Pharmacological chaperones (PC) are small molecules that can stabilize the conformation of a mutant protein, allowing it to pass the quality control system of the ER. To date all successful PCs have also been competitive inhibitors. Screening for Hex A inhibitors in a library of 1040 FDA-approved compounds identified pyrimethamine (PYR) as the most potent inhibitor. Cell lines from 10 late-onset Tay-Sachs (11 α-mutations, 2 novel), and 7 Sandhoff (9 β-mutations, 4 novel) disease patients, were cultured with PYR at concentrations corresponding to therapeutic doses. Cells carrying the most common late-onset mutation, αG269S, showed significant increases in residual Hex A activity, as did all 7 of the β-mutants tested. Cells responding to PC-treatment included those carrying mutants resulting in reduced Hex heat stability and partial splice junction mutations of the inherently less stable α-subunit. PYR, which binds to the active site in domain II, was able to function as PC even to domain I β-mutants. We concluded that PYR functions as a mutation-specific PC, variably enhancing residual lysosomal Hex A levels in late-onset GM2 patient cells.
GM2 gangliosidosis (GM2, OMIM 230700), is a clinically heterogeneous inherited neurodegenerative disorder characterized by progressive deterioration of motor, cerebral and spinocerebellar function caused by deficiency of lysosomal β-hexosaminidase A. Normal human tissues contain two major β-hexosaminidase (Hex) isozymes, Hex A and Hex B. Hex A is a heterodimer made up of α and β subunits. These subunits have nearly identical three dimensional structures and similar active sites. They are encoded by two evolutionarily related genes, HEXA (15q23–q24) and HEXB (5q13), respectively. Hex B is a homodimer made up of two identical β-subunits. A third minor, unstable Hex isozyme, Hex S, is comprised of two α-subunits and is only unequivocally detectable in tissues from patients with the Sandhoff disease variant (SD, OMIM 268800) of GM2. SD results from HEXB mutations producing abnormal or deficient β-subunits. Thus SD is associated with combined deficiency of both Hex A (αβ) and Hex B (ββ) activities. On the other hand, Tay-Sachs disease variant (TSD; OMIM 272800) is caused by HEXA mutations resulting in abnormal or deficient α-subunits, which only affects Hex A levels. Mutations affecting GM2A (5q31.3–q33.1), encoding the non-catalytic GM2 activator protein (Activator), results in the third very rare AB-variant form of GM2 (OMIN 272750) (1).
In humans, only the Hex A isozyme catalyzes the removal of the β-GalNAc residue from the non-reducing terminal end of GM2 ganglioside, but it requires the Activator as a substrate-specific co-factor for the reaction (1). The synthetic substrate, 4-methylumbelliferyl-(2-acetamido-2-deoxy)- β-D-glucopyranoside (MUG), is hydrolyzed by both the α- and β-active sites and therefore, is used to measure total Hex activity. A newer, more specific synthetic substrate is 4-methylumbelliferyl-7-(6-sulfo-2-acetamido-2-deoxy)- β-D-glucopyranoside (MUGS). Its negatively charged 6-sulfate group has been shown to interact with the same positively charge binding pocket, found only in the α-active site, that binds the sialic acid residue of GM2 ganglioside (2-4). Thus, this substrate most closely mimics the natural substrate. However, MUGS hydrolysis is Activator-independent and thus, it is turned over more rapidly by Hex S (in SD samples) than by Hex A (5).
GM2 is characterized by a wide spectrum of clinical presentations. The most severe forms are the infantile or acute TSD and SD, associated with <0.5% of normal Hex A activity, resulting in rapid neurodegeneration, and culminating in death in infancy. At the other end of the spectrum are the late-onset forms, which are subdivided into juvenile or sub-acute and adult or chronic forms (6). These are usually associated with residual Hex A activities, ~1–10% of normal (7). Patients with juvenile GM2 usually present with evidence of neurodeterioration starting after one year of age, experiencing a slower rate of progression than patients with the infantile forms (8). Patients with adult-onset forms may present with spinocerebellar, psychiatric and/or peripheral neuropathies, which do not significantly decrease life expectancy in some cases (9). The rate of disease progression and severity has been found to correlate roughly with the level of residual Hex A activity. Generally, a clinical disease does not develop unless residual Hex A activity is <10% of normal (10). Thus, only a low level of residual Hex A activity is apparently needed to prevent or reverse substrate-storage in this condition.
Pharmacological chaperones (PC) are low molecular weight compounds that stabilize the native conformation of a mutant enzyme in the ER, allowing it to escape aggregation and premature degradation by the ER-associated degradation pathway (ERAD). The properly folded mutant enzyme, stabilized by the PC can then be transported to the lysosome, increasing the residual enzyme activity of the cells (11). Most PCs have also been competitive inhibitors of their target enzyme (12). Once the PC-enzyme complex reaches the lysosome, the large amounts of stored substrate(s) are believed to displace the PC and take over stabilization of the mutant enzyme (13). The PC approach has been shown to enhance the residual activity levels of five different mutant lysosomal enzymes causing chronic forms of the lysosomal storage diseases, GM2 (14), GM1 gangliosidosis (15), Fabry (16), Gaucher (11), and Morquio B diseases (17).
A major challenge to the exploitation of this phenomenon for the treatment of disease is identifying potential PCs from among the thousands of existing chemicals and drugs. High throughput screening (HTS) of small-molecule libraries has been used to identify specific enzyme inhibitors with PC-potential (18,19).
We report here the application of a small-molecule library screening approach to search for novel inhibitors of lysosomal Hex A with the potential to treat late-onset variants of GM2 by functioning as PCs. We undertook a manual screening for Hex inhibitors in a 1040-compound library of Food Drug Administration (FDA) approved drugs obtained from National Institute of Neurological Diseases and Stroke (NINDS). Pyrimethamine (PYR) was discovered to have considerable PC-potential. The evaluation of PYR as a PC was performed using fibroblast cell lines from 17 patients with juvenile and adult GM2. Ten of the 11 different HEXA mutations, and 5 of the 9 different HEXB mutations, we identified in patients and reported previously. Each of the mutant cell lines from our patients responded differently to PYR, as well as to a carbohydrate-based PC, N-acetyl glucosamine–thiazoline (NGT), which we reported previously (14). In addition, the different responses of mutants to PYR brought interesting insights into the role of the 2 domains present in each subunit (αand β) in the folding and assembly of the Hex dimers.
Experimental Procedures
Study subject
Fibroblast cell lines were obtained from 17 patients with late-onset forms of GM2. Patients were from two Genetic Metabolic centers, and informed consents and assents approved by relevant research ethic boards were obtained from each patient.
Chemical reagents and antibodies
The NINDS drug library was received from MicroSource Discovery Systems in a 96-well format; one drug per well at a concentration of 10 mM diluted in dimethylsulfoxide (DMSO), sealed under nitrogen. Plates were stored at −20°C. The following fluorogenic substrates, all purchased from Sigma, 4-methylumbelliferyl-β-D-galactopyranoside (MUβGal) and 4-methylumbelliferyl phosphate (MUP), MUG and MUGS were used to assay the lysosomal enzymes βGal, AcPhos, total Hex, and Hex A/Hex S, respectively. Enzymatic reactions were stopped using 2-amino-2-methyl-1-propanol (MAP at 0.1M, pH 10.5). Purified Hex A and Hex B, which were used in kinetic characterization of PYR, were extracted from human placenta as described previously (21). QIAGEN kits for DNA and RNA extraction from fibroblasts were used. Molecular kits for PCRs and RT-PCRs were purchased from Invitrogen Inc.. Specific oligonucleotides for sequencing of genomic and cDNA were synthesized by The Center of Applied Genomics from Hospital for Sick Children. Rabbit polyclonal antibodies against human Hex A (Western blot), sheep polyclonal IgG against human β-subunit (used for immunoprecipitation, IP, and cell immunofluorescence) were prepared as previously described (22). Mouse IgG monoclonal against Lamp-1 was purchased from Iowa Hybridoma Bank. Secondary antibodies, donkey anti-sheep IgG and chicken anti-mouse IgG were purchased from Molecular Probes Inc.. Stock solutions of NGT, compound provided by Dr. S. Withers from University of British Columbia, were prepared by dissolving it in DMSO, (4 mg/mL) or in water (10 mg/mL). PYR, purchased from Sigma, was dissolved in DMSO (stock solution of 4 mg/mL), or ethanol, ETOH, (stock solution of 0.1 mg/mL). For the IP, Gamma beads purchased from Amersham Biosciences (UK) were used. Steel wool #0000 (International Steel Wool, Mexico), FeCl2 and FeCl3 (Sigma), and dextran T4000 (Amersham Biosciences, UK) were used to prepare lysosomes by magnetic chromatography. Tritiated GM2 ganglioside, [3H]-GM2 (10 mol%), cholesterol (20 mol%), phosphatidyl inositol (20 mol%), phosphatidyl choline (50 mol%), polycarbonate Liposo-Fast filter (Avestin 100 nm), AG3X4 (acetate form) resin (Biorad), Concanavalin A spheroid beads (Amersham Biosciences, UK), and recombinant Activator were used for the natural substrate assay. Chemical cross-linking was performed using dithiobis (DSP), succinimidylpropionate as a cross-linker reagent. Stock solution of DSP was dissolved in DMSO at 6 mM.
