

Abstract
Mouse embryonic fibroblasts derived from Nrf2 −/− mice (N0) and Nrf2 +/+ mice (WT) have been used to characterize both basal and diquat (DQ)-induced oxidative stress levels and to examine Nrf2 activation during exposure to DQ-generated superoxide anion. Microarray analysis revealed that N0 cells have similar constitutive mRNA expression of genes responsible for the direct metabolism of reactive oxygen species but decreased expression of genes responsible for the production of reducing equivalents, repair of oxidized proteins and defense against lipid peroxidation, compared to WT cells. Nonetheless, the basal levels of ROS flux and oxidative damage biomarkers in WT and N0 cells were not different. Diquat dibromide (DQ), a non-electrophilic redox cycling bipyridylium herbicide, was used to generate intracellular superoxide anion. Isolated mitochondria from both cell lines exposed to DQ produced equivalent amounts of ROS, indicating a similar cellular capacity to generate ROS. However, N0 cells exposed to DQ for 24-hr exhibited markedly decreased cell viability and aconitase activity as well as increased lipid peroxidation and glutathione oxidation, relative to WT cells. 2′,7′-Dichlorofluorescein fluorescence was not increased in WT and N0 cells after 30-min of DQ exposure. However, increased levels of ROS were detected in N0 cells but not WT cells after 13-hr of DQ treatment. Additionally, total glutathione concentrations increased in WT, but not N0 cells following a 24-hr exposure to DQ. DQ exposure resulted in activation of an antioxidant response element-luciferase reporter gene, as well as induction of Nrf2-regulated genes in WT, but not N0 cells. Thus the enhanced sensitivity of N0 cells does not reflect basal differences in antioxidative capacity, but rather an impaired ability to mount an adaptive response to sustained oxidative stress.
Keywords: Keywords: Nrf2, oxidative stress, adaptive response, cytotoxicity, lipid peroxidation, superoxide anion, protein oxidation, glutathione, antioxidative enzymes
Introduction
Numerous studies have linked Nrf2-deficiency with increased sensitivity to electrophile stress. For example, Nrf2-deficient mice (N0) have been demonstrated to exhibit increased acetaminophen hepatotoxicity[1], butylated hydroxytoluene pneumotoxicity[2], N-nitrosobutyl(4-hydroxybutyl)amine-induced bladder carcinogenesis[3] and benzo(a)pyrene-induced forestomach carcinogenesis[4]. Additionally, recent studies are beginning to illustrate that Nrf2-deficiency leads to increased toxicity during oxidative conditions or following exposure to agents in which oxidative stress has been implicated in the toxicological response. Exposure to 95% oxygen resulted in increased lung toxicity in N0 mice compared to nrf2 +/+ (WT) mice[5]. Nrf2-defcient mice also exhibited increased levels of 8-oxo-7,8-dihydro-2′-deoxyguanosine, a biomarker of oxidative DNA damage, following exposure to cigarette smoke and diesel exhaust[6, 7]. Additionally, nrf2-deficient mice display increased oxidative damage during allergen-driven airway inflammation[8], as well as endotoxin-mediated septic shock[9].
The transcription factor NF-E2-related factor 2 (Nrf2) belongs to the Cap-N-Collar family of transcription factors and recognizes antioxidant response elements (ARE) present in the promoter regions of target genes[10]. Nrf2 is normally sequestered in the cytosol by interaction with Keap1, preventing nuclear accumulation of Nrf2 and leading to proteolysis of Nrf2 by the ubiquitin-proteosome pathway[11, 12]. Activation of Nrf2 occurs by dissociation of Nrf2 from Keap1 following exposure to electrophiles and reactive oxygen species or following phosphorylation of Nrf2 by kinases[13]. These events result in translocation of Nrf2 to the nucleus, dimerization with small Maf proteins and transactivation of target genes[14].
Nrf2 has been shown to have a role in the regulation of inducible expression of a number of enzymes important in protection against reactive oxygen species. Transcript levels of glutathione reductase (Gsr), peroxiredoxin-1 (Prdx1), thioredoxin-2 (Txn2), thioredoxin reductase-1 (Txnrd1), the regulatory and catalytic subunits of glutamate-cysteine ligase (Gclm, Gclc), catalase (Cat), Cu/Zn superoxide dismutase (Sod1), and glutathione peroxidase (Gpx) gene expression were increased in tissues of WT mice administered chemopreventive agents such as 3H-1,2-dithiole-3-thione and sulforaphane [15, 16]. No induction of these genes was observed in N0 mice. Additionally, the expression of several genes important in the production of reducing equivalents, including UDP-glucose dehydrogenase (Ugpdh), malic enzyme (Mod1), and glucose-6-phosphate dehydrogenase (G6pd) were increased in various tissues of WT mice, but not N0 mice [15, 16].
On the other hand, microarray analyses comparing gene expression in WT and N0 mice revealed that constitutive expression of several direct-acting antioxidant genes, such as Sod1, Cat, Txn2, Gsr, and Txnrd1 was not appreciably affected by Nrf2 genotype [16]. By contrast, the expression of Gpx, glutathione-S-transferase α4 (Gstα4) and ferritin light chain subunit 1 (Ftl1) has been shown to be higher in WT mice, compared to N0 mice, implicating a role for Nrf2 in the basal expression of these genes [16, 17]. Additionally, Nrf2 has been shown, via microarray analysis of WT and N0 mice, to regulate the constitutive expression of genes important in the production of NADPH, such as Mod1, and G6pd, that are necessary for the function of antioxidant enzymes[16, 17]. Collectively, these studies illustrated that Nrf2 plays an important role in an inducible antioxidative response but the role of Nrf2 in maintaining constitutive expression of antioxidative genes, and hence basal oxidative tone, may be minor relative to other regulatory pathways.
A previous study revealed that N0 mouse embryonic fibroblasts (MEF) cells exhibit increased cytotoxicity, relative to WT MEF cells, following exposure to menadione, a redox-cycling quinone, [18]. However, menadione is also an electrophile and is a substrate for NADPH-quinone oxidoreductase-1 (NQO1), an Nrf2-regulated enzyme that catalyzes an obligatory two-electron reduction of quinones[19]. Therefore, it is likely that differences in cytotoxicity were due to increased expression of NQO1 in WT relative to N0 MEF cells. In fact, it was shown that N0 cells exhibited 60% less NQO1 activity than WT cells[18]. Other redox-cycling chemicals exist that are not electrophiles, but rather are intracellular generators of superoxide anion. Diquat dibromide (DQ) is a bipyridylium herbicide that redox cycles, at the mitochondria, to produce superoxide anion[20]. DQ is a pure redox cycler in that it is not electrophilic and prior studies have shown that DQ is only minimally metabolized in rodents[21]. Using DQ as a model superoxide generating system is a more appropriate approach since any differences in cell response witnessed after exposure to DQ can be directly attributed to differences in oxidative stress as opposed to differences in electrophile metabolism. Additionally, DQ exposure results in the intracellular generation of ROS, thereby eliminating the effects of media components on the extracellular delivery of oxidative compounds.
