

Abstract
We previously showed that infection of rat oligodendrocytes by ultraviolet light-inactivated mouse hepatitis virus (MHV) resulted in apoptosis, suggesting that the apoptosis is triggered during cell entry. To further characterize the earliest apoptotic signaling events, here we treated cells with an antibody specific to the MHV receptor prior to and during virus infection or with an antibody specific to MHV spike protein following virus binding. Both treatments blocked virus infection and apoptosis, indicating that virus–receptor binding is necessary but not sufficient for the apoptosis induction. Furthermore, virus infection significantly increased the formation of the “death–receptor complexes” consisting of Fas, Fas-associated death domain and procaspase-8, but did not induce the complexes involving the tumor necrosis factor receptor and its associated death domain, demonstrating the specific activation of the Fas signaling pathway. Moreover, virus infection did not alter the abundance of the individual proteins of the complexes, suggesting that the activation of the Fas signaling pathway was at the post-translational level. Treatment with a Fas/Fc chimera, which blocks Fas-Fas ligand-mediated apoptosis, inhibited the formation of the complexes and blocked the activation of caspase-8 and apoptosis in MHV-infected cells. It also inhibited the release of cytochrome c from mitochondria and the activation of caspase-9. These results demonstrate that oligodendrocyte apoptosis is triggered by MHV infection during cell entry through the activation of the Fas signaling pathway.
Keywords: Murine coronavirus, Mouse hepatitis virus, Oligodendrocyte, Apoptosis, Signaling pathway, Fas activation
Introduction
Apoptosis is an important biologic process that regulates homeostasis, tissue development and the immune system (Nijhawan et al., 2000, Roulston et al., 1999, Wyllie, 1980). Initiation of apoptosis usually follows cascades of signaling events or apoptotic pathways upon receipt of a variety of apoptotic signals. The most common apoptotic pathway is the “death” receptor-mediated signaling pathway, which involves interactions of apoptotic factors with cell surface molecules such as Fas (CD95)/Fas-ligand (Susin et al., 1997) and tumor necrosis factor receptor (TNFR)/TNF. Activation of the Fas or TNFR signaling pathway facilitates the recruitment of adaptor proteins such as Fas-associated death domain (FADD) or TNFR-associated death domain (TRADD) proteins, which in turn recruit initiator caspases (such as caspases-8/10) to form a “death–receptor complex” or “pre-apoptosome” (Hsu et al., 1996). The procaspases in the pre-apoptosome undergo cleavage to form active caspases. Activation of initiator caspases further activates effector caspases (i.e., caspases-3/6/7), ultimately leading to apoptosis (Cohen, 1997, Hsu et al., 1996, Nijhawan et al., 2000, Strasser et al., 2000). Another well-characterized apoptotic pathway is the mitochondria-mediated pathway, which can be activated by either extracellular or intracellular death signals (Strasser et al., 2000, Vaux et al., 1988). Activation of the mitochondrial pathway triggers cytochrome c release, which then interacts with Apaf-1. The cytochrome c/Apaf-1 complex in turn activates the initiator caspase-9 and subsequently the effector caspase-3, leading to apoptosis (Cohen, 1997).
Apoptosis can be triggered by diverse intrinsic and extrinsic signals, including virus infection. Many viruses have the ability to induce apoptosis in the host cells, which is one of the cytolytic properties of viral infections. Murine coronavirus mouse hepatitis virus (MHV) is an enveloped RNA virus that can infect rodents and cause enteritis, hepatitis and central nervous system (CNS) diseases. Depending on the genetic background of the virus strains and the host, the CNS diseases caused by MHV infection can vary significantly, ranging from acute fatal encephalitis to persistent infections with or without chronic demyelination (Stohlman et al., 1999). The precise mechanism of CNS demyelination caused by MHV infection is not known, although experimental evidence strongly suggests that the immune components play an important role in initiation and progression of a robust CNS demyelination disease following MHV infection (Dandekar and Perlman, 2002, Fleming et al., 1993, Houtman and Fleming, 1996, Wang et al., 1990, Wu et al., 2000, Wu and Perlman, 1999). In addition, apoptotic oligodendrocytes have been detected in the vicinity of demyelinating lesions of MHV-infected mice and rats (Barac-Latas et al., 1997, Schwartz et al., 2002, Wu and Perlman, 1999). We previously observed that cultured rat oligodendrocytes were susceptible to MHV infection but that MHV replication was severely restricted (Liu et al., 2003). Moreover, infection of the oligodendrocytes by MHV resulted in apoptotic cell death and the apoptosis was caspase dependent (Liu et al., 2003). Subsequently, we found that the mitochondrial signaling pathway and caspase-8 were also activated by MHV infection (Liu et al., 2006). However, the upstream signal(s) that triggers the caspase-8 activation and apoptosis has not been determined.