Screening for competitive Hex inhibitors
The NINDS library of 1040 FDA-approved compounds was screened in order to identify potential PCs for Hex A. The screening was performed using wild-type Hex B, and the common fluorescent MU-based artificial substrate, MUG. The library was screened in duplicate; with test compounds at a final concentration of 20 μM. From the original 96-well plates, compounds were initially diluted to 10 mM by addition of DMSO. Real-time Hex assays were performed in final volumes of 100 μL, containing 1mM of each drug, 12 ng of purified Hex B diluted in citrate phosphate buffer (CP, pH 4.1) and 25 μL of MUG (0.4 mM). Real-time fluorometric assays were performed using a Gemini EM Microplate Spectrofluorometer (Molecular Devices), with excitation at 345 nm, and emission at 450 nm, every 2 s during 20 minutes incubation period at 37°C. Mean Vmax was then obtained and expressed in Relative Fluorescence Units (RFU)/s using standardized SoftMax® Pro Software coupled to the spectrofluorometer. The values described on a replicate screen plot represent ratios related to mean of the Vmax obtained from the control DMSO sample (Fig. 1).
Fig. 1.

Replicate plot of a screen of the NINDS FDA-approved library of 1,040 small molecules for Hex A inhibitors. The graphic represents a replicate plot of residual Hex A activity in the presence of individual compounds from the NINDS library, i.e. each set of replicates define an X,Y coordinate. The zone outlined (dashed line) at 40% was set as the “inhibitor compound area,” which contained 4 compounds.
Mutation identification
Most mutations were identified and reported in a previous studies from our group (20,23,24). For mutations of cell lines, 26649, 32429 and 39997, total genomic DNA and cDNA were isolated from fibroblasts by routine techniques (25). PCR amplification was performed using primers described previously (24,26). RT-PCR from extracted total RNA followed by specific PCR amplification from cDNA with oligonucleotides previously described were performed (26). Fragments were sequenced with the use of ABI 377 and 3700 sequence analysers.
Tissue culture conditions and enzyme assays
Fibroblast cell lines were cultured to confluency. The culture medium used, α–minimal essential medium (α-MEM) with 10% of fetal calf serum (FCS) and antibiotics, was then replaced by the same medium containing filter-sterilized (Millipore-0.45 μm) PYR or NGT at the described concentrations. For the initial experiments, 10 cm-culture plates of each cell line were treated with PYR and NGT diluted in DMSO at final concentrations of 20, 10 and 5 μg/mL. In follow-up experiments, each cell line was treated with PYR at 3.0, 1.5, 0.5 and 0.1 μg/mL, and NGT, at 300, 150 and 75 μg/mL. Control plates containing only solvents, ETOH (used to dissolve PYR) and water (used to dissolve NGT) added to culture media were also established. After 5 days of incubation in drug-containing media, culture medium was removed; cells were washed twice with phosphate buffered saline (PBS, 1x) and harvested. Cell pellets were re-suspended in NaH2PO4 (10mM, pH 6.0), containing 5% glycerine, and lysed by freezing-thawing on dry ice. Hex assays using MUG or MUGS were performed. Lysates were diluted 10-fold by addition of 20 mM CP buffer (pH 4.1). For all lysosomal enzyme assays, stock solutions of the substrates dissolved in the same CP buffer of MUG (3.2 mM), MUGS (3.2 mM), MUP (10mM) and MUβGal (0.56 mM), were used. Assays were carried out by addition of 100 μL of substrate solution (final volume 200 μL), and incubation at 37 °C for 1 h for all artificial substrate used, except from MUG where the incubation time was 15 min. Reactions were stopped by addition of 1.5 mL 0.1 M MAP (pH 10.5), and fluorescence was measured as described above (22). Enzyme activities were calculated in nmoles of 4-methylumbelliferone (MU)-hydrolyzed/hr/mg of protein. The relative activities of total Hex, Hex A, and Hex S were expressed as ratios of corresponding samples from treated versus control cells. Residual Hex A % activities were calculated based on those of wild-type cell line lysate, assayed concomitantly with the mutant cell lines (range of Hex A activity 3,500–8,500 MU nmoles/hr/mg of protein).
Immunoselection assays for Hex A and Hex S
For measurement of residual Hex activities in SD cell lines (β-mutants), Hex A and Hex B (residual Hex B activity is generally undetectable in SD cells) were immunoprecipitated by solid-phase IP with polyclonal sheep anti β-subunit IgG as previously described (22,27,28). Residual Hex A activity was determined by measurement of Hex activity from immunoprecipitated phase (containing antibody bound to Hex A) with MUGS as substrate. Hex S activity was determined by measurement of enzyme activity in the IP supernant, also using MUGS as substrate.
Western blot analysis
The total protein contained in clarified lysates was determined by the Lowry method (29). Aliquots of lysates containing 20 μg total protein were diluted 1:1 with 1X standard Laemmli buffer containing 50 mM dithiothreitol (DTT) and heating at 65°C for 15 min. Each sample was then subjected to SDS-PAGE on a 10% bisacrylamide gel, and transferred to nitrocellulose. The nitrocellulose was then incubated with a rabbit anti human Hex A antibody as previously described (23). Blots were developed using chemiluminescent substrate according to the manufacturer’s protocol (Amersham Biosciences, UK). Bands were visualized, recorded and their optical density quantitated using a high sensitivity documentation system (Fluorchem 8000) consisting of a cooled CCD camera coupled with software from Alpha Innotech Corp., USA.
Purification of iron-dextran-labelled lysosomes by magnetic chromatography
Lysosomal fractions were prepared from fibroblasts grown for 5 days in media lacking or containing PYR, 3 μg/mL, using a previously described procedure (14,30). Solid-phase IP of the LYSO and PNS fractions were performed as described above.
Immunofluorescence labelling
Cells were grown on coverslips, fixed with 100% methanol for 15 min, and then blocked with FCS 10% for 30 min. After washing twice with PBS, the cells were incubated in the presence of anti β-subunit (diluted 1:400) and anti-Lamp-1 (diluted 1:500) antibodies at room temperature for 1 hour. The cells were then immunostained with suitable secondary antibodies cited earlier (Molecular Probes) at room temperature for 1 h. Confocal laser scanning microscopy on Zeiss LSM 510 confocal system was performed. All images were taken with 100 × 1.4 numerical apertures (NA) and 63 × 1.4 NA Apochromat objective (Zeiss). All image processing was performed using the Zeiss LSM 5 image examiner software.
Natural substrate assay
Tritiated GM2 ganglioside, [3H]-GM2, containing liposomes were prepared as previously described (4). In order to concentrate Hex A, as well as other soluble lysosomal enzymes, 3.5–3.8 g of total cell lysate protein was incubated with Concanavalin A beads (30 μL of drain solution) overnight at 4°C. Beads were then washed with PBS buffer three times and assayed with final volume of 100 μL, which contained CP (20 mM; pH 4.1), BSA (50 μg/mL), 2.5 μg of recombinant Activator isolated from transformed E. coli as described (31), and 20 nMoles of [3H]-GM2 contained in negatively charged liposomes. The incubation period of 18 hs, as well as further procedure for stopping reaction and preparation for liquid scintillation counting analysis, were performed as previously described (4).
Chemical cross-linking with dithiobis (DSP). (succinimidylpropionate)
Total lysate protein from mutant cells grown in medium containing PYR (3.0 μg/mL) or solvent (ETOH) were adjusted to a concentration of 0.5 mg/mL with NaH2PO4 (10mM, pH 6.0), containing sufficient DSP to give a final concentration of 0.9 mM. After 15 min incubation at 37°C, the reaction was quenched by the addition of 2.2 volumes of Tris-HCl, pH 7.5, to bring the mixture to 100 mM. The protein samples (5μg, normal control, 10 or 20 μg mutant cell lines) were then mixed with the 2X Laemmli sample buffer (1:1, containing no reducing agent), heated at 65°C for 15 min and separated by SDS-PAGE (10% gel) for Western blot analysis and quantitation (see above).