The present study directly characterized both basal and DQ-induced oxidative stress levels in MEF cells derived from WT and N0 mice and examined Nrf2 activation during exposure to DQ-generated superoxide anion as a response to sustained oxidative challenge. We determined that antioxidant gene expression is minimally lower in N0 MEF cells and that basal oxidative tone is not appreciably different by transcription factor genotype. Initially, both WT and N0 cells can efficiently metabolize DQ-generated ROS but, over time, exposure to DQ resulted in increased levels of ROS and oxidative damage in N0 cells, but not WT cells. Additionally, DQ exposure led to increased activation of an ARE-luciferase reporter gene and increased expression of antioxidative genes in WT but not N0 cells. Taken together, these results illustrate that Nrf2 regulates an adaptive cytoprotective response against sustained exposure to superoxide anion.
Materials and Methods
Reagents
DQ, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT), dimethyl sulfoxide (DMSO), butylated hydroxytoluene (BHT), trichloroacetic acid, thiobarbituric acid, 1,1,2,2-tetraethoxypropane, reduced glutathione (GSH), oxidized glutathione (GSSG), dansyl chloride, n-butanol, luminol, β-nicotinamide adenine dinucleotide phosphate (NADP+), β-nicotinamide adenine dinucleotide phosphate, reduced (NADPH) and isocitrate dehydrogenase (NADP+-dependent) were obtained from Sigma-Aldrich (St. Louis, MO, USA). γ-glutamylglutamate (γ-EE) was obtained from Fisher Scientific (Hampton, NH, USA). Iscove’s modified Dulbecco’s media (IMDM) and 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-DCF-DA) were obtained from Invitrogen (Carlsbad, CA, USA) while Complete protease inhibitor cocktail was obtained from Roche Diagnostics (Indianapolis, IN, USA). IQ SYBR Green Supermix and iScript cDNA synthesis kits were obtained from Bio-Rad (Hercules, CA, USA). GeneJuice transfection reagent was purchased from EMD Biosciences (San Diego, CA, USA). All other chemicals and solvents were of analytical/reagent grade or higher.
Instrumentation
All spectroscopy measurements were performed using a Spectramax 190 microtiter plate spectrophotometer (Molecular Devices, Sunnyvale, CA, USA). Microarray GeneChips were scanned with the GeneArray scanner (Agilent Technologies, Palo Alto, CA, USA) and hybridization was done using a GeneChip Hybridization Oven 640 (Affymetrix). Chemiluminescence was analyzed using a Berthold Biolumat LB9505 luminometer (Berthold Detection Systems, Oak Ridge, TN, USA) as well as a Turner Designs TD-20/20 luminometer (Turner Designs, Sunnyvale, CA, USA). Flow cytometry analysis was performed using an Epics Elite flow cytometer (Beckman Coulter, Fullerton, CA, USA). HPLC analysis was accomplished using a Hewlett Packard 1050 chromatography system (Agilent, Palo Alto, CA, USA). A Hewlett Packard 1046 programmable fluorescence detector (Agilent, Palo Alto, CA, USA) was used for fluorescence detection of HPLC eluents. Quantitative RT-PCR analysis was performed using a Bio-Rad ICycler and My iQ detector (Bio-Rad, Hercules, CA, USA).
Cell Culture
MEF cells derived from WT and N0 mice (ICR background) were cultured in Iscove’s modified Dulbecco’s medium containing 10% fetal bovine serum and antibiotics (IMDM)[22]. Unless indicated otherwise, N0 and WT cells were plated in 6 cm dishes (5×105 cells/dish) and incubated overnight prior to DQ exposures.
Cell Viability
MEF cells were plated in 96-well plates (5×103 cells/well) and incubated overnight. The media was replaced with IMDM containing phosphate buffered saline (PBS) or various concentrations of DQ (1.6 – 500 μM) and incubated for 24-hr at 37ºC, 5% CO2. Cell viability was assessed by the MTT reduction assay as previously described[23]. Briefly, MTT in PBS was added to each well (final concentration = 0.5 mg/ml) followed by a 3-hr incubation. The absorbance of the resulting MTT formazan, dissolved in DMSO, was measured at 540 nm.
Oligonucleotide Microarray
Total RNA, from WT and N0 cells, was purified using the RNeasy Mini kit (Qiagen, Valencia, CA) after isolation using TRIzol reagent (Life Technologies, Inc., Grand Island, NY). Murine Genome MOE 430A GeneChip arrays (Affymetrix, Santa Clara, CA), which contain probes for detecting ~14,500 well-characterized genes and 4371 expressed sequence tags (ESTs), were used to probe the RNA obtained from the cells using the procedure below. Briefly, double-stranded cDNA was synthesized from total RNA with SuperScript Choice System (Invitrogen) by using oligo(dT)24 primers with a T7 RNA polymerase promoter site added to its 3′ end (Genset Corp., La Jolla, CA). The isolated cDNA was then labeled to generate biotinylated cRNA in vitro and amplified using the BioArray T7 RNA polymerase labeling kit (Enzo, Farmingdale, NY). After purification of the cRNA by RNeasy Mini kit, cRNA were fragmented at 94°C for 35 min. cRNA was used in a hybridization mixture containing herring sperm DNA (0.1 mg/ml; Promega Corp., Madison, WI), plus bacterial and phage cRNA controls (1.5 pmol of BioB, 5 pmol of BioC, 25 pmol of BioD, and 100 pmol of Cre) to serve as internal controls for hybridization efficiency as directed by the manufacturer (Affymetrix). Aliquots of the mixture were hybridized onto the array for 18 h at 45°C. Each array was washed, stained with streptavidin-phycoerythrin (Molecular Probes, Eugene, OR), amplified with biotinylated anti-streptavidin antibody (Vector Laboratory, Burlingame, CA) and scanned to obtain image and signal intensities.
Scanned output files were analyzed using Affymetrix GCOS (GeneChip Operating Software) and independently normalized to an average intensity of 500. Further analyses were done as described previously[16] by performing 9 pair-wise comparisons for each group (WT, n = 3 vs. N0, n = 3). Only those altered genes that appeared in at least 6 of the 9 comparisons were selected for further analyses to limit the number of false positives. Mann-Whitney pair-wise comparison test in Data Mining Tool was used to rank the results based on the P value of each identified change in transcript level. Multiple probes matched to the same gene were removed after retaining the probe set that exhibited the highest magnitude fold-change.
Mitochondrial ROS production
Luminol chemiluminescence of mitochondria isolated from WT and N0 MEF cells exposed to 50 μM DQ was monitored as previously described[24]. Briefly, confluent WT and N0 MEF cells were harvested by scraping into PBS and homogenized using a dounce homogenizer. The lysate was centrifuged (1,000 × g, 10-min) and the resultant supernatant was centrifuged (9,000 × g, 7-min). The pellet was washed with PBS and centrifuged (9,000 × g, 7-min). The pellet was resuspended in respiratory control buffer (RCB: 70 mM sucrose, 220 mM mannitol, 2 mM HEPES, 2.5 mM KH2PO4, 0.5 mM EDTA, 2.5 mM MgCl2 and 0.1% (w/v) BSA). Chemiluminescence from a constant volume of isolated mitochondria suspended in 5 μg/ml horseradish peroxidase, 5 μM luminol, 2.5 mM succinate and RCB with and without 50μ M DQ was monitored at 37°C for 40-min. Protein concentrations were determined using the Bradford protein assay (Bio-Rad, CA, USA).