In the current study, we further characterized the upstream apoptotic signaling pathway that is activated in the differentiated CG-4 cell line of rat oligodendrocyte by MHV infection. Our results indicate that virus–receptor binding is necessary but not sufficient for the apoptosis induction and that fusion between virus envelope and cell membranes mediated by the viral spike protein during cell entry is likely the trigger of the apoptosis. Furthermore, we found that MHV infection induced the formation of pre-apoptosomes, which consist of Fas, FADD and procaspase-8. Inhibition of the Fas signaling pathway by a soluble receptor blocked the pre-apoptosome formation, caspase-8 activation and apoptosis. Thus, our results demonstrate that MHV-induced oligodendrocyte apoptosis is mediated via the activation of the Fas signaling pathway.
Results
Virus–receptor interaction is necessary but not sufficient for the induction of oligodendrocyte apoptosis
We previously showed that UV-inactivated MHV could still induce oligodendrocyte apoptosis, indicating that the apoptosis is likely induced during cell entry (Liu et al., 2003). This finding also suggests that the activation of the apoptotic pathway is either triggered by direct interaction between virion surface proteins and the viral receptor during attachment and/or during fusion between viral envelope and cell membranes. To test these possibilities, we treated cells with a monoclonal antibody (termed CC1) specific to the MHV receptor (Dveksler et al., 1991) during the 1-h infection and then determined its effect on apoptosis induction. We first determined the virus titers following treatment of cells with the CC1 antibody. The result confirmed the effectiveness of the CC1 antibody treatment in blocking virus infection as evidenced by a decrease of approximately 4 log10 in virus titer (Fig. 1A). In a parallel experiment, when apoptosis was measured by propidium iodide (PI) staining, sub-G0/G1 cells were 19.1% and 18.3% for the live- and UV-inactivated viruses, respectively, in CC1 antibody-treated samples. These results were similar to that in CC1-treated and mock-infected cells (18.4%). However, in mock-treated and virus-infected samples, more than 60% of the cells were apoptosis-positive (Fig. 1B). Similar results were obtained from annexin V staining (Fig. 1C). These results indicate that the virus–receptor interaction is required for the induction of apoptosis.
Fig. 1.

Inhibition of MHV-induced oligodendrocyte apoptosis by treatment of cells with a neutralizing anti-MHV receptor antibody. (A) Effect of the MHV receptor antibody on virus infection. Cells were mock-treated or treated with the anti-MHV receptor antibody CC1 at 37 °C for 30 min and were infected with MHV in the presence of the CC1 antibody for 1 h. Virus titers were determined at 24 h p.i. by plaque assay and were expressed as means ± SD from three independent experiments. (B) Analysis of apoptosis by propidium iodide (PI) staining. Cells were either treated with the CC1 antibody or mock-treated as described for panel A and were infected with live- or UV-inactivated MHV. At 48 h p.i., cells were stained with PI and subjected to flow cytometric analysis. The bar in each graph represents the sub-G0/G1 population of cells (indicated as a percentage) that have the lowest intensity of PI staining. Data are representative of at least three independent experiments. (C) Analysis of apoptosis by detecting annexin V binding. The experiments were performed as in panel B, except that the detection of annexin V binding on the cell surface was carried out at 12 h p.i. with the annexin V-EGFP assay kit. Annexin V-positive cells was quantified by flow cytometry and indicated as percentage in each graph. For statistical analysis, data from three independent experiments were analyzed and compared with the mock controls. FL1-H, fluorescence intensity.
To further determine whether virus–receptor binding alone is sufficient for the apoptosis induction, we performed a complementary experiment in which the virus was pre-bound to the cells at 4 °C for 1 h. Infected cells were then washed with cold PBS to remove unbound viruses and incubated with fresh medium in the presence of a neutralizing monoclonal antibody specific to the viral spike protein (Fleming et al., 1983, Ramakrishna et al., 2003) or IgG2b (as an isotype control) at 4 °C for 1 h. The culture was then moved to 37 °C for a period of time as indicated. At 24 h p.i., culture medium was harvested for determining virus titer and cells for evaluating apoptosis. As shown in Fig. 2A, treatment with the spike neutralizing antibody reduced virus titer by ≈ 3 log10, indicating that the neutralizing antibody was able to block virus entry even after virus attachment. Significantly, this treatment inhibited MHV-induced apoptosis by 87–93% as demonstrated by both PI and annexin V analyses as compared to those treated with the control IgG2b (Figs. 2B and C). Taken together, these results demonstrate that virus–receptor binding is necessary but not sufficient for the induction of oligodendrocyte apoptosis. It is noted that the cells used for this set of experiments had a lower passage and therefore exhibited a lower background when assayed for apoptosis.
Fig. 2.

Inhibition of MHV-induced oligodendrocyte apoptosis by treatment with a neutralizing antibody against the MHV spike protein. The experiments were carried out essentially the same as described in the legend of Fig. 1, except the initial treatment with the antibody as noted below. Cells were mock-infected or infected with live- or UV-inactivated MHV at 4 °C for 1 h. Virus-bound cells were washed with cold PBS twice and the neutralizing monoclonal antibody specific to the spike protein of MHV, termed J2.6, or IgG2b (as isotype control) was added to the culture and incubated at 4 °C for another hour. The culture was then incubated at 37 °C for 1 h to allow virus entry. Virus titers were determined only for live virus-infected culture at 24 h p.i. and were expressed as means ± SD from three independent experiments (A). Apoptosis was analyzed with PI staining at 48 h p.i. (B) or by detecting annexin V binding at 12 h p.i. (C) as described in the legend of Fig. 1.