Heat denaturation assays
The effect of PYR on the stability of the Hex A containing a mutant β-subunit (βR505Q) was determined by heat denaturation experiments. Lysates from mutant cell lines containing 100–150 μg of protein were added to preheated CP buffer (20 mM; pH 4.1) with 0.3% of heat-treated BSA (Sigma) and PYR with a final concentration of 3.0 μg/mL. Aliquots of 100 μL were removed at fixed intervals of 0, 5, 10 and 30 min at 37°C and put on ice. The fraction of heated non-heated aliquots were put in tube with anti-β antibodies bound to Gamma beads, and IP procedure was followed (as described above). Mutant lysate samples containing only ETOH (PYR solvent) were also tested as a control for the PYR exposed lysates.
Statistical analysis
The statistical test, Z’ factor was used to measure the quality of the assay and its applicability to screening (32). This single statistic takes into consideration both signal-to-noise and reproducibility. Assays with a Z’-statistic >0.5 are robust enough to identify enhancement of enzyme activity reliably (32). Where applicable, data are expressed as the mean ± SEM. Comparisons of parametric data was analysed in the use of conventional parametric statistical methods as 2-tailed Student’s t-test.
RESULTS
Identification and characterization of PYR as a hexosaminidase inhibitor
The results of screening each of the 1040 small molecules in the NINDS library done in duplicate, for inhibition of Hex activity are shown in Figure 1. Each of the two replicates is defined by a X and Y pair of coordinates on the graph. Thus, consistent replicates fall on, or near the diagonal line (Z’=0.54) (32). Compounds within the box were considered “hits”, potentially effective as PCs, and were selected for secondary screening.
Two inhibitory compounds were identified as potential PCs, PYR and thioguanine (TGN) (Fig. 2). Secondary screening of PYR showed it had an IC50 of 5–13 μM for the Hex isozymes at pH 4.3 (Fig. 2A). On the other hand, TGN had a significantly higher IC50 of 170 μM (Fig. 2B). Unlike PYR, TGN was toxic to the fibroblast cell lines tested at the concentrations needed for it to act as a PC. Thus, PYR was chosen for further study. Kinetic examinations demonstrated that PYR behaved as a competitive inhibitor of Hex A with a Ki of 13 μM at pH 4.5 (Fig. 2C). Interestingly, PYR was found to have a pKHA of 6.5 with an IC50~ 2 μM at pH 6.5 (Fig. 2D). Thus, PYR would be least effective as an inhibitor in the acidic environment of the lysosome, but would bind maximally at the neutral pH of the ER, where optimal PC-activity is desired. Other compounds considered to be possible PC candidates failed to show competitive inhibition properties in a secondary screening and were not studied further (Fig. 1).
Fig. 2.

(A, B) Characterization of two inhibitors identified in the screen. The IC50 curves for the two “hits”, PYR (A) and thioguanine (TGN, B). (C) PYR behaves kinetically as a classical competitive inhibitor of Hex. (D) Maximal inhibition of Hex A by PYR was at pH 6.5, while the transition state analog, NGT, showed a maximum inhibition at pH of 4.5.
PYR as a pharmacological chaperone for Hex A
Initially, two fibroblast cell lines from TSD patients were selected for testing of PYR, αG269S/IVS6+1G>A (#32540), and αR178H/R178H (#27986) (Table I). The total relative Hex (A and B) activity (MUG) showed no significant increases in both treated cell lines (data not shown). However, when MUGS was used as the substrate, the αG269S/IVS6+1G>A cell line showed a statistically significant increase in Hex A activity, over 3-fold, with the highest concentration of PYR added to the culture media, while NGT at the same concentration (20 μg/mL) showed a smaller increase (Fig. 3A). As expected, the αR178H/R178H cell mutant, which carries an active site mutation affecting substrate binding, also known as the B1 variant (33,34), showed no effect above that of the DMSO control for either PYR or NGT (Fig. 3B). Interestingly, DMSO was found to be a weak competitive inhibitor of Hex (Ki of 170±20 μM), and to have mild chaperone activity, complicating the initial interpretation of the data. For this reason, we used ethanol (ETOH) as a solvent of PYR in further experiments. The final ETOH concentration was 0.03% in culture media, and did not affect the Hex activity (Methods).
Table I.
Fibroblast cell lines tested with their respective mutations in HEXA (TSD variant), HEXB (SD variant) genes, phenotype and residual Hex A activities.
| Patient cell line numberA | Mutation 1 | Phenotype | Mutation 2 | Phenotype | % residual Hex A |
|---|---|---|---|---|---|
|
|
|||||
|
TSD variants
|
|||||
| 27991 | R178H | Juvenile | c.1510delC | Infantile | 1.7 |
| 27986 | R178H | Juvenile | R178H | Juvenile | 3.0 |
| 27985 | R178H | Juvenile | Y277XB | InfantileD | 2.3 |
| 28236 | R178H | Juvenile | R499C | Juvenile | 2.2 |
| 27989 | G269S | Adult | c.1278insTATC | Infantile | 5.3 |
| 32540 | G269S | Adult | IVS6+1G>A | Juvenile | 6.4 |
| 32664 | R499H | Juvenile | c.1278insTACT | Infantile | 3.3 |
| 7638 | R499H | Juvenile | IVS9+1G>A | Infantile | 2.4 |
| 28237 | R499H | Juvenile | IVS11+1G>A | Infantile | 2.0 |
| 26649 | IVS9+1G>A | Infantile | IVS8-7G>AB | AdultD | 3.7 |
|
SD variants
|
|||||
| 1303 | C137YB | JuvenileD | C137YB | JuvenileD | 1.3 |
| 32429 | T150LB,C | InfantileD | P417L | Adult | 2.6 |
| 30037 | G353RB | InfantileD | IVS12-26G>A | Juvenile | 3.5 |
| 3585 | P417L | Adult | Δ16kbE | Infantile | 3.6 |
| 2400 | P504S | Adult | Δ16kbE | Infantile | 12.7F |
| 32045 | R505Q | Adult | IVS11+5G>AB | InfantileD | 4.2 |
| 39997 | R505Q | Adult | Δ16kbE | Infantile | 5.32 |
Patients’ cell lines are labeled according to the storage number from our Tissue Culture Laboratory.
Novel mutations.
This specific mutation was originally identified by Dr. John O’Brien.
Predicted phenotype of novel mutations based on clinical data and residual Hex A activity levels.
Large deletion of promoter-exon5 previously described (48).
The residual Hex A activity is apparently above the critical threshold, because this mutation was shown to specifically decrease the ability of the residual Hex A to bind and thus, hydrolyze the GM2 ganglioside/Activator complex by ~3-fold (natural substrate assay) (23).
Fig. 3.

PYR enhances Hex A enzyme levels in some TSD patient cell lines. Two mutant TSD fibroblast cell lines, αG269S/IVS6+1G>A (A) and αR178H/R178H (B), were treated with PYR and NGT, at 20, 10 and 5 μg/mL, dissolved in DMSO. Hex A activities using MUGS as a substrate were determined. The relative fold-increase of Hex A activity was calculated based on the activity of cell lines treated only with DMSO. The mature (lysosomal) α-subunit protein levels seen in Western blots (beneath histograms) correlated with the relative increase of Hex A observed in the αG269S/IVS6+1G>A mutant (A). However, the cells containing the αR178H/R178H active site mutations failed to show any increases in relative Hex A activity or changes in α-subunit protein levels (B). αp, indicates the α-subunit precursor, αm, the mature lysosomal α-subunit. *P < 0.01, **P < 0.001.
The PC effect of PYR was also specific for Hex; the activities of acid phosphatase (AcPhos) and β-galactosidase (βGal) were not enhanced in αG269S/ins1278TACT mutant cell line, while Hex A levels were enhanced at levels ≥0.1 mM (Fig. 4).
Fig. 4.

Specificity of PYR as a PC for Hex A. Relative levels of MU-substrate hydrolysis by acid phosphatase (AcPhos, substrate MUP), Hex A (substrate, MUGS) and βGal (substrate, MUβGal) in one of TSD cell lines, αG269S/c1278insTACT, incubated with increasing concentrations of PYR (units mM).
Assembly, genotyping and enzymatic phenotyping of fibroblast cell lines from TSD and SD patients
The genotypes of the various cell lines used to evaluate PYR are shown in Table I. The localizations in the crystallographic structure of Hex A of the different missense mutations examined in this study are shown in Figure 5 (2). Clinical features of the juvenile GM2 patients were reported previously by our group (20). The genotype and residual Hex A activity against MUGS of each cell line are given in Table I. These activities, with the exception of the βP504S-containing line, were within the expected range, based on previous correlations made between natural substrate hydrolysis and clinical phenotypes (7).
Fig. 5.