Intracellular ROS detection
Intracellular ROS generation was assessed by flow cytometry detection of CM-DCF-DA fluorescence in viable cells[25]. WT and N0 MEF cells were exposed to 50 μM DQ for 13-hr or 30-min, followed by a 30-min incubation with IMDM containing 50 μM DQ and 10 μM CM-DCF-DA. The cells were harvested by trypsinization. The harvested cells were washed with PBS and resuspended in PBS containing 5 μg/ml propidium iodide (PI). The mean fluorescence intensity of viable cells (PI-negative cells) was measured using an excitation wavelength of 488 nm and emission wavelengths of 525 nm (CM-DCF-DA) and 675 nm (PI).
Aconitase activity
MEF cells were exposed to 50 μM DQ for 24-hr and harvested by scraping into PBS containing Complete protease inhibitor cocktail. Aconitase activity of sonicated cell lysates was determined as previously described with some modifications[26]. Briefly, sonicated cell lysate was added to reaction buffer (50 mM Tris-HCl pH 7.4, 5 mM sodium citrate, 0.6 mM MnCl2, 0.2 mM NADP+ and1 U/ml isocitrate dehydrogenase (NADP+-dependent)). The generation of NADPH, following a 10-min. incubation period at room temperature was measured by monitoring absorbance at 340 nm and activity was determined using a NADPH extinction coefficient of 6.2 mM−1 cm−1. Protein concentrations were determined using the Bradford protein assay (Bio-Rad, CA, USA).
Lipid Peroxidation
MEF cells were exposed to 50 μM DQ for 24-hr, and harvested by scraping into PBS containing 0.5% butylated hydroxytoluene (BHT). Lipid peroxidation of sonicated cell lysates was assessed by HPLC analysis of thiobarbituric acid substances (TBARS) as previously described with some modifications[27]. Briefly, equal volumes of cell lysate and trichloroacetic acid (20%) were mixed. Four volumes of 0.67% (w/v) thiobarbituric acid were added and this solution was incubated at 95ºC for 45-min. TBARS were extracted with n-butanol and the organic layer was injected on a Supelcosil LC-18 HPLC column (4.6 × 250 mm, Supelco, PA, USA) with a mobile phase consisting of 50 mM potassium phosphate pH 6.4, 52.5% methanol and a flow rate of 1 ml/min. TBARS were detected by fluorescence (ex. 515 nm, em. 553) and quantified using a standard curve of 1,1,2,2-tetraethoxypropane dissolved in PBS containing 0.5% BHT.
Glutathione measurements
The concentration of reduced glutathione (GSH) and oxidized glutathione (GSSG) was determined as previous described[28]. Briefly, MEF cells were exposed to 50 μM DQ for 24-hr and harvested by scraping into 5% perchloric acid, 0.2 M boric acid and 10 μM γ-EE. The lysate was centrifuged (12,000 × g, 10 min) and the pellet was resuspended in 1 M potassium hydroxide and protein concentrations were determined using the Bradford protein assay (Bio-Rad, CA, USA). Iodoacetic acid (14.8 mg/ml) was added to the supernatant. The pH of the supernatant was adjusted to 9.0 and samples were incubated for 20-min. at room temperature. Dansyl chloride was added to the supernatant and incubated overnight in the dark. Dansylated GSH, GSSG and γ-EE were extracted with chloroform and the organic layer was injected on a Hypersil aminopropyl column (4.6 × 250 mm, Thermo, NJ, USA). The dansylated eluents were detected by fluorescence (ex. 335 nm, em. 515 nm). GSH and GSSG were quantified based on the peak area of the γ-EE internal standard.
ARE-luciferase reporter gene activity
MEF cells, seeded at a density of 6 × 105 in a 10 cm culture dish, were transfected with 5.0 μg of pRBGP2[29] and 0.5 μg of pRLTK-ΔARE, a RLTK plasmid in which the ARE was deleted, using GeneJuice transfection reagent. Briefly, media was replaced with serum-free media and plasmids incubated with serum-free media containing GeneJuice was added to each dish. Cells were incubated for 4-hr followed by addition of a 1:1 volume of media containing 20% serum and incubated for 16-hr. The media was then replaced with 10% serum media and incubated overnight. The cells were trypsinized and seeded into 6-well culture plates at a density of 1.5 × 105 and incubated overnight. The media was replaced with media containing 50 μM DQ, 10 μM D3T or PBS and incubated for 13-hr. Cells were harvested and luciferase activity measurements were performed using the Promega Dual Luciferase Reporter Assay System (Promega, Madison, WI, USA) according to the manufacturer’s directions.
Quantitative RT-PCR analysis
MEF cells were exposed to 50 μM DQ for 13-hr and total RNA was extracted using VERSAGENE RNA purification columns (Gentra, Minneapolis, MN, USA) according to the manufacturer’s directions. 1 μg of cDNA was synthesized from 1 μg of total RNA using the iScript cDNA synthesis kit according to the manufacturer’s directions. Quantitative RT-PCR was performed using iQ SYBR green supermix. Primers for genes encoding Gclc (5′-GCACGGCATCCTCCAGTTCCT, 3′-TCGGATGGTTGGGGTTTG-TCC)[30], Gclm (5′-ACCTGGCCTCCTGCTGTGTG, 3′-GGTCGGTGAGCTGTGGG-TGT) [31], Gsr (5′-CAGGGATGGTGACTTAGCCTCAGCAACC, 3′-CAGRGAGCCCCA-CGGTCCCAATAGGG), Prdx1 (5′-CTGCCAAGTGATTGGCGCTTCTGTGGATTCTC, 3-TCCAGCCAGCTGGACACACTTCACC) and hypoxanthine guanine phosphoribosyl transferase (Hprt) (5′-GCTGGTGAAAAGGACCTCTC, 3′-CACAGGACTAGAACACCTGC) were synthesized and used for specific gene expression analysis. The specificity of the amplification products was confirmed using melting curve analysis. A standard curve consisting of serial dilutions of cDNA was performed with each analysis in order to calculate PCR efficiency and relative quantification was calculated using the method of Pfaffl[32].
Statistical analysis
Data are expressed as mean ± SE from at least three independent experiments. Unless stated otherwise, Student’s t test, for experiments with two groups, and ANOVA with Student-Neuman Kuels analysis, for experiments with three or more groups, was used to evaluate statistically significant differences.
Results
Cell Viability
Cell viability following a 24-hr exposure to increasing doses of DQ was assessed in order to discern whether Nrf2 genotype affects sensitivity to DQ exposure. N0 cells displayed a 4.5-fold increased sensitivity (IC50, μM) to DQ (Figure 1), compared to WT cells (N0, 37.9 ± 4.6; WT, 179.5 ± 17.5; p < 0.01). This finding indicated that N0 cells are more susceptible to toxicity of superoxide anion and warranted further analysis of the basal and adaptive antioxidative capacities of these cells, as well as the extent of oxidative stress in these cells following DQ exposure.
Figure 1.