Increased formation of pre-apoptotic complex in MHV-infected oligodendrocytes
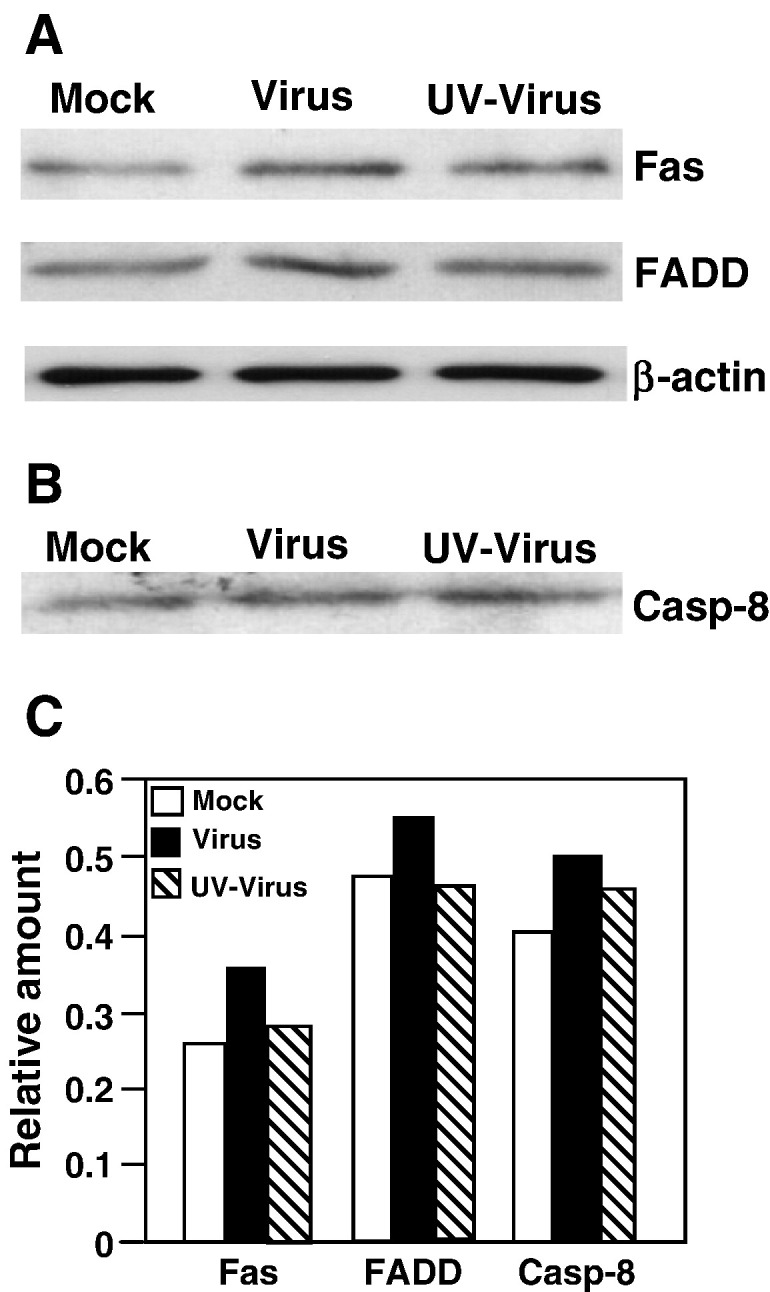
The above results suggest that the apoptotic signals are likely activated in the vicinity of cell surface during entry. As discussed in the introduction, there are two major apoptotic signals, the Fas/FasL and the TNFR/TNF, at the cell surface. To determine whether these apoptotic pathways are triggered by MHV infection, we analyzed by Western blot the abundance of the proteins that can potentially form into a pre-apoptotic complex. The results showed that the amounts of Fas, FADD, caspase-8 and TRADD were virtually unchanged following virus-infection at 24 h p.i. as compared with those of mock-infected cells (Figs. 3A and B), indicating that MHV infection did not activate the Fas or TNFR signaling pathway at the transcriptional and translational levels. We then performed co-immunoprecipitation of cell lysates with an antibody specific to one of the components and detected the interacting partners with antibodies to the other components in Western blot analysis. Interestingly, significantly (> 3-fold) more FADD and Fas were co-immunoprecipitated by the antibody specific to caspase-8 in both live- and UV-inactivated viruses-infected cells at 24 h p.i. as compared to those of mock-infected cells (Figs. 3C and F). Similarly, more caspase-8 protein (≈ 3-fold) was coprecipitated by the antibody specific to FADD in virus-infected cells than in mock-infected cells (Figs. 3D and F). In contrast, the amount of TRADD that was co-immunoprecipitated by the caspase-8 antibody remained similar in virus-infected and mock-infected cells (Figs. 3E and F). These results indicate that MHV infection increased the formation of the Fas/FADD/pro-caspase-8 complex but not the TNFR/TRADD/pro-caspase-8 complex, suggesting the specific activation of the Fas signaling pathway by MHV infection. In addition, to determine the kinetics of the activation of the Fas signal pathway, we analyzed the pre-apoptotic complex formation from 6 to 18 h p.i. by co-immunoprecipitation with the anti-FADD antibody and Western blot analysis with the anti-caspase-8 antibody. We found that caspase-8 protein was significantly increased in the complexes at 12 h p.i. and thereafter, but not at 6 and 8 h p.i. (Fig. 3G).