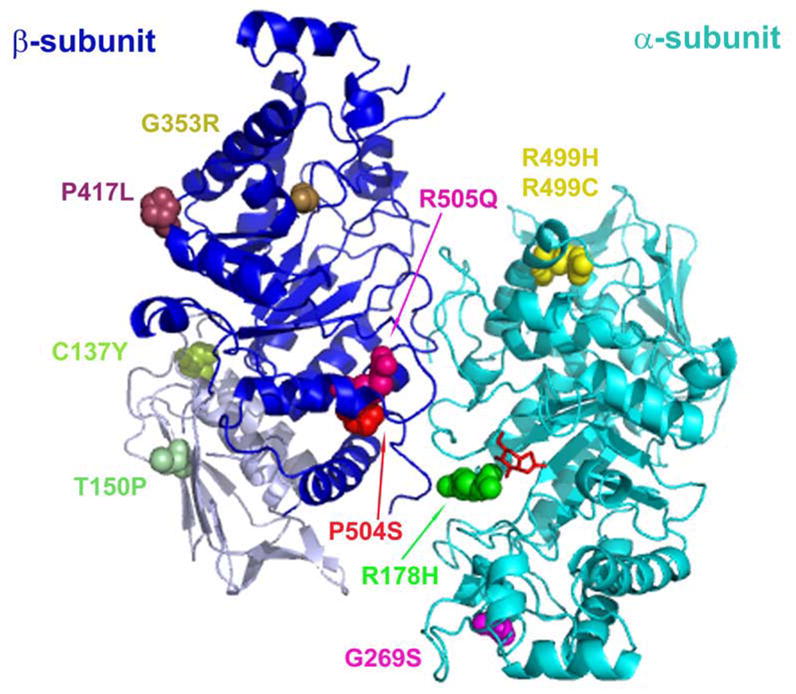
Missense mutations identified in TSD and SD patients localized onto a three-dimensional Hex A ribbon diagram (2). Aminoacid residue alterations are highlighted in color and shown in a space filled format. Two novel mutations (βC137Y and βT150P) are located in domain I (βA50-P201; light gray) of the β-subunit (blue). NGT is shown as a red stick format bound in the active site of the α-subunit (cyan).
Responses of different TSD and SD mutants to PYR and NGT at concentrations known to be non-toxic
At concentrations of PYR achievable in cerebrospinal fluid, thus available to neuronal cells of patients receiving therapeutic doses of the drug for malaria and toxoplasmosis, residual Hex A levels of two late-onset TSD cell lines, αG269S/c.1278insTACT and αIVS9+1G>A/IVS8-7G>A, were enhanced (Fig. 6A and b, respectively). The αG269S mutants showed lower relative increases (Fig. 6A) than those observed using higher concentrations of PYR (Fig. 3A). On other hand, NGT, at 300 μg/mL, which is non-toxic in mice (unpublished data), showed up to an 8-fold relative increase (Fig. 6A). The residual Hex A from the αIVS9+1G>A/IVS8-7G>A cell line was also chaperoned by both PYR and NGT (Fig. 6B). Interestingly, with this cell line the relative fold increases with PYR was higher than the one obtained with 100-fold higher concentrations of NGT (Fig. 6B). The increase in the Hex A activity was confirmed by the increased levels of the mature α-subunit (αm) in the respective Western blots shown (Fig. 6A and B). Maturation of the Hex subunits has been shown to occur after dimerization in the late endosome/lysosome (35–37).
Fig. 6.
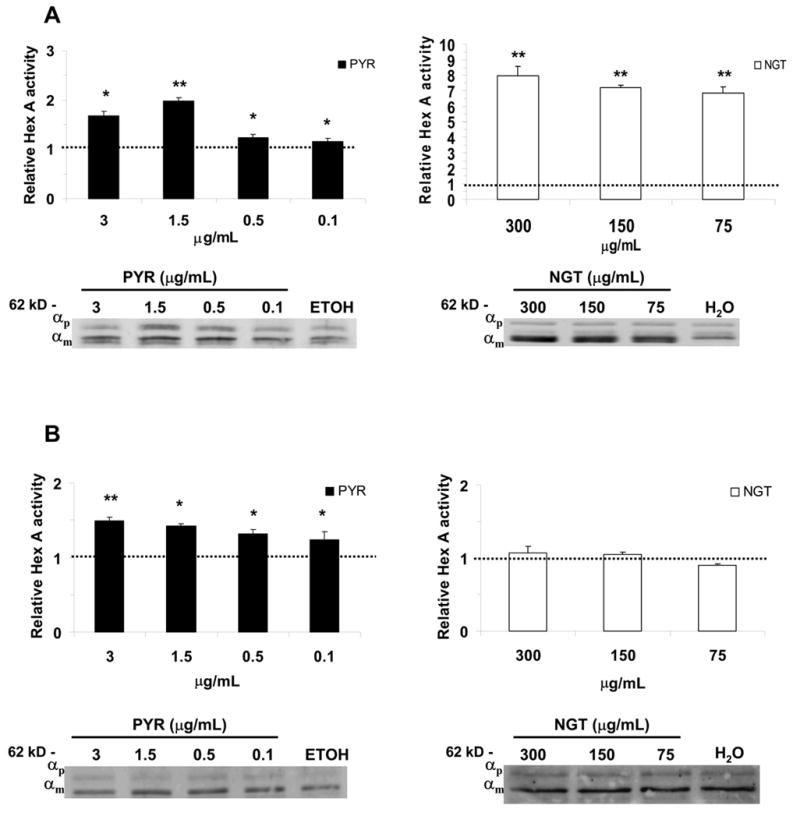
TSD or α-mutants cell lines which responded to PYR and NGT. The αG269S/c1278insTACT (A) and the αIVS9+1G>A/IVS8-7G>A (B) are shown with their relative Hex A activity and α-subunit protein levels at different treatment regimens of PYR and NGT. The fold-increase in activities was calculated based on the activity measured in control cell lines treated only with the dissolvent, i.e. ETOH for PYR-treated and H2O, for NGT-treated cells (in Methods). αp, indicates the α-subunit precursor, αm, the mature, lysosomal α-subunit. *P < 0.01, **P < 0.001.
Some α-mutants, αR178H, αR499H and αR499C (allelic to R178H mutation), show no increase of Hex A when treated with either PYR or NGT (Fig. 7A and B). The αR499H (Fig. 7A), a common mutation in Western Europeans, is associated with an α-subunit mutant with decreased solubility that tends to aggregate in the ER (38). Western blots confirmed that there were also no increases in the protein level of mature α-subunit (Fig. 7A and B).
Fig. 7.

Some TSD or α-mutants showed no relative increase in residual Hex A activity when exposed to PYR. PYR-treatment had no effect on cell lines containing αR499H, αR499C and αR178H mutations. The αR499H/c.1278insTACT mutant cell line (A) and αR178H/c.1510delC (B) are shown here in order to illustrate this observation. αp, indicates the α-subunit precursor, αm, the mature lysosomal α-subunit in Western blots.
All seven of the late-onset SD cell lines showed some degree of relative increase in residual Hex A (and Hex S) activity with PYR or NGT (Table II and Fig. 8). IP was used to separate the residual Hex A from Hex S in lysates obtained from treated β-mutants (Methods). The highest relative increase of Hex A with PYR was seen in cells with the βR505Q mutant allele. The βR505Q/Δ16kb mutant showed the best response to PYR of all 7 SD cell lines (Fig. 8). The response to PYR was even greater than the response to NGT (Fig. 8). Interestingly, the βR505Q/IVS11+5G>A cell line showed a smaller, although significant increase (up to 4-fold) of residual Hex A with PYR (Table II). The cell line βC137Y/C137Y also showed a significantly large increase in relative Hex A activity with PYR as compared to NGT (Table II). This cell line also had the lowest starting residual Hex A activity (Table I). On other hand, a cell line with a mutation in the adjacent residue, βP504S/Δ16kb, showed higher relative Hex A increases with NGT than with PYR. The cell line βT150L/P417L showed a 2.7-fold relative increase in Hex A activity with PYR, which was a better response than the 1.6 and 1.5-fold relative increases observed for the βP417L/Δ16kb and βG353R/IVS12-26G>A cell lines, respectively (Table II). Data from Western blots correlated with the increased Hex A activities as shown in the β-mutants from Table II (data not shown).
Table II.
Six of seven the SD or β-mutant cell lines with their relative activities of Hex A and Hex S after treatment with PYR or NGT.
| β-mutant | βC137Y/C137Y | βT150P/P417L | βG353R/IVS12-26G>A | βP417L/Δ16kb | βR505Q/IVS11+5G>A | βP504S Δ16kb | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|||||||||||||
|
Isoforms
|
Hex A | Hex S | Hex A | Hex S | Hex A | Hex S | Hex A | Hex S | Hex A | Hex S | Hex A | Hex S | |
| PYRA | 4.7+0.1D | 6.7±0.2D | 2.7±0.9C | 10.8±0.9D | 1.5±0.1D | 3.2±0.1D | 1.6±0.1D | 4.0±0.2D | 3.7±0.5C | 3.9±0.4D | 1.9±0.1D | 2.2±0.1C | |
| NGTB | 4.8±1.5C | 4.7±0.1D | 2.1±0.1D | 10.3±1.9C | 1.6±0.1D | 1.0±0.1 | 1.5±0.1D | 3.4±0.1D | 4.6±0.1C | 5.4±0.1D | 4.7±0.1D | 3.1±0.4D | |
3.0 μg/mL,
300 μg/mL,
P < 0.05.