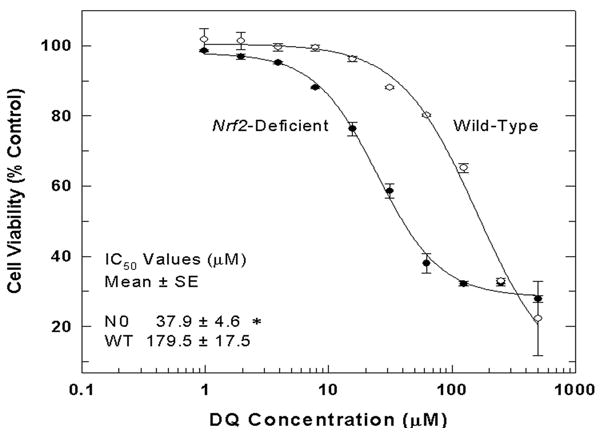
N0 cells display increased sensitivity to superoxide anion following exposure to DQ. WT and N0 cells were treated with increasing doses of DQ for 24-hr and cell viability was assessed by the MTT reduction assay. Mean cell viability measurements are expressed as the percentage of vehicle-treated control. Values represent the mean ± SE of 4 independent experiments. * p < 0.01 compared to WT.
Constitutive gene expression in WT and N0 cells
Microarray analysis of constitutive gene expression was performed in order to assess whether N0 cells exhibited altered expression of antioxidative genes. As a whole, WT cells were shown to have modestly increased expression (fold-change, WT/N0) of a few antioxidant genes (Figure 2) including glutathione peroxidase 4 (gpx4, 2.0), peroxiredoxin 3 (Prdx3, 2.2), thioredoxin 2 (Txn2, 1.7), and ferritin light chain subunit 1 (Ftl1, 1.6). Additionally, modest increases in the expression of genes responsible for the production of GSH and reducing equivalents (Figure 2) including glutamate cysteine ligase, catalytic subunit (Gclc, 2.6), malic enzyme (Mod1, 1.9), and 3-phosphoglucuronate dehydrogenase (Pgd, 1.7) were demonstrated in WT cells, relative to N0 cells. The basal expression in WT cells of biliverdin reductase B (Blvrb, 5.3) and heme oxygenase 1 (Hmox1, 2.0), enzymes contributing to the production of bilirubin, and glutathione-S-transferase α4 (Gsta4, 719), an enzyme responsible for the conjugation of 4-hydroxynonenal, was substantially higher than N0 cells (Figure 2). These findings of limited differences in gene expression suggested that the constitutive antioxidative capacity of WT cells was not likely to be substantially different from that of N0 cells.
Figure 2.
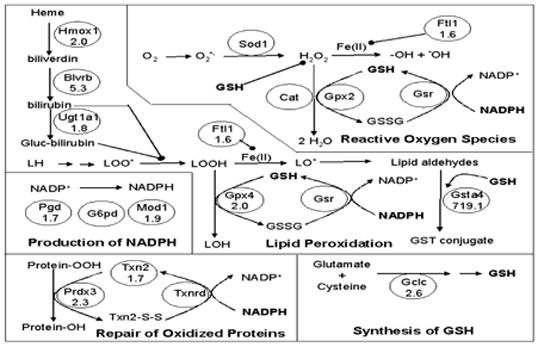
Functional representation of constitutive expression of antioxidative genes and genes responsible for the production of reducing equivalents in WT and N0 cells. Microarray analysis of mRNA expression was performed on WT and N0 cells. White ovals with no fold-change represent genes that have been previously reported to be regulated by Nrf2 in vivo, but for which no differential expression was detected in the present study. White ovals with a fold-change reported represent genes that have been previously reported to be regulated by Nrf2 and displayed statistically significant increased expression in WT cells compared to N0 cells. Grey ovals with a fold change reported represent genes that have not previously been reported to be regulated by Nrf2 and displayed statistically significant increased expression in WT cells compared to N0 cells. Mean fold-change values are expressed as the ratio of expression in WT cells relative to N0 cells. Only statistically significant differences (p<0.05) are reported.
ROS analysis
Since N0 cells exhibit markedly increased sensitivity to DQ, the mitochondrial capacity to generate ROS during DQ exposure was assessed in order to rule out inherent differences in the cellular capacity to generate ROS as an explanation for the differential cytotoxicity. Isolated mitochondria from both WT cells (6.6 ± 2.1 × 109) and N0 cells (10.0 ± 2.1 × 109) produced equivalent luminol chemiluminescence (AUC/mg protein) during exposure to vehicle (Figure 3, p = 0.2). Increased chemiluminescence was observed in isolated mitochondria from both WT cells (21.8 ± 5.8 × 109, p < 0.05) and N0 cells (25.8 ± 6.9 × 109, p < 0.05) during exposure to 50 μM DQ (Figure 3). The amount of chemiluminescence during DQ treatment was equivalent in isolated mitochondria prepared from WT and NO cells (p=0.6). These data indicated that mitochondria from WT and N0 cells possessed similar capacity to generate ROS from DQ and therefore, differential ROS flux is not a primary determinant for the differential sensitivity to DQ.
Figure 3.
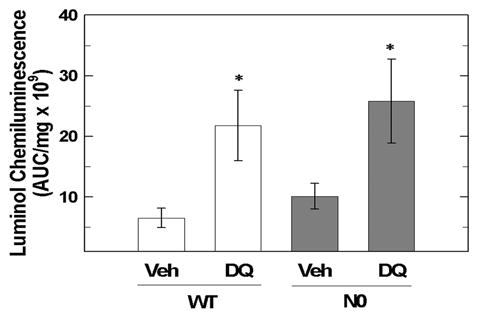
Isolated mitochondria from WT and N0 cells possess similar capacity to generate ROS. Mitochondria isolated from WT and N0 cells were exposed to 50 μM DQ or vehicle for 40-min and luminol chemiluminescence monitored. Mean area under the curve (AUC) readings were normalized to the protein content of the isolated mitochondria. Values represent the mean ± SE of 4 independent experiments. * p < 0.05, compared to respective vehicle treated group.
The intracellular levels of ROS at 30-min and 13-hr of DQ treatment were measured using flow cytometric analysis of DCF fluorescence in order to ascertain whether ROS levels changed over increasing time of DQ exposure. No difference in DCF fluorescence (mean fluorescence intensity) at 30-min and 13-hr of exposure to vehicle between WT (3.5 ± 0.3 and 3.6 ± 0.1, respectively) and N0 cells (4.4 ± 1.3 and 5.0 ± 0.7, respectively) was observed, supporting the view that basal antioxidative capacity does not differ by genotype(Figure 4). Similarly, exposure to DQ for 30 minutes did not result in increased ROS in either WT cells (3.5 ± 0.6) or N0 cells (4.4 ± 0.7). However, DCF-fluorescence was substantially increased in N0 cells (8.1 ± 0.8, p<0.05), but not WT cells (3.7 ± 0.1), at 13-hr of exposure to 50 μM DQ (Figure 4). The DCF fluorescence data indicated that both basal levels of ROS and ROS levels at 30-min of DQ treatment were not different between WT and N0 cells. However, the concentration of ROS increased in N0 but not WT cells with increasing time of DQ exposure.
Figure 4.