Fig. 3.

Induction of death receptor complex formation by MHV infection. (A) Western blot analysis of protein abundance. Cells were infected with live (Virus)- or UV-inactivated MHV (UV virus) at an m.o.i. of 10 or mock-infected (Mock). At 24 h p.i., cells were lysed and an equivalent amount of proteins in each sample was analyzed by Western blot using antibodies specific to the individual proteins as indicated at the right of the panel. β-Actin was used as an internal control. (B) Quantification of the protein bands shown in panel A. The intensity of each band was determined by densitometry and was expressed as relative amount to the β-actin control in each lane, which was set as 1-fold. (C) Detection of complex formation by co-immunoprecipitation and Western blot. Cells were infected and lysed as described in panel A. Cell lysates were used for precipitation with an antibody specific to caspase-8. The immunocomplexes were resolved by SDS–PAGE and analyzed by Western blot with an antibody specific to the individual proteins as indicated at the right of the panel. Note that the lysates for each sample used in the immunoprecipitation were approximately 24 times more than those used in the Western blot analysis shown in panel A. (D) The experiment was performed as in panel C, except that the anti-FADD antibody was used for immunoprecipitation and the anti-caspase-8 antibody for Western blot. (E) The experiment was performed as in panel C using the same anti-caspase-8 antibody for immunoprecipitation except that the anti-TRADD antibody was used for Western blot analysis. (F) Quantification of the protein bands shown in panels C, D and E as described in panel B. (G) Kinetics of the complex formation in virus-infected and mock-infected cells. The complexes were precipitated by an antibody specific to FADD and were detected by the presence of caspase-8 with an antibody specific to caspase-8 in Western blot analysis. β-Actin was used as a control for protein loading.
To establish the biological relevance of the complex formation and the apoptotic signaling, we treated the cells with the Fas/Fc chimera prior to and during virus infection. Because the chimera contains the ectodomain of Fas and the Fc region of an immunoglobulin (in place of the membrane-anchoring domain of Fas), it functions as a soluble receptor that is able to block Fas-ligand-mediated apoptosis when it is added to the cell culture medium (Cheng et al., 1994). At 24 h p.i., the ability of the complex formation was then determined by the combination of co-immunoprecipitation and Western blot analysis. As expected, treatment of cells with the Fas/Fc chimera significantly blocked the formation of the Fas/FADD/pro-caspase-8 complex to the levels similar to those in mock-infected cells (Fig. 4 ). These results thus establish that the Fas signaling pathway is activated by MHV infection.
Fig. 4.
Inhibition of death receptor complex formation by the Fas/Fc chimera. (A and B) Cells were treated with the Fas/Fc chimera 1 h prior to and during virus infection. Cells were infected with live (Virus)- or UV-inactivated virus (UV virus) or mock-infected (Mock). Cell lysates were subjected to immunoprecipitation with an antibody to caspase-8 (A) or to FADD (B). The immunocomplexes were resolved by SDS–PAGE and analyzed by Western blot with an antibody specific to the individual proteins shown on the right of the panels. β-Actin was used as a control for protein loading. (C) Quantification of the protein bands shown in panels A and B. The intensity of each band was determined by densitometry and was expressed as relative amount to the β-actin control in each lane, which was set as 1-fold.
Activation of caspase-8 and induction of apoptosis in oligodendrocytes by MHV infection
Although we previously showed that caspase-8 was activated in oligodendrocytes by MHV infection (Liu et al., 2006), its biological relevance to the observed apoptosis has not been directly established. Here we extended these studies by treating cells with a caspase-8 inhibitor and examining the apoptosis. We found that caspase-8 was significantly activated by both live- and UV-inactivated viruses at 24 h p.i. (≈ 17-fold increase over mock-infected cells) and that treatment of cells with the caspase-8 inhibitor almost completely blocked caspase-8 activation (p < 0.01) (Fig. 5A). We then performed PI- and annexin V staining in infected oligodendrocytes. As shown in Fig. 5B, 16.2% of the cells were in sub-G0/G1 phase in mock-infected cells as revealed by PI staining followed by flow cytometric analysis, whereas 59.6% and 61.5% of the cells were in sub-G0/G1 phase when the cells were infected with live- and UV-inactivated viruses, respectively. When the cells were treated with the caspase-8 inhibitor, the percentage of sub-G0/G1 cells decreased to 31% and 32.3%, respectively, for cells infected with live- and UV-inactivated viruses, but it was still significantly higher than that for mock-infected cells (10.5%). The differences between each group were statistically significant (p < 0.01). A similar result was obtained when the cells were stained with annexin V at 12 h p.i. (Fig. 5C). These results indicate that MHV-induced apoptosis of oligodendrocytes is mediated at least in part through the activation of caspase-8. The data also indicate that the activation of caspase-8 is most likely mediated during virus entry and does not require viral replication since both live- and UV-inactivated viruses were capable of activating the caspase-8 and inducing apoptosis to a similar extent.