P < 0.01.
Fig. 8.

PYR increases the enzyme activity and protein level of the α and β subunits of Hex A in the βR505Q/Δ16kb cell line. Histograms show relative activity enhancement of the lysate (total Hex A and S), as well as Hex A and S separated by IP (Methods). Western blots of the samples treated with PYR and NGT also document an increase in protein levels of the mature, lysosomal α- and β– subunits in the total lysate of treated cells. α/βp, indicates the α-and β-subunit precursors; αm and βm indicate the mature (lysosomal) α- and β-subunits, respectively. *P < 0.05; **P < 0.01.
Hex A is increased in a lysosomal enriched fraction of PYR-treated cells
In order to confirm the cellular localization of Hex A in PYR-treated and untreated cell lines, we loaded lysosomes with ferrous-dextran colloid, and performed a magnetic fractionation from which, we obtained a post nuclear supernatant (PNS), and an enriched lysosomal (LYSO) fraction, as previously described (14,30). After separating Hex A from Hex S by IP, the LYSO as well PNS fractions of one PYR-treated cell line (βC137Y/C137Y) showed significantly higher absolute Hex A and Hex S activities than the untreated controls (Table III).
Table III.
Increased Hex A and S activities in the enriched lysosomal fraction, LYSO, in relation to postnuclear supernatant (PNS) fraction in a domain I β-mutant, βC137Y/C137Y, cell line treated with PYR.
| Regimen | PYRB | Control (ETOH only) | |||
|---|---|---|---|---|---|
|
|
|||||
|
Fractions
|
PNS | LYSO | PNS | LYSO | |
| Hex AB,C | 125A | 205A | 80.5 | 97.4 | |
| Hex SD | 122A | 237A | 75.5 | 117 | |
| LysateD | 409A | 1140A | 173 | 385 | |
P < 0.05.
3.0 μg/mL,
The difference in Hex A activity between the PNS and LYSO fractions of PYR-treated cells was significantly higher than the difference observed in control cells (P < 0.01),
Units: hydrolyzed MU-nmoles/hr/mg total protein.
Mutant Hex β-subunits of Hex A are present in the lysosomes of PYR- treated SD cells
To further confirm that enhanced Hex A levels in SD cells was lysosomal, we localized Hex A by virtue of its mutant β-subunit in one of our high responding SD cell lines, βR505Q/IVS11+5G>A, treated with PYR (3.0 μg/mL). Immunofluorescence microscopy, using sheep anti-β-subunit IgG and mouse anti-Lamp-1 IgG, co-localization the β-subunit (green) of Hex A with Lamp1 (red) only in treated cells (Fig. 9, A-F). In control cells lines treated only with solvent (Fig. 9, G-L), β-subunit-staining was difficult to detect even in the ER.
Fig. 9.

Cellular localization of the mutant β-subunit-containing Hex isozymes in a patient fibroblast cell line, βR505Q/IVS11+5G>A. Cells were grown in the presence of PYR (3.0 μg/mL; A-F) or the solvent (ETOH) alone (control; G-L). The images in panels D-F represent selected areas of the same cell seen in panels A-C, but with higher magnification in order to demonstrate co-localization of the β-subunit of Hex (soluble protein in the lysosomal lumen) and Lamp-1 (integral lysosomal membrane protein) in lysosomes, versus the control cell, (G-I and J-L at higher magnification). Scale bars, red (10 μm) and magenta (2 μm).
PYR enhances natural substrate hydrolysis
PYR-treatment was confirmed to enhance both natural substrate (Activator/[3H]-GM2 complex) and MUGS hydrolysis in a αG269S chronic TSD cell line (Fig. 10).
Fig. 10.
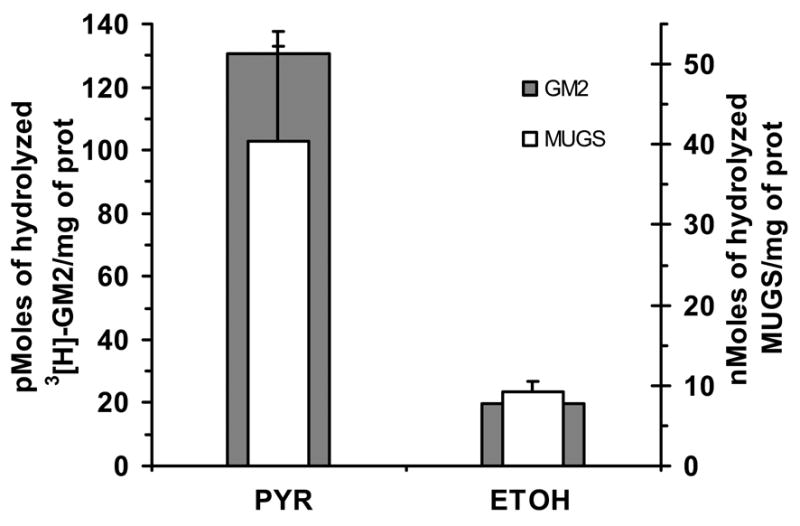
Natural and artificial substrate activities are increased in a αG269S mutant TSD cell line grown in the presence of PYR (3 μg/mL). The level of Hex-enhancement as measured by natural substrate, 3[H]-GM2 ganglioside and recombinant Activator (gray bars), hydrolysis was similar to that observed with the α-active-site-specific artificial substrate, MUGS (white bars).
PYR mechanism of function on Hex
In order to investigate the mechanism which PYR promotes the observed increases in Hex A activity two experiments were conducted. Firstly, lysates from two mutants, αG269S, a mutation that is located well away from the subunit-subunit interface, and βR505Q, located at the interface (Fig. 5), were exposed to a cross-linking reagent, DSP. PYR was shown to stabilize dimers and monomers in both α- and β-mutant cell lines (Fig. 11A). Optical density analysis (Methods) documented a two-fold increase in both dimer and monomer bands in the treated αG269S TSD cells (Fig. 11A). However, the treated βR505Q/Δ16kb cells showed a larger, 15-fold, increased in their Hex dimer as compared to a 4-fold increase in their monomer bands. These data indicate that in the former case PYR (and NGT) are primarily stabilizing mutant monomers, while in the later case the effect is primarily on dimers. Secondly, NGT has previously been shown to protect αG269S Hex A from heat denaturation (39), and we now demonstrate that PYR (3 μg/mL) also increases the thermostability the Hex A containing the βR505Q mutation, from a half-life of 7.6±3.7 min at 37°C to 24.9±5.3 min (Fig. 11B).
Fig. 11.
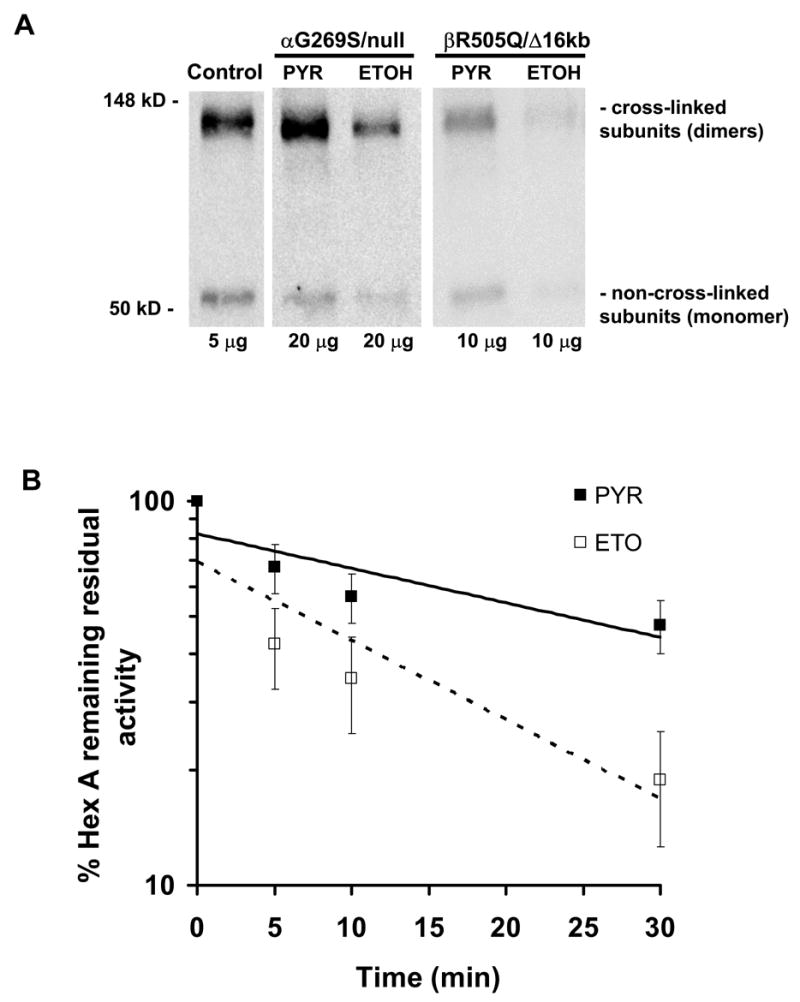
Effects of growth in PYR on the levels of monomeric and dimeric subunits of Hex; (A) Western blots from a normal, an α-G296S/null and a βR505Q/Δ16kb mutant cell line grown with or without PYR in their media. (B) Log scale of the percentage of remaining Hex A activity from βR505Q/Δ16kb mutant cell lysate exposed to PYR or solvent (ETOH) over a course of 30 minutes at 37°C.