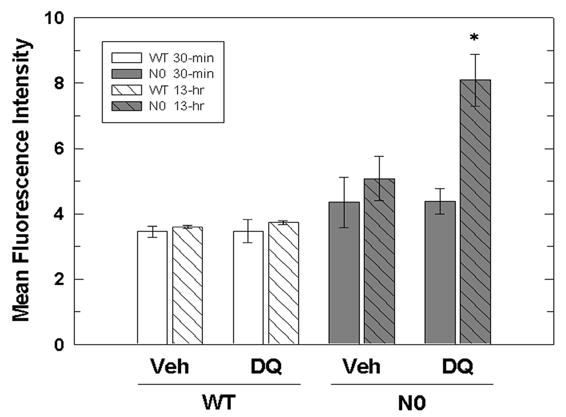
Intracellular ROS levels in N0 cells increase with duration of DQ treatment. WT and NO cells were treated with 50 μM DQ or vehicle for 30-min (solid) and 13-hr (hatched). The media was replaced with media containing 50 μM DQ and 10 μM CM-DCFDA and the cells were incubated for 30 min. DCF fluorescence of viable cells was determined by flow cytometry. The data are expressed as mean fluorescence intensity of viable cells. Values represent the mean ± SE of three independent experiments. * p < 0.05 compared to WT and N0 vehicle-treated groups.
Biomarkers of oxidative stress
Aconitase activity was measured as a biomarker of protein oxidation because aconitase requires ferric iron (Fe(III)) for enzymatic activity. Upon exposure to superoxide anion, the iron is reduced to ferrous iron (Fe(II)) resulting in release of Fe(II) and loss of enzymatic activity. The basal aconitase specific activity (mU/mg) in WT cells (1.4 ± 0.1) and N0 cells (1.7 ± 0.4) was not different (Figure 5a). However, exposure to 50 μM DQ for 24 hours resulted in decreased aconitase specific activity in N0 cells (0.8 ± 0.1, p < 0.05) but not WT cells (1.2 ± 0.02, p = 0.5). These results indicated that protein oxidation increased in N0 but not WT cells following DQ exposure.
Figure 5.
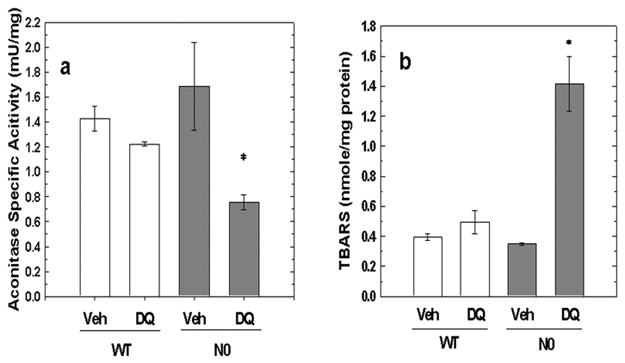
Biomarker analyses of WT and N0 cells following DQ treatment. WT and N0 cells were treated with 50 μM DQ or vehicle for 24-hr. A. Aconitase activity in WT and N0 cells. The aconitase specific activity of sonicated cell lysates was determined. Aconitase specific activity is expressed as mU activity/mg protein. Values represent the mean ± SE of 3 independent experiments. * p < 0.05, compared to N0 vehicle-treated group. B. Lipid peroxidation in WT and N0 cells. The concentration of TBARS in sonicated cell lysates was determined. TBARS are expressed as nmole/mg protein. Values represent the mean ± SE of 3 independent experiments. * p < 0.05, compared to all groups.
TBARS were measured as a biomarker of lipid oxidation. The basal levels of TBARS (nmole/mg protein) in WT cells (0.39 ± 0.02) and N0 cells (0.35 ± 0.01) were similar (Figure 5b). Exposure to 50 μM DQ for 24 hours resulted in increased TBARS in N0 cells (1.41 ± 0.18, p < 0.01) but not WT cells (0.50 ± 0.08). These findings indicated that although basal levels of lipid peroxidation in WT and N0 cells were not different, DQ exposure resulted in substantial lipid peroxidation in N0 but not WT cells.
Glutathione concentrations
The levels of GSH, GSSG and total glutathione before and after exposure to DQ were measured in order to assess whether increased GSH oxidation, increased GSSG reduction or increased de novo synthesis of GSH occurred following exposure to DQ. No differences in the basal concentrations (nmole/mg protein) of GSH (WT, 34.8 ± 1.3; N0, 38.4 ± 1.9), GSSG (WT, 2.0 ± 0.1; N0, 2.5 ± 0.3) and total glutathione (WT, 39.3 ± 1.2; N0, 43.5 ± 2.3) as well as basal GSH oxidation ([GSH]/[GSSG]; WT, 17.7 ± 1.0; N0, 15.9 ± 1.4) were detected (Table 1). However, exposure to 50 μM DQ for 24-hours resulted in a 50% increase in GSH content (Table 1) in WT cells (52.7 ± 2.5, p<0.05) and a 40% decrease in GSH content in N0 cells (24.4 ± 3.8, p<0.05). The concentration of GSSG in WT cells (3.6 ± 0.5, p=0.4) was not affected by DQ treatment while the GSSG concentration in N0 cells (7.4 ± 1.5, p<0.05) increased 3-fold following DQ exposure as reflected in a lower GSH/GSSG ratio in N0 cells (4.1 ± 1.0, p < 0.05) but not WT cells (15.5 ± 1.4) following a 24-hr DQ exposure (Table 1). Also, total glutathione levels increased in WT cells (59.9 ± 3.4, p<0.05) but not N0 cells (39.2 ± 4.9, p = 0.6) following DQ treatment (Table 1). The increased total glutathione was witnessed in WT cells as early as 13-hr of DQ treatment while no increase or decrease was seen in N0 cells at any time of DQ treatment (data not shown). These findings indicated that significant GSH oxidation occurred in N0 but not WT cells following DQ exposure while reduction of GSSG and de novo synthesis of GSH occurred in WT but not N0 cells following DQ exposure.
Table 1.
GSH, GSSG and total glutathione concentrations following DQ treatmenta.
| Genotype | Treatment | [GSH] | [GSSG] | [GSH]/[GSSG] | Total Glutathione |
|---|---|---|---|---|---|
| WT | Veh | 34.8 ± 1.3 | 2.0 ± 0.1 | 17.7 ± 1.0 | 39.3 ± 1.2 |
| DQ | 52.7 ± 2.5 * | 3.6 ± 0.5 | 15.5 ± 1.4 | 59.9 ± 3.4 * | |
| N0 | Veh | 38.4 ± 1.9 | 2.5 ± 0.3 | 15.9 ± 1.4 | 43.5 ± 2.3 |
| DQ | 24.4 ± 3.8 * | 7.4 ± 1.5 * | 4.1 ± 1.0 * | 39.2 ± 4.9 |
WT and NO cells were treated with 50 μM DQ or vehicle for 24-hr. Concentrations are expressed as nmole/mg protein. Values represent the mean ± SE of 6 independent experiments.
p < 0.05, compared to respective vehicle-treated group.