Fig. 5.

Activation of caspase-8 and apoptosis in oligodendrocytes by MHV infection. (A) Caspase-8 activity. Cells were infected with live- or UV-inactivated MHV at an m.o.i. of 10 or mock-infected (mock) and were either mock-treated (DMSO) or treated with caspase-8 inhibitor (C-8-IN). At 24 h p.i., caspase-8 activity in the cell lysates was detected with a caspase colorimetric protease assay kit using specific substrates as described in Materials and methods. The caspase-8 activity from virus-infected cells was expressed as means ± SD from three independent experiments and as fold increase over that from mock-infected control, which was set as 1-fold. (B) Analysis of apoptosis by PI staining. Cells were infected and treated as described in panel A. At 48 h p.i., cells were stained with PI and subjected to flow cytometric analysis. The bar in each graph represents the sub-G0/G1 population of cells (indicated as a percentage) that have the lowest intensity of PI staining. Data are representative of at least three independent experiments. (C) Analysis of apoptosis by detecting annexin V binding. The experiments were performed as in panels A and B, except that the detection of annexin V binding on the cell surface was carried out at 12 h p.i. with the annexin V-EGFP assay kit. Annexin V-positive cells were quantified by flow cytometry and indicated as percentage in each graph. For statistical analysis, data from three independent experiments were analyzed and compared to those of the controls (mock-treated or mock-infected). FL1-H, fluorescence intensity.
Activation of caspase-8 by MHV-infection is mediated via the activation of Fas
To determine whether the activation of caspase-8 is mediated through the upstream Fas signaling pathway, cells were pre-treated with a Fas/Fc chimeric protein for 1 h. Pre-treated cells were infected with MHV at an m.o.i. of 10 in the presence of the Fas/Fc chimera. Following infection for 1 h, the inoculum was removed and replaced with fresh medium. Caspase-8 activity and apoptosis were evaluated. As shown in Fig. 6A, treatment of cells with the Fas/Fc chimera almost completely blocked the activation of caspase-8 activity induced by infections with both live- and UV-inactivated viruses (p < 0.001). Similarly, the number of sub-G0/G1-phase cells decreased significantly in virus-infected cells (from 65.1% to 29.4% for live-virus-infected cells and from 64.9% to 28.1% for UV-inactivated virus-infected cells) following the chimera treatment (p < 0.01) (Fig. 6B). There was also a slight decrease in cell numbers in sub-G0/G1 phase for mock-infected cells (from 13.9% to 8.1%, Fig. 2B), which may suggest that spontaneous apoptosis of oligodendrocytes can also be inhibited by Fas/Fc treatment. Consistent with the PI staining, when apoptosis was analyzed by annexin V staining at 12 h p.i., apoptotic cells reduced from 76.6% and 74.7% to 22.9% and 24.3%, respectively, for live-virus- and UV-inactivated virus-infected cells by the Fas/Fc chimera treatment (p < 0.01) (Fig. 6C). We thus conclude that the activation of caspase-8 activity and the induction of apoptosis were mediated via the activation of the Fas signaling pathway by MHV infection, although these experiments cannot exclude the involvement of other signaling pathways (see below).
Fig. 6.

Inhibition of caspase-8 activity and apoptosis by the Fas/Fc chimera. The experiments were performed exactly as described in the legend of Fig. 5, except that the cells were treated with Fas/Fc chimera instead of the caspase-8 inhibitor.
Inhibition of the mitochondrial signaling pathway by treatment of cells with the Fas/Fc chimera
We recently showed that the mitochondrial pathway was activated by MHV-infection (Liu et al., 2006). To further determine whether MHV-induced mitochondrial signaling is dependent on the activation of the Fas signaling pathway, we treated cells with the Fas/Fc chimera and determined the relocalization of cytochrome c and the caspase-9 activity. We found that, without the treatment with the Fas/Fc chimera, the ratios of cytoplasmic to mitochondrial fraction for cytochrome c were 3.6:1 and 3.4:1 for live- and UV-inactivated viruses, respectively. When the cells were treated with the Fas/Fc chimera, the ratios were reversed to 1:3.6 and 1:3.5 for the two viruses, respectively, which were similar to that in mock-infected cells (the ratio was 1:4) (Fig. 7A). Likewise, treatment of cells with the Fas/Fc chimera drastically blocked the activation of caspase-9 (more than 6-fold reduction in caspase-9 activities for both viruses) (Fig. 7B). These results indicate that the activation of the mitochondrial apoptotic pathway by MHV infection is largely dependent on the activation of the Fas signaling pathway in oligodendrocytes.
Fig. 7.