DISCUSSION
This study demonstrated that PYR, functions as a PC for several α- and β-mutants affecting Hex A (Figs. 6 and 8, Table II). The αG269S mutant, the most prevalent mutation encountered in late-onset, adult GM2 (TSD) (40,41), showed significant response to PYR. An early pulse-chase study demonstrated that the majority of mutant αG269S-precursors remain primarily as monomers in patients’ cells and are degraded. Only low level mature α-subunits were found in the lysosome where they were always associated with β-subunits (35), confirming that dimerization is necessary for transport from the ER to lysosomes. It was later demonstrated that this apparent defect in association could be substantially overcome by over expressing the mutant α-subunit along with the normal human β-subunit in co-transfected COS cells, but the resulting Hex A was unstable at 37° C. Additionally, when the same mutation was made in the aligned Gly of the more stable β-subunit, there was little effect on the levels of expression of Hex B (22). Taken along with the new crystal structure of Hex A (2), these data indicate that the mutation destabilizes the folded α-monomer, accelerating its clearance by ERAD, which results in a diminished pool of α-monomers available for heterodimer formation. We also noted that, at high PYR concentrations, >0.1 mM, the inhibitory effects of the drug on Hex became evident, while at the same time, there was no effect on the activities of two other lysosomal enzymes (Fig. 4). This indicates that, when applied to patients, dosages must be carefully evaluated to maximize PYR’s PC activity, while minimizing its inhibitory effect.
Another α-mutant, αIVS9+1G>A/IVS8-7G>A, showed a small, but significant increase in Hex A levels with PYR (Fig 6B). Interestingly, in these cells, PYR treatment produced a higher relative level of Hex A activity than NGT, an effect that was not observed with αG269S mutants (Fig. 6A). This observation suggests that, in addition to missense mutations, some splice mutants may be treatable by PC therapy. Moreover, the αIVS9+1G>A mutation has been shown to produce no normal mRNA (42,43), so all the residual Hex A activity in this cell line originates from the novel αIVS8-7G>A mutation we have identified. Since properly spliced mRNA from this allele would have the wild-type sequence, we concluded that stabilizing the pool of normal α-monomer in the ER, i.e. increasing the half-life and thus the concentration of α-monomers, leads to an increase in heterodimer formation. This model is also consistent with the previously published biochemical data on the in vivo effects of the αG269S mutant (see above) and the cross-linking studies we now report (Fig. 11A).
On other hand, the Hex A levels of other α-mutant cell lines from our collection, αR178H, αR499H (Fig. 7) and αR499C (data not shown), fail to be enhanced by PYR treatment. The αR178H mutation, i.e. the B1 variant affects an active site residue (Fig. 5) (33,44) and would not be expected to respond. The αR499H and αR499C mutants were previously shown to be retained and form aggregates in ER (38,45). This illustrates that PCs can be mutation-specific, regardless of the mutant proteins common degradation pathway.
Among the β-mutants, the βR505Q mutation is located at or near the subunit-subunit interface (Fig. 5) (3). When βR505Q, occurs with the β16kb mutation, (cell line #39997), the highest relative increase of residual Hex A activity with PYR treatment was seen (Fig. 8). However, the βR505Q/IVS11+5G>A mutant (#32045) showed a smaller increase in both Hex A and S activities (Table II). In addition, the fibroblast cell line from an affected sibling of this patient produced the same levels of enhancement (data not shown). Thus, the observed differences in response to PC treatment may not be solely due to the identity of the βmutations, but may also be influenced by other factors, e.g. components of the ER quality control system.
Treatment of the βP504S/Δ16kb cell line with PYR resulted in a ~2-fold increase of Hex A activity. However, NGT produced an even higher level of enhancement in this cell line, 4.7-fold. The βP504S mutation is unique in its effect on Hex A. It was shown to reduce the specific activity of the mutant Hex A towards its natural substrate by ~3-fold as compared to MUGS; thus explaining its apparently high residual MUGS activity (23). Interestingly, βP504S is adjacent to βR505Q, which is enhanced better by PYR than NGT. Both residues are near the subunit-subunit interface contained in domain II, which buries a surface area of 2694 Å2 in each monomer and forms a large groove into which the Activator/GM2 ganglioside complex can be docked. Pro504 introduces a kink into helix α8, which is required for proper packing of the helix against two loops that interact directly with the docked Activator (3). Arg505 forms a salt bridge and some hydrogen bonds, which are needed to stabilize the surfaces of the dimer interface once they are buried (2). Like the αG269S substitution, βP504S and also βR505Q generate a more heat labile form of mutant Hex A (46), which indicates that monomer stability also affects the stability of the dimer. Such mutations appear to respond well to PYR treatment.
The βG353R mutation found in one of the β-mutant cell lines (#30037) is located in a very well conserved amino acid sequence, GGDE, found in all members of family 20 glycoside hydrolases (47). The Glu residue in this sequence is the catalytic acid group, and the Asp residue is involved in stabilizing the reaction intermediate (47). This cell line had βIVS12-26G>A as the second allele, which was shown to result in inefficient splicing in another juvenile SD cell line, reported to produce ~3% of normal Hex A activity (48,49), Thus, it is likely that the 3.5% of normal Hex A activity that we found in our patient’s cells (Table I) is the result of the splice junction mutation and not the missense mutation.
One interesting observation was the different responses of Hex A and Hex S to the PC-treatment (Table II). In general, cells treated with PYR showed higher relative increases of Hex S, as compared with Hex A, again suggesting that PYR is more effective than NGT in stabilizing the wild type α-subunit. As a consequence, the α-subunit could then act as a chaperone for the mutant β subunit, rather than the other way around as with the wild-type subunits (35,37).
One SD cell line was homozygous for a novel βC137Y mutation in domain I (Fig. 5). This cell line responded to PYR treatment with a relatively high increase in residual Hex A activity. Since protein domains are believed to fold independently of each other (50–52), and domain II contains both the active site and the subunit-subunit interface, it is possible that the heterodimer can form without domain I. If this is the case, the increased stability of the dimer resulting from PC treatment, might allow domain I additional time to achieve its proper conformation before being directed to ERAD. Regardless of the mechanism, this is the first example of a PC appearing to enhance the folding of a domain adjacent to the domain containing its site of binding. This phenomenon was not observed in the βT150P cell line that also had a novel domain I mutation in heterozygozity with the previously reportedβP417L, (24,26) (Fig. 5 and Table II). In the previous reports, βP417L allele was present with the Δ16kb null allele and was shown to affect mRNA splicing. Thus, it conferred the residual Hex A activity we observed (24,26). Testing the βP417L/Δ16kb cell line with PYR or NGT resulted in similar enhancements (Table II) as were achieved in the βT150P/P417L cells. Thus, as for domain II mutants, not all domain I mutants can benefit from PC treatment.
PYR, [2,4 diamino 5-(4-chlorophenyl)-6-ethylpyrimidine], was originally developed as a dihydrofolate reductase inhibitor, which is used for treatment of parasitic diseases, including chloroquine-resistant malaria and toxoplasmosis (53,54). PYR is an orally administered drug, with a well-studied pharmacokinetic profile (55). Studies have shown that 12–26% of serum levels cross the blood-brain barrier (BBB) (56). Thus to test PYR as a PC for mutant forms of Hex A, we used PYR concentrations corresponding to levels achieved in the nervous system by administration of routine therapeutic doses. This is a critical observation, as in late-onset GM2, along with many other lysosomal storage diseases, symptoms are principally related to the effect of the metabolic abnormality on the brain; the BBB still remains the main obstacle for different types of emerging therapeutic interventions for these inherited metabolic diseases (12).