Effects of DQ on Gene Expression
Activation of an ARE-containing luciferase reporter gene in WT and N0 cells was assessed in order to demonstrate that exposure to DQ-generated superoxide anion results in activation of Nrf2 enhanced transcription of ARE-containing genes and consequently, cytoprotection. Exposure to 50 μM DQ resulted in a 2.5-fold increase in luciferase activity, relative to vehicle-treatment (p < 0.05, Figure 6), in WT but not N0 cells. Similarly, exposure to D3T, serving as a positive control, resulted in 4.5-fold increase in luciferase activity (p < 0.05) in WT but not N0 cells. These results indicated that exposure to DQ-generated superoxide anion results in increased Nrf2 transcriptional activation of ARE-containing genes.
Figure 6.
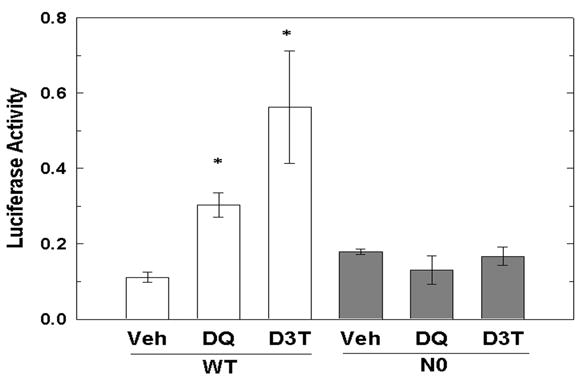
ARE-luciferase reporter gene transcription increases with DQ treatment in WT but not N0 cells. WT and N0 cells were transfected with pRBGP2 and pRLTK-ΔARE, exposed to PBS, 50 μM DQ or 10 μM D3T for 13-hr and luciferase activities were determined. Luciferase activity is expressed as firefly luciferase activity normalized to renilla luciferase activity. Values represent mean ± SE of 3 independent experiments. * p < 0.05, compared to vehicle-treated group.
Quantitative RT-PCR analysis of gene expression following exposure of WT and N0 cells to DQ was performed in order to confirm the luciferase reporter gene activity data and to demonstrate increased transcription of Nrf2-regulated genes following DQ exposure. DQ exposure resulted in a greater fold-induction (treated/vehicle) of the expression of Gclc (WT, 2.1 ± 0.3; N0, 0.7 ± 0.2, p < 0.05), Gclm (WT, 4.1 ± 0.3; N0 1.4 ± 0.4, p < 0.05), Gsr1 (WT, 2.1 ± 0.3; N0 1.0 ± 0.1, p < 0.05) and Prdx1 (WT, 2.3 ± 0.2; N0 1.0 ± 0.1, p < 0.05) in WT cells than N0 cells (Table 2). These findings indicated that exposure to DQ-generated superoxide anion resulted in increased expression of Nrf2-regulated genes in WT but not N0 cells.
Table 2.
Induction of selected Nrf2-regulated genes following DQ treatmenta.
| Gene | WT | N0 |
|---|---|---|
| Gclc | 2.1 ± 0.3 * | 0.7 ± 0.2 |
| Gclm | 4.1 ± 0.3 * | 1.4 ± 0.4 |
| Gsr1 | 2.1 ± 0.3 * | 1.0 ± 0.1 |
| Prdx1 | 2.3 ± 0.2 * | 1.0 ± 0.1 |
WT and NO cells were treated with 50 μM DQ or vehicle for 13-hr. Data are expressed as fold-induction of gene transcripts (treated/vehicle). Values represent the mean ± SE of 3 independent experiments.
p < 0.05, compared to WT.
Discussion
Our study clearly illustrated that Nrf2-deficiency resulted in increased cytotoxicity following exposure to DQ-generated superoxide anion. N0 MEF cells were approximately 5-fold more sensitive to cell death than WT MEF cells following DQ exposure. The sensitivity of N0 MEF cells to DQ-generated superoxide anion was greater than the previously reported sensitivity of cardiac fibroblasts exposed to ROS, using a xanthine/xanthine oxidase (X/XO) system. Zhu, et al.[33] demonstrated that cardiac fibroblasts isolated from N0 mice were 1.7-fold more sensitive to ROS generated from X/XO, compared to cardiac fibroblasts from WT mice. The increased cytotoxicity seen in our study using DQ to generate superoxide anion illustrated the value of using an intracellular generator of superoxide anion since extracellular generation or delivery of ROS may be subject to extracellular defenses or spurious interactions with components of the culture media. Differences in duration of superoxide anion flux may also account for the differential effects of the two superoxide generating systems.
A possible simple explanation for the increased sensitivity of N0 cells was that N0 mitochondria, a target organelle of DQ[34], possessed an enhanced capacity to generate superoxide. Therefore, it was important to note that isolated mitochondria from both WT and N0 cells produced equivalent amounts of ROS during DQ exposure, thereby eliminating differential inherent DQ metabolic capacities as an explanation for the increased cytotoxicity of N0 cells.
Differential basal antioxidative capacity between WT and N0 cells or the ability of WT, but not N0 MEF cells to mount an adaptive response to DQ-mediated superoxide anion represented two alternative explanations for the increased cytotoxicity of N0 cells following exposure to DQ-generated superoxide anion. We characterized both constitutive gene expression and basal oxidative stress in WT and N0 cells to determine whether Nrf2-deficiency resulted in a reduced basal antioxidative capacity. Our study revealed that the expression of genes responsible for the direct metabolism of ROS was not decreased in N0 cells. On the other hand, N0 cells displayed modestly lower expression of genes responsible for lipid peroxidation defense, repair of oxidized protein, and NADPH and GSH synthesis. However, these differences in constitutive gene expression did not result in altered levels of oxidative stress biomarkers in quiescent N0 cells. Basal levels of ROS, lipid peroxidation, and GSH oxidation were similar in WT and N0 cells. Interestingly, both genotypes had similar total glutathione content revealing that Nrf2 does not play a substantive role in regulating constitutive GSH synthesis in these cells. This finding contradicted a previous study that demonstrated that Nrf2-deficient primary fibroblasts exhibited decreased basal total glutathione content[35]. However, the primary fibroblasts used in that study were derived from C57BL6 mice while our MEF cells were derived from an ICR background. It was possible that this discrepancy may be due to strain difference, the use of different methodologies to detect glutathione or adaptation of our MEF cells to long-term culture. Overall, our results excluded differential basal antioxidative capacity as a primary determinant for the increased sensitivity of N0 cells to DQ treatment.
Since Nrf2 genotype did not alter basal oxidative stress, our results indicated that an adaptive response in WT, but not N0 cells was responsible for decreased cytotoxicity of WT cells following exposure to DQ-generated superoxide anion. Exposure to DQ resulted in greater loss of aconitase activity, increased lipid peroxidation, and increased GSH oxidation in N0 but not WT cells. Also, increased ROS levels were detected in N0 cells at 13-hr, but not 30-min of DQ treatment. These results suggested that the antioxidative capacity of N0 cells diminished over time while the antioxidant capacity of WT cells increased with increasing duration of exposure to DQ-generated ROS, representing an adaptive response in WT, but not N0 cells.