Effect of Fas/Fc treatment on the release of cytochrome c from mitochondria during MHV infection. (A) Cells were infected with live-virus (Virus) or UV-inactivated virus (UV-V) or mock-infected (Mock) and were either untreated (−) or treated with the Fas/Fc chimera. At 24 h p.i., cells were harvested and the cytosolic (cytosol.) and mitochondrial (mito.) fractions were separated by differential centrifugation using cytochrome c release assay kit as described in Materials and methods. Proteins were separated by SDS–PAGE (10% gel), transferred to nitrocellulose membranes and detected by Western blot analysis. Cytochrome c was detected in Western blot with a cytochrome c-specific antibody included in the kit. The amount of each protein band was quantified by densitometric analysis with the UPV software. The efficiency of release of cytochrome c from mitochondrial fraction into cytosolic fraction was expressed as a ratio of cytochrome c in the cytosolic fraction to mitochondrial fraction (C:M) shown at the bottom of the panel. (B) Inhibition of caspase-9 activity by the Fas/Fc chimera. Cells were infected with MHV and treated with the Fas/Fc chimera as described in panel A. At 12 h p.i., caspase-9 activity in the cell lysates was detected with a caspase colorimetric protease assay kit using specific substrates as described in Materials and methods. The caspase-9 activity from virus-infected cells was expressed as means ± SD from three independent experiments and as fold increase over that from mock-infected control, which was set as 1-fold.
Discussion
The mechanism by which MHV induces oligodendrocyte apoptosis is currently unknown. We have begun to address this issue in the current study by determining which events in the virus life cycle trigger the Fas signaling pathway. Because infection with UV-inactivated virus can activate apoptosis (Liu et al., 2003; this work), it is most likely that events during virus entry trigger the apoptosis. Therefore, we have specifically sought to determine whether the initial step of virus entry (i.e., virus–receptor binding), the second step (i.e., virus-cell fusion) or both trigger the observed apoptosis. We employed two complementary approaches. The first was to use a neutralizing antibody to the viral receptor. Treatment of cells with the receptor antibody demonstrated that virus–receptor binding was necessary for triggering the apoptosis but that receptor–antibody binding did not induce apoptosis since uninfected cells did not exhibit apoptosis even though the cells were treated with the receptor antibody (ligand) (Fig. 1). This interpretation is supported by the second experiment in which apoptosis was also blocked by a neutralizing anti-spike protein antibody even though the virus was pre-bound to the receptor (Fig. 2). This suggests that receptor binding alone is not sufficient for the apoptosis induction. Therefore, the second step (i.e., fusion) is likely involved in triggering the apoptosis. Consistent with this interpretation is our previous finding that when the endosomal pH was elevated by ammonium chloride, chloroquine or bafilomycin A1, all of which do not block virus–receptor binding but block virus-endosomal membrane fusion for the pH-dependent mutant MHV, OBLV60 (Gallagher et al., 1991), apoptosis was inhibited (Liu et al., 2003). In addition, the neutralizing antibody was shown to also inhibit virus-cell fusion (Fleming et al., 1983, Ramakrishna et al., 2003). Taken together, these findings suggest that the virus fusion during entry likely initiates the apoptotic signal, i.e., Fas activation. However, the mechanistic detail of this activation remains to be further investigated.
In this study, we have demonstrated that infection of the differentiated oligodendrocyte cell line with MHV resulted in activation of caspase-8 activity and apoptosis whereas treatment of cells with the caspase-8 inhibitor suppressed caspase-8 activation and the apoptosis (Fig. 5), indicating that the caspase-8 signaling pathway is involved in MHV-induced oligodendrocyte apoptosis. Furthermore, we have identified that Fas is the most upstream apoptotic signal that is activated by MHV infection. This conclusion is supported by three pieces of evidence. First, the activation of caspase-8 and apoptosis following MHV infection were blocked by treatment of cells with an inhibitory soluble receptor (Fas/Fc chimera) prior to and during virus infection (Fig. 6). Second, there was a significant (> 3-fold) increase of the death receptor complexes consisting of Fas, FADD and procaspase-8 following MHV infection, which were completely blocked by treatment of cells with the Fas/Fc soluble receptor (Fig. 3, Fig. 4). The death receptor complexes began to increase at 12 h p.i. (Fig. 3G), at which time point apoptosis began to be detectable, albeit at a low level (Liu et al., 2003). Third, the release of cytochrome c from mitochondria into cytoplasm or the activation of caspase-9 caused by MHV infection was prevented by treatment of cells with the Fas/Fc soluble receptor (Fig. 7). These findings thus establish that the Fas activation is the primary apoptotic signal triggered by MHV infection. Interestingly, although caspase-8 can be usually activated by both Fas and TNFR, the TNFR signal pathway did not appear to be activated by MHV infection. In no instance was an increased amount of TRADD detected in the death receptor complexes by co-immunoprecipitation and Western blot analysis (Fig. 3). Thus, it appears that MHV infection specifically activates the Fas signaling pathway in the oligodendrocytes. It is noted that the relatively high background of the pre-apoptotic complexes in mock-infected cells shown in Fig. 3, Fig. 4 was likely due to two chief reasons. First, the total protein amounts for each sample in the co-immunoprecipitation experiment were > 20 times higher than those used in the Western blot alone (see Fig. 3A). It is very likely that if a much lower amount of the proteins were used for immunoprecipitation, the background protein complexes for the mock-infected cells would be below the detectable level. Second, terminally differentiated CG-4 oligodendrocytes usually undergo apoptosis at a low level (below 20%), which may contribute to the background complex formation. Nevertheless, the increase in the amount of the Fas/FADD/caspase-8 complexes following virus infection was significant under the same experimental conditions.