The potential clinical impact of the PYR acting as a PC may be estimated from the level of enhancement of the residual Hex A activity in patients’ tissues compared with the critical threshold approximation of 5–10% of normal (10). In Table IV, we calculate the % of residual Hex A based on the maximum increases that were achieved with either PYR or NGT treatment. Five mutant cell lines, αG269S/c1278insTACT, αG269S/IV6+1G>A, βP504S/Δ16kb, βR505Q/IVS11+5G>A and βR505Q/Δ16kb showed increases in residual Hex A activity over the 10% critical threshold. Similar treatment of other cell lines significantly increased residual Hex A levels, but failed to produce levels over 10% of normal. Given the wide range of clinical phenotypes associated with a very narrow range of residual enzyme activities, even these smaller enhancements might be expected to be clinically relevant and slow the rate of progressive neurodeterioration in GM2. NGT, in general, preformed better as a PC, but at much higher concentrations than PYR. However, while PYR is an FDA-approved drug with extensive data about its pharmacokinetics and safety, studies on the toxicity of NGT in animals are still in progress.
Table IV.
Fibroblast cell lines with relative increases of residual Hex A activity in the presence of PYR and NGT.
| Patient cell line numberA | Mutation 1 | Mutation 2 | % of residual Hex A | % of Hex A with PYR | % of Hex A with NGT |
|---|---|---|---|---|---|
| TSD or α-mutants |
HEXA mutations
|
||||
| 27989 | G269S | c.1278insTATC | 5.3 | 9.5 | 41.9 |
| 32540 | G269S | IV6+1G>A | 6.5 | 10.4 | 46.8 |
| 26649 | IVS9+1G>A | IVS8-7G>AB | 3.7 | 5.5 | -D |
| SD or β-mutants |
HEXB Mutations
|
||||
| 32045 | R505Q | IVS11+5G>AB | 4.2 | 15.5 | 19.3 |
| 39997 | R505Q | Δ16kb | 5.3 | 41.9 | 19.0 |
| 1303 | C137YB | C137YB | 1.3 | 6.1 | 6.2 |
| 30037 | G353RB | IVS12-26G>A | 3.5 | 5.2 | 5.6 |
| 32429 | T150PB | P417L | 2.6 | 7.0 | 5.4 |
| 3585 | Δ16kb | P417L | 3.6 | 5.8 | 5.4 |
| 2400 | P504S | Δ16kb | 12.7C | 23.1 | 59.7 |
Patients’ cell lines are labeled according to the storage number from our Tissue Culture laboratory.
Novel mutations.
βP504S mutation reduces the ability of residual Hex A to bind the GM2 ganglioside/Activator complex by 3-fold as previously described (23)□.
NGT failed to show any significant increases in either Hex A activity or α-mutant protein level (Fig. 6b).
In conclusion, PYR was identified, and shown to function as a PC for mutant cell lines from patients presenting with a late-onset form, i.e. juvenile or adult, of GM2. Our studies on the effects of PYR-treatment on α and β-mutants have provided interesting insights into the independence of protein domains during the folding and subunit assembly stages of Hex in the ER. We have shown that screening of a library of FDA-approved drugs for competitive inhibitors of lysosomal enzymes is a feasible and practical approach to find potential PC for mutants causing devastating inherited metabolic disorders, such as GM2. The drugs represented in the library are already in current use for other medical purposes, indicating that they already passed extensive testing for toxicity, both in animals and humans. This considerably decreases the time and expense associated with conventional drug development, bringing potentially effective therapies to patients more rapidly. Clinical trials using such drugs could also provide a proof-of-principle for the concept of PC therapy, which would encourage investments into development and testing more optimized PC-molecules.
Nonstandard abbreviations used
- Activator
GM2 activator protein
- AcPhos
acid phosphatase
- α-MEM
α–minimal essential medium
- BBB
blood-brain-barrier
- βGal
β-galactosidase
- BSA
bovine serum albumin
- CP
citrate phosphate buffer
- DMSO
dimethylsulfoxide
- DSP
dithiobis- succinimidylpropionate
- DTT
dithiothreitol
- ERAD
ER-associated degradation
- ETOH
ethanol
- FCS
fetal calf serum
- FDA
Food Drug Administration
- GalNAc
N-acetylgalactosamine
- GM2
GM2 gangliosidosis
- [3H]-GM2
tritiated GM2 ganglioside
- Hex
hexosaminidase
- HTS
high-throughput screening
- IP
immunoprecipitation
- Lamp-1
lysosomal associated membrane protein-1
- LYSO
lysosomal fraction
- PBS
phosphate buffered saline
- PC
pharmacological chaperone
- PNS
postnuclear supernant fraction
- PYR
Pyrimethamine
- MAP
2-amino-2-methyl-1-propanol
- MU
methylumbelliferone
- MUβGal
4-methylumbelliferyl-β-D-galactopyranoside
- MUG
4-methylumbelliferyl-(2-acetamido-2-deoxy)-β-D-glucopyranoside
- MUGS
4-methylumbelliferyl-7-(6-sulfo-2-acetamido-2-deoxy)-β-D-glucopyranoside
- MUP
4-methylumbelliferyl phosphate
- NGT
N-acetylglucosamine-thiazoline
- NINDS
National Institute of Neurological Disorders and Stroke
- SD
Sandhoff disease
- TGN
thioguanine
- TSD
Tay-Sachs disease
Footnotes
* We acknowledge the technical assistance of Amy Leung, Daphne Benedict and Scott Bukovac. We are thankful to Richard Bagshaw and Dr. John Callahan for helpful and critical suggestions. We thank Dr. Corien Verschuuren-Bemelmans from Groningen, Netherlands, who made available one of patient’s cell lines studied. We also thank one of the patients, who donated his own cell line, banked by Dr John O’Brien at Mayo Clinic, Rochester, MN. This research was funded by Canadian Institutes of Health Research (CIHR), National Institutes of Health (NIH), the Uger Estate and Life for Luke Foundation. The salary of G.M. was partially supported by a donation from Amicus Therapeutics USA.
References
- 1.Hirabayashi Y, Li YT, Li SC. J Neurochem. 1983;40(1):168–175. doi: 10.1111/j.1471-4159.1983.tb12667.x. [DOI] [PubMed] [Google Scholar]
- 2.Lemieux MJ, Mark BL, Cherney MM, Withers SG, Mahuran DJ, James MN. J Mol Biol. 2006;359(4):913–929. doi: 10.1016/j.jmb.2006.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mark BL, Mahuran DJ, Cherney MM, Zhao D, Knapp S, James MN. J Mol Biol. 2003;327(5):1093–1109. doi: 10.1016/s0022-2836(03)00216-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sharma R, Bukovac S, Callahan J, Mahuran D. Biochim Biophys Acta. 2003;1637(1):113–118. doi: 10.1016/s0925-4439(02)00221-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hou Y, Tse R, Mahuran DJ. Biochemistry. 1996;35:3963–3969. doi: 10.1021/bi9524575. [DOI] [PubMed] [Google Scholar]
- 6.Gravel R, Kaback M, Proia R, Sandhoff K, Suzuki K, Suzuki K. The GM2 gangliosidosis. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Basis of Inherited Disease. 8. McGraw-Hill; New York: 2001. [Google Scholar]
- 7.Leinekugel P, Michel S, Conzelmann E, Sandhoff K. Hum Genet. 1992;88(5):513–523. doi: 10.1007/BF00219337. [DOI] [PubMed] [Google Scholar]
- 8.Maegawa GH, Stockley T, Tropak M, Banwell B, Blaser S, Kok F, Giugliani R, Mahuran D, Clarke JT. Pediatrics. 2006;118(5):e1550–1562. doi: 10.1542/peds.2006-0588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neudorfer O, Pastores GM, Zeng BJ, Gianutsos J, Zaroff CM, Kolodny EH. Genet Med. 2005;7(2):119–123. doi: 10.1097/01.gim.0000154300.84107.75. [DOI] [PubMed] [Google Scholar]