As previously mentioned, a number of Nrf2-inducible enzymes responsible for the metabolism of ROS have been identified. The absence of increased ROS in WT cells at 13-hr of DQ exposure suggested that increased expression of these enzymes occurred, allowing for increased detoxication of ROS in WT, but not N0 MEF cells. Additionally, the concentration of total glutathione was increased in WT, but not N0 cells following DQ exposure. It has been well documented that the expression of Gclc, a gene responsible for the catalysis of the rate limiting step in GSH synthesis, increases following exposure to Nrf2-activating agents[36]. In fact, Wakabayashi, et al.[22] reported that administration of sulforaphane, an activator of Nrf2 signaling, resulted in a 50% increase in total GSH in WT but not N0 MEF cells. Therefore, our data suggested that DQ-generated ROS acted as an Nrf2-activating agent and induced an adaptive response leading to increased de novo synthesis of GSH.
A number of studies have used inducers of the Nrf2 pathway to ameliorate toxicity mediated exposure to oxidative stressors. For instance, Shih, et al.[37] demonstrated that Nrf2 pathway activation by tert-butylhydroquinone resulted in decreased brain toxicity in WT, but not N0 mice, following to exposure to ROS generated during 3-nitroproprionic acid-induced mitochondrial uncoupling. Also, tert-butylhydroperoxide-induced oxidative stress was repressed by triterpenoid activation of Nrf2 signaling in WT, but not N0 MEF cells[38]. Our data suggested that reactive oxygen species produced by DQ redox-cycling acted as an Nrf2-inducing agent resulting in an adaptive response in WT MEF cells only.
DQ-generated superoxide anion resulted in increased expression of an ARE-luciferase reporter gene as well as endogenous target genes, in an Nrf2-dependent manner. Increased expression of GSH synthesis genes, Gclc and Gclm, as well as antioxidative genes, Gsr1 and Prdx1, was demonstrated in WT compared to N0 cells following a 13-hr DQ exposure. Increased expression of Gclc, Gclm and Gsr1 represented a plausible mechanism for the increased GSH and low GSSG concentrations demonstrated in WT cells following DQ exposure. Similarly, a lack of induction of these genes may account for the increased cytotoxicity of DQ demonstrated in N0 cells. It is important to note that this adaptive response, in WT cells, occurred at DQ concentrations in which minimal toxicity (> 85% cell viability) was witnessed illustrating the importance of Nrf2 in maintaining cellular oxidative homeostasis under conditions of mild oxidative stress. Taken together, our findings clearly indicated that exposure to DQ-generated ROS, in itself, leads to activation of Nrf2 and increased transcription of Nrf2-regulated genes representing an Nrf2-dependent adaptive response protecting against toxicity of superoxide anion and its products.
DQ-generated superoxide anion is readily metabolized in cells to hydrogen peroxide by enzymatic dismutation, via Cu/Zn or Mg superoxide dismutase, or non-enzymatic disproportionation of two superoxide anion molecules. Two possible explanations exist for the mechanism of H2O2-mediated Nrf2 activation including direct reaction of H2O2 with Keap1 or activation of kinase signaling pathways by H2O2. It has been proposed that H2O2 may directly react with reactive cysteine residues in Keap1 resulting in release of Nrf2 since H2O2 has high reactivity to protein thiol groups and this type of regulation of thiol-containing proteins is seen in prokaryotes[39]. Several oxidative stress responsive signaling pathways have been shown to positively regulate Nrf2 resulting in activation of Nrf2. Huang, et al.[40] demonstrated that protein kinase C can specifically phosphorylate Nrf2 resulting in release of Nrf2 from Keap1. Also, activation of phophsphatidylinositol 3-kinase (PI3K) by tert-butylhydroquinone-induced oxidative stress resulted in activation of Nrf2 following depolymerization of actin filaments[41]. Additionally, Nrf2 activation was repressed by PI3K inhibitors[41]. Activation of Nrf2 by these mechanisms may be responsible for the adaptive response seen in WT, but not N0 MEF cells following DQ treatment.
Oxidative stress has been implicated in the etiology of a variety of conditions including inflammatory diseases, neurodegenerative conditions, cancer and following exposure to xenobiotics[42–44]. Although several redox-sensitive transcription factors, such as nuclear factor-κB and activating protein 1, influence the development of oxidative diseases[45, 46], our study clearly illustrated the role of Nrf2 in regulating an adaptive response leading to protection against ROS. The results of this and other studies demonstrate that an Nrf2-mediated adaptive antioxidative response represents an effective innate mechanism protecting organisms against oxidative stress. Therefore, it is likely that the Nrf2-regulated signaling pathway serves as a powerful molecular target for the prevention of oxidative diseases.
Acknowledgments
This work was supported by the following grants: NIEHS training grant, T32ES07141, RO1 CA94076, P50 CA058184, HL081205, NIEHS center grant P30 ES038819, The Multi-Center AIDS Cohort Study (MACS): U01-AI35042 and The Center For AIDS Research (CFAR): I1P30-AI42855-1042.
References
- 1.Enomoto A, Itoh K, Nagayoshi E, Haruta J, Kimura T, O'Connor T, Harada T, Yamamoto M. Toxicol Sci. 2001;59:169–177. doi: 10.1093/toxsci/59.1.169. [DOI] [PubMed] [Google Scholar]
- 2.Chan K, Kan YW. Proc Natl Acad Sci U S A. 1999;96:12731–12736. doi: 10.1073/pnas.96.22.12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iida K, Itoh K, Kumagai Y, Oyasu R, Hattori K, Kawai K, Shimazui T, Akaza H, Yamamoto M. Cancer Res. 2004;64:6424–6431. doi: 10.1158/0008-5472.CAN-04-1906. [DOI] [PubMed] [Google Scholar]
- 4.Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K, Yamamoto M, Talalay P, Kensler TW. Proc Natl Acad Sci U S A. 2001;98:3410–3415. doi: 10.1073/pnas.051618798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cho HY, Jedlicka AE, Reddy SP, Kensler TW, Yamamoto M, Zhang LY, Kleeberger SR. Am J Respir Cell Mol Biol. 2002;26:175–182. doi: 10.1165/ajrcmb.26.2.4501. [DOI] [PubMed] [Google Scholar]