We previously reported that the activation of the mitochondrial apoptotic pathway was triggered in part through the activation of caspase-8 and the cleavage of Bid, but it was also in part independent of the caspase-8 pathway (Liu et al., 2006). The latter conclusion was based on the finding that cytochrome c release and caspase-9 activity were only partially inhibited by the treatment with the caspase-8 inhibitor (Liu et al., 2006). Here we showed that treatment with the caspase-8 inhibitor also only partially blocked the apoptosis induced by virus infection (from ≈ 75% to ≈ 35% annexin V-positive), which was still much higher than that in mock-infected, untreated cells (≈ 17% annexin V-positive) (Fig. 5C). However, higher concentrations of the caspase-8 inhibitor, which completely inhibited the caspase-8 activity, did not significantly increase the effectiveness on inhibiting apoptosis (data not shown). Thus, it is possible that MHV-induced apoptosis is via both caspase-8-dependent and -independent pathways. This may be explained in part by the results that show that treatment of cells with the Fas/Fc soluble receptor exerted greater inhibitory effect on apoptosis (≈ 70% reduction) than that with the caspase-8 inhibitor (≈ 55% reduction) when apoptosis was measured with annexin V staining at an earlier time (at 12 h p.i.) (compare Fig. 6C with Fig. 5C). This may suggest that the activation of the Fas signaling pathway not only activates the caspase-8 pathway but also some other downstream pathways. Although we have not yet examined the other pathways, one candidate pathway that can be potentially triggered upon Fas activation is the Daxx. Like FADD, Daxx binds to the death domains of Fas and activates a FADD-independent death pathway that involves the stress-activated c-Jun NH2-terminal kinase (JNK) (Yang et al., 1997). Phosphorylated JNK can either activate p53 or inhibit Bcl-2, leading to apoptosis via the mitochondrial pathway. This may explain why the caspase-8 inhibitor cannot completely block the translocation of Bax and the mitochondrial apoptotic pathway induced by MHV-infection (Liu et al., 2006). Such a possibility warrants further investigation.
Based on the current and previous findings (Liu et al., 2003, Liu et al., 2006, Liu and Zhang, 2005), we propose the following model for the activation of the oligodendrocyte apoptosis by MHV infection (Fig. 8 ). During the initiation of virus infection, the spike proteins interact with MHV receptor, which then induces cleavage and conformational change of the spike protein, leading to fusion between viral envelope and cytoplasma membrane. The fusion and/or post-fusion events trigger the activation of the Fas apoptotic signaling pathway by a yet undefined mechanism. Activation of the Fas signaling pathway facilitates the recruitment of FADD and procaspase-8 into the death receptor complexes, leading to activation of the caspase-8. Activation of caspase-8 further activates the effector caspases, leading to apoptosis and the cleavage of Bid. Cleaved Bid then translocates to mitochondria, leading to the activation of the mitochondrial apoptotic pathway, which in turn leads to the release of cytochrome c and activation of caspase-9. The activation of caspase-9 further activates the effector caspases, leading to apoptosis and the mitochondria via a positive feedback mechanism. MHV infection of oligodendrocytes may also activate an alternative, minor (Daxx → JNK → p53 → Bax → mitochondria) pathway following the activation of Fas, although much of this pathway remains to be established experimentally. It is worth noting, however, that the findings from in vitro-differentiated CG-4 cell lines might not faithfully reflect on the in vivo situation, which requires further testing in animals.
Fig. 8.

Proposed model for the activation of oligodendrocyte apoptosis by MHV infection. (a) Interaction between the viral S protein and MHV receptor (MHVR). (b) Fusion between viral envelope and cell membrane. (c) Integration of viral envelope proteins (E, M, S, S2) into the cell membrane after fusion. (d) Activation of the Fas signal pathway by virus–cell fusion and the post-fusion complexes. This is likely the major pathway leading to activation of the caspase-8 and mitochondrial apoptotic pathways. (e) An alternative pathway of the activation of the Fas signaling. Solid lines with arrows indicate the steps (pathways) that are supported by experimental data, whereas dashed lines with arrows denote the hypothetic pathways with little or no experimental data. The following blocking reagents were used in the experiments to establish the individual steps of the pathways: the CC1 (anti-MHVR antibody), αS (anti-S protein antibody), Fas/Fc (chimeric soluble receptor), C8IN (caspase-8 inhibitor), C9IN (caspase-9 inhibitor) and Bcl-2 or Bcl-xL.