- 10.Conzelmann E, Sandhoff K. Dev Neurosci. 1983;6(1):58–71. doi: 10.1159/000112332. [DOI] [PubMed] [Google Scholar]
- 11.Sawkar AR, Cheng WC, Beutler E, Wong CH, Balch WE, Kelly JW. Proc Natl Acad Sci U S A. 2002;99(24):15428–15433. doi: 10.1073/pnas.192582899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Desnick RJ, Schuchman EH. Nat Rev Genet. 2002;3(12):954–966. doi: 10.1038/nrg963. [DOI] [PubMed] [Google Scholar]
- 13.Desnick RJ. Journal of Inherited Metabolic Disease. 2004;27(3):385–410. doi: 10.1023/B:BOLI.0000031101.12838.c6. [DOI] [PubMed] [Google Scholar]
- 14.Tropak MB, Reid SP, Guiral M, Withers SG, Mahuran D. J Biol Chem. 2004;279(14):13478–13487. doi: 10.1074/jbc.M308523200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tominaga L, Ogawa Y, Taniguchi M, Ohno K, Matsuda J, Oshima A, Suzuki Y, Nanba E. Brain Dev. 2001;23(5):284–287. doi: 10.1016/s0387-7604(01)00216-9. [DOI] [PubMed] [Google Scholar]
- 16.Fan JQ, Ishii S, Asano N, Suzuki Y. Nat Med. 1999;5(1):112–115. doi: 10.1038/4801. [DOI] [PubMed] [Google Scholar]
- 17.Matsuda J, Suzuki O, Oshima A, Yamamoto Y, Noguchi A, Takimoto K, Itoh M, Matsuzaki Y, Yasuda Y, Ogawa S, Sakata Y, Nanba E, Higaki K, Ogawa Y, Tominaga L, Ohno K, Iwasaki H, Watanabe H, Brady RO, Suzuki Y. Proc Natl Acad Sci U S A. 2003;100(26):15912–15917. doi: 10.1073/pnas.2536657100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verkman AS, Lukacs GL, Galietta LJ. Curr Pharm Des. 2006;12(18):2235–2247. doi: 10.2174/138161206777585148. [DOI] [PubMed] [Google Scholar]
- 19.Arnold LA, Estebanez-Perpina E, Togashi M, Shelat A, Ocasio CA, McReynolds AC, Nguyen P, Baxter JD, Fletterick RJ, Webb P, Guy RK. Sci STKE. 2006;2006(341):13. doi: 10.1126/stke.3412006pl3. [DOI] [PubMed] [Google Scholar]
- 20.Maegawa GHB, Stockley T, Tropak M, Banwell B, Blaser S, Kok F, Giugliani R, Mahuran D, Clarke JTR. Pediatrics. 2006;118(5):e1550–1562. doi: 10.1542/peds.2006-0588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mahuran D, Lowden JA. Can J Biochem. 1980;58(4):287–294. doi: 10.1139/o80-038. [DOI] [PubMed] [Google Scholar]
- 22.Brown CA, Mahuran DJ. Am J Hum Genet. 1993;53(2):497–508. [PMC free article] [PubMed] [Google Scholar]
- 23.Hou Y, McInnes B, Hinek A, Karpati G, Mahuran D. J Biol Chem. 1998;273(33):21386–21392. doi: 10.1074/jbc.273.33.21386. [DOI] [PubMed] [Google Scholar]
- 24.McInnes B, Potier M, Wakamatsu N, et al. J Clin Invest. 1992;90(2):306–314. doi: 10.1172/JCI115863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sambrook J, David W. Molecular cloning: a laboratory manual. 3. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 2001. [Google Scholar]
- 26.Wakamatsu N, Kobayashi H, Miyatake T, Tsuji S. J Biol Chem. 1992;267(4):2406–2413. [PubMed] [Google Scholar]
- 27.Sagherian C, Poroszlay S, Vavougios G, Mahuran D. Biochem Cell Biol. 1993;71(7–8):340–347. doi: 10.1139/o93-051. [DOI] [PubMed] [Google Scholar]
- 28.Sagherian C, Thorner P, Mahuran D. Biochem Biophys Res Commun. 1994;204(1):135–141. doi: 10.1006/bbrc.1994.2436. [DOI] [PubMed] [Google Scholar]
- 29.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. J Biol Chem. 1951;193(1):265–275. [PubMed] [Google Scholar]
- 30.Rodriguez-Paris JM, Nolta KV, Steck TL. J Biol Chem. 1993;268(12):9110–9116. [PubMed] [Google Scholar]
- 31.Smiljanic-Georgijev N, Rigat B, Xie B, Wang W, Mahuran DJ. Biochim Biophys Acta. 1997;1339(2):192–202. doi: 10.1016/s0167-4838(97)00002-2. [DOI] [PubMed] [Google Scholar]
- 32.Zhang JH, Chung TD, Oldenburg KR. J Biomol Screen. 1999;4(2):67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 33.Ohno K, Suzuki K. J Neurochem. 1988;50(1):316–318. doi: 10.1111/j.1471-4159.1988.tb13266.x. [DOI] [PubMed] [Google Scholar]
- 34.Tanaka A, Ohno K, Sandhoff K, et al. Am J Hum Genet. 1990;46(2):329–339. [PMC free article] [PubMed] [Google Scholar]
- 35.d’Azzo A, Proia RL, Kolodny EH, Kaback MM, Neufeld EF. J Biol Chem. 1984;259(17):11070–11074. [PubMed] [Google Scholar]
- 36.Hasilik A, Neufeld EF. J Biol Chem. 1980;255(10):4937–4945. [PubMed] [Google Scholar]
- 37.Proia RL, d’Azzo A, Neufeld EF. J Biol Chem. 1984;259(5):3350–3354. [PubMed] [Google Scholar]
- 38.Paw BH, Moskowitz SM, Uhrhammer N, Wright N, Kaback MM, Neufeld EF. J Biol Chem. 1990;265(16):9452–9457. [PubMed] [Google Scholar]
- 39.Tropak MB, Blanchard J, Withers SG, Brown E, Mahuran D. Chem Biol, submitted. 2006 doi: 10.1016/j.chembiol.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaback MM. Eur J Pediatr. 2000;159(Suppl 3):S192–195. doi: 10.1007/pl00014401. [DOI] [PubMed] [Google Scholar]
- 41.Navon R, Proia RL. Science. 1989;243(4897):1471–1474. doi: 10.1126/science.2522679. [DOI] [PubMed] [Google Scholar]
- 42.Akerman BR, Zielenski J, Triggs-Raine BL, Prence EM, Natowicz MR, Lim-Steele JS, Kaback MM, Mules EH, Thomas GH, Clarke JT, et al. Hum Mutat. 1992;1(4):303–309. doi: 10.1002/humu.1380010407. [DOI] [PubMed] [Google Scholar]
- 43.Akli S, Chomel JC, Lacorte JM, Bachner L, Kahn A, Poenaru L. Hum Mol Genet. 1993;2(1):61–67. doi: 10.1093/hmg/2.1.61. [DOI] [PubMed] [Google Scholar]
- 44.Kytzia HJ, Sandhoff K. J Biol Chem. 1985;260(12):7568–7572. [PubMed] [Google Scholar]
- 45.Mules EH, Hayflick S, Dowling CE, Kelly TE, Akerman BR, Gravel RA, Thomas GH. Hum Mutat. 1992;1(4):298–302. doi: 10.1002/humu.1380010406. [DOI] [PubMed] [Google Scholar]
- 46.Bolhuis PA, Ponne NJ, Bikker H, Baas F, Vianney de Jong JM. Biochim Biophys Acta. 1993;1182(2):142–146. doi: 10.1016/0925-4439(93)90134-m. [DOI] [PubMed] [Google Scholar]
- 47.Henrissat B, Bairoch A. Biochem J. 1993;293(Pt 3):781–788. doi: 10.1042/bj2930781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Neote K, McInnes B, Mahuran DJ, Gravel RA. J Clin Invest. 1990;86(5):1524–1531. doi: 10.1172/JCI114871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakano T, Suzuki K. J Biol Chem. 1989;264(9):5155–5158. [PubMed] [Google Scholar]
- 50.Murzin AG, Brenner SE, Hubbard T, Chothia C. J Mol Biol. 1995;247(4):536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 51.Apic G, Gough J, Teichmann SA. Bioinformatics. 2001;17(Suppl 1):S83–89. doi: 10.1093/bioinformatics/17.suppl_1.s83. [DOI] [PubMed] [Google Scholar]
- 52.Teichmann SA, Rison SC, Thornton JM, Riley M, Gough J, Chothia C. Trends Biotechnol. 2001;19(12):482–486. doi: 10.1016/s0167-7799(01)01813-3. [DOI] [PubMed] [Google Scholar]
- 53.Leport C, Chene G, Morlat P, Luft BJ, Rousseau F, Pueyo S, Hafner R, Miro J, Aubertin J, Salamon R, Vilde JL. J Infect Dis. 1996;173(1):91–97. doi: 10.1093/infdis/173.1.91. [DOI] [PubMed] [Google Scholar]
- 54.Weiss LM, Luft BJ, Tanowitz HB, Wittner M. Am J Trop Med Hyg. 1992;46(3):288–291. doi: 10.4269/ajtmh.1992.46.288. [DOI] [PubMed] [Google Scholar]
- 55.Buxton ILO. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. 11. McGraw-Hill Books; New York: 2006. Pharmacokinetics and pharmacodynamics: the dynamics of drug absorption, distribution, action and elimination. [Google Scholar]
- 56.Weiss LM, Harris C, Berger M, Tanowitz HB, Wittner M. J Infect Dis. 1988;157(3):580–583. doi: 10.1093/infdis/157.3.580. [DOI] [PubMed] [Google Scholar]