- 6.Rangasamy T, Cho CY, Thimmulappa RK, Zhen L, Srisuma SS, Kensler TW, Yamamoto M, Petrache I, Tuder RM, Biswal S. J Clin Invest. 2004;114:1248–1259. doi: 10.1172/JCI21146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aoki Y, Sato H, Nishimura N, Takahashi S, Itoh K, Yamamoto M. Toxicol Appl Pharmacol. 2001;173:154–160. doi: 10.1006/taap.2001.9176. [DOI] [PubMed] [Google Scholar]
- 8.Rangasamy T, Guo J, Mitzner W, Roman J, Singh A, Fryer A, Yamamoto M, Kensler T, Tuder R, Georas S, Biswal S. J Exp Med. 2005;202:47–59. doi: 10.1084/jem.20050538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, Biswal S. J Clin Invest. 2006;116 doi: 10.1172/JCI25790. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu X, Kensler T. Mutat Res. 2005;591:93–102. doi: 10.1016/j.mrfmmm.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 11.Furukawa M, Xiong Y. Mol Cell Biol. 2005;25:162–171. doi: 10.1128/MCB.25.1.162-171.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dinkova-Kostova AT, Holtzclaw WD, Kensler TW. Chem Res Toxicol. 2005;18:1779–1791. doi: 10.1021/tx050217c. [DOI] [PubMed] [Google Scholar]
- 13.Kobayashi M, Yamamoto M. Antioxid Redox Signal. 2005;7:385–394. doi: 10.1089/ars.2005.7.385. [DOI] [PubMed] [Google Scholar]
- 14.Motohashi H, Katsuoka F, Engel JD, Yamamoto M. Proc Natl Acad Sci U S A. 2004;101:6379–6384. doi: 10.1073/pnas.0305902101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwak MK, Wakabayashi N, Itoh K, Motohashi H, Yamamoto M, Kensler TW. J Biol Chem. 2003;278:8135–8145. doi: 10.1074/jbc.M211898200. [DOI] [PubMed] [Google Scholar]
- 16.Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, Biswal S. Cancer Res. 2002;62:5196–5203. [PubMed] [Google Scholar]
- 17.Lee JM, Shih AY, Murphy TH, Johnson JA. J Biol Chem. 2003;278:37948–37956. doi: 10.1074/jbc.M305204200. [DOI] [PubMed] [Google Scholar]
- 18.Kwak MR-G, Wakabayashi MKN, Kensler TW. Methods Enzymol. 2004;382:414–423. doi: 10.1016/S0076-6879(04)82022-6. [DOI] [PubMed] [Google Scholar]
- 19.Fahey ATD-K, Stephenson JWKK, Talalay P. Methods Enzymol. 2004;382:243–258. doi: 10.1016/S0076-6879(04)82014-7. [DOI] [PubMed] [Google Scholar]
- 20.Bonneh-Barkay D, Reaney SH, Langston WJ, Di Monte DA. Brain Res Mol Brain Res. 2005;134:52–56. doi: 10.1016/j.molbrainres.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 21.Sun IAC, Lindeke YPB, Moldeus P. Pharmacol Toxicol. 1990;66 doi: 10.1111/j.1600-0773.1990.tb00768.x. [DOI] [PubMed] [Google Scholar]
- 22.Wakabayashi N, Dinkova-Kostova AT, Holtzclaw WD, Kang MI, Kobayashi A, Yamamoto M, Kensler TW, Talalay P. Proc Natl Acad Sci U S A. 2004;101:2040–2045. doi: 10.1073/pnas.0307301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Page M, Bejaoui N, Cinq-Mars B, Lemieux P. Int J Immunopharmacol. 1988;10:785–793. doi: 10.1016/0192-0561(88)90001-x. [DOI] [PubMed] [Google Scholar]
- 24.Li Y, Zhu H, Trush MA. Biochim Biophys Acta. 1999;1428:1–12. doi: 10.1016/s0304-4165(99)00040-9. [DOI] [PubMed] [Google Scholar]
- 25.Sakon S, Xue X, Takekawa M, Sasazuki T, Okazaki T, Kojima Y, Piao JH, Yagita H, Okumura K, Doi T, Nakano H. Embo J. 2003;22:3898–3909. doi: 10.1093/emboj/cdg379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gardner PR, Nguyen DD, White CW. Proc Natl Acad Sci U S A. 1994;91:12248–12252. doi: 10.1073/pnas.91.25.12248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Agarwal R, Chase SD. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;775:121–126. doi: 10.1016/s1570-0232(02)00273-8. [DOI] [PubMed] [Google Scholar]
- 28.Jones DP. Methods Enzymol. 2002;348:93–112. doi: 10.1016/s0076-6879(02)48630-2. [DOI] [PubMed] [Google Scholar]
- 29.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leung L, Kwong M, Hou S, Lee C, Chan JY. J Biol Chem. 2003;278:48021–48029. doi: 10.1074/jbc.M308439200. [DOI] [PubMed] [Google Scholar]
- 31.Johnson DA, Andrews GK, Xu W, Johnson JA. Journal of Neurochemistry. 2002;81:1233–1241. doi: 10.1046/j.1471-4159.2002.00913.x. [DOI] [PubMed] [Google Scholar]
- 32.Pfaffl M. Nucleic Acids Res. 29(9):e45. doi: 10.1093/nar/29.9.e45. 29 (2001) 2002–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu H, Itoh K, Yamamoto M, Zweier JL, Li Y. FEBS Lett. 2005;579:3029–3036. doi: 10.1016/j.febslet.2005.04.058. [DOI] [PubMed] [Google Scholar]
- 34.Wong RC, Stevens JB. J Toxicol Environ Health. 1986;18:393–407. doi: 10.1080/15287398609530880. [DOI] [PubMed] [Google Scholar]
- 35.Chan JY, Kwong M. Biochim Biophys Acta. 2000;1517:19–26. doi: 10.1016/s0167-4781(00)00238-4. [DOI] [PubMed] [Google Scholar]
- 36.McWalter GK, Higgins LG, McLellan LI, Henderson CJ, Song L, Thornalley PJ, Itoh K, Yamamoto M, Hayes JD. J Nutr. 2004;134:3499S–3506S. doi: 10.1093/jn/134.12.3499S. [DOI] [PubMed] [Google Scholar]
- 37.Shih AY, Imbeault S, Barakauskas V, Erb H, Jiang L, Li P, Murphy TH. J Biol Chem. 2005;280:22925–22936. doi: 10.1074/jbc.M414635200. [DOI] [PubMed] [Google Scholar]
- 38.Liby K, Hock T, Yore MM, Suh N, Place AE, Risingsong R, Williams CR, Royce DB, Honda T, Honda Y, Gribble GW, Hill-Kapturczak N, Agarwal A, Sporn MB. Cancer Res. 2005;65:4789–4798. doi: 10.1158/0008-5472.CAN-04-4539. [DOI] [PubMed] [Google Scholar]
- 39.Velayutham M, Villamena FA, Navamal M, Fishbein JC, Zweier JL. Chem Res Toxicol. 2005;18:970–975. doi: 10.1021/tx049687h. [DOI] [PubMed] [Google Scholar]
- 40.Huang HC, Nguyen T, Pickett CB. J Biol Chem. 2002;277:42769–42774. doi: 10.1074/jbc.M206911200. [DOI] [PubMed] [Google Scholar]
- 41.Kang KW, Lee SJ, Park JW, Kim SG. Mol Pharmacol. 2002;62:1001–1010. doi: 10.1124/mol.62.5.1001. [DOI] [PubMed] [Google Scholar]
- 42.Seril D, Liao J, Yang G, Yang C. Carcinogenesis. 2003;24:353–362. doi: 10.1093/carcin/24.3.353. [DOI] [PubMed] [Google Scholar]
- 43.Moreira PI, Smith MA, Zhu X, Nunomura A, Castellani RJ, Perry G. Ann N Y Acad Sci. 2005;1043:545–552. doi: 10.1196/annals.1333.062. [DOI] [PubMed] [Google Scholar]
- 44.Barthel A, Klotz LO. Biol Chem. 2005;386:207–216. doi: 10.1515/BC.2005.026. [DOI] [PubMed] [Google Scholar]
- 45.Liu H, Colavitti R, Rovira I, Finkel T. Circ Res. 2005;97:967–974. doi: 10.1161/01.RES.0000188210.72062.10. [DOI] [PubMed] [Google Scholar]
- 46.Na H, Surh Y. Mol Carcinog. 2006;45:368–380. doi: 10.1002/mc.20225. [DOI] [PubMed] [Google Scholar]