Materials and methods
Cells, virus and reagents
The CG-4 cell is a permanent, undifferentiated type 2 oligodendrocyte/astrocyte progenitor cell that was originally established during a primary neural cell culture derived from the brain of newborn Sprague–Dawley rat pups (1 to 3 days post-natal) (McNulty et al., 2001). Under a conditional culture medium, CG-4 cell maintains its undifferentiated progenitor phenotype indefinitely (Liu et al., 2003). Under a defined culture condition, CG-4 cells differentiate into mature oligodendrocytes as evidenced by the appearance of myelin basic protein (Liu et al., 2003). Mature oligodendrocytes are susceptible to MHV infection. However, virus replication was severely restricted in the mature oligodendrocytes; only < 1% of the cells exhibited strong expression of viral antigens at 24 h p.i. (Liu et al., 2003). Mature oligodendrocytes were thus used for virus infection throughout this study. The mouse astrocytoma DBT cells (Hirano et al., 1974) were cultured in Eagles Minimum Essential Medium (EMEM) and used for virus propagation and virus plaque assay (Liu et al., 2003). Mouse hepatitis virus (MHV) strain JHM was obtained from Dr. Michael Lai's laboratory (University of Southern California School of Medicine). All virus preparations were purified through sucrose cushion and the virus titer was determined by plaque assay on DBT cells as described previously (Liu et al., 2003). A multiplicity of infection (m.o.i.) of 10 was used throughout this study. Caspase-8 inhibitor (2-Ile-Glu-Thr-Asp-CH2F) was purchased from CalBiochem and dissolved in dimethyl sulfoxide (DMSO). Its cytotoxicity was determined in oligodendrocytes and a non-cytotoxic concentration was used for all subsequent experiments. The recombinant rat Fas/Fc chimera (Cat.# 2159-FA, R and D Systems, Inc.) was prepared in phosphate-buffered saline (PBS) and was used at a final concentration of 1 μg/ml. A monoclonal antibody specific to MHV receptor CC1 was kindly provided by Dr. Kay Holmes (University of Colorado at Denver) (Dveksler et al., 1991). Monoclonal antibodies to MHV spike protein J2.6 and J7.2 were a gift of Dr. John Fleming (University of Wisconsin at Madison), and their properties were characterized as described (Fleming et al., 1983, Ramakrishna et al., 2003). Mouse IgG2b was purchased from Sigma and was used as an isotype control for the anti-spike antibody J2.6.
Assay for caspase activity
The caspase assay was carried out essentially as described previously (Liu et al., 2006). Briefly, cells were grown in 60-mm dishes and either mock-infected with EMEM or infected with MHV at an m.o.i. of 10. Cells were collected at various time points post-infection (p.i.) by scraping, split into a 96-well plate and spun down at 500×g for 5 min. Following removal of the supernatant, lysis of cells and determination of caspase activities were carried out with a fluorometric caspase assay kit (caspase-9 activity assay kit #QIA72 and caspase-8 activity assay kit #QIA71) according to the manufacturer's instruction (Oncogene Research Products). The fluorescence was read with a fluorescent plate reader (Spectra Max M2, Molecular Device) capable of measuring excitation at ∼ 400 nm and emission at ∼ 505 nm.
Ultraviolet (UV) inactivation of viruses
To inactivate the virus by UV light, purified virus was diluted to a concentration of 107 plaque forming units (PFU) per ml in serum-free medium. Aliquots of 1 ml were added to each well of the 6-well tissue culture plate. The plates were placed on ice at a distance of 14 cm from the UV light in a UV Crosslinker (Fisher Scientific) and exposed to UV light at energy of 1200 μW/ms for 30 min. Viral inactivation was confirmed by titration of samples before and after UV exposure and by the absence of cytopathic effect (CPE) after inoculation of DBT cell monolayers with the UV-irradiated virus samples. Preliminary experiments indicated that a 10-min exposure to such UV light completely inactivated MHV infectivity.
Detection of subdiploid cell population
Apoptotic nuclei containing subdiploid DNA content were determined by propidium iodide staining and flow cytometric analysis as described previously (Liu et al., 2003). Briefly, at various time points p.i., adherent and floating cells were collected and centrifuged at 1000×g for 5 min. Pelleted cells were resuspended in phosphate-buffered saline (PBS), centrifuged and then fixed with 70% ice-cold ethanol and incubated overnight at 4 °C. Cells were centrifuged, resuspended in PBS and incubated with 50 μg/ml of propidium iodide and 100 μg/ml of RNase A (Sigma). Aliquots of 106 cells were subjected to flow cytometric analysis with a FACS Calibur Flow Cytometer equipped with an argon-ion laser (488 nm), and the data were acquired and analyzed with CELLQuest software (Becton Dickinson).
Assay for apoptosis with annexin V
Annexin V binding assay was carried out as described (Liu et al., 2006). Briefly, cells were infected with MHV at an m.o.i. of 10 or mock-infected as a control. At 12 h p.i., approximately 1 × 105 cells were collected by centrifugation. To determine apoptosis, cells were resuspended in 500 μl of 1× annexin V binding buffer and incubated with 1 μl of the annexin V-enhanced green fluorescence protein (EGFP) at room temperature for 5 min in the dark using the assay kit according to the manufacturer's instruction (Bio Vision Inc., Mountain View, Ca). Cells were then analyzed by flow cytometry using the FL1 channel for detection of annexin V-EGFP.
Cytochrome c release assay
The cytochrome c release apoptosis assay kit (Calbiochem, Cat# QIA87) was used for detection of cytochrome c according to the manufacturer's instruction as described (Liu et al., 2006). Briefly, cells were collected, resuspended in cytosol extraction buffer and homogenized in an ice-cold tissue grinder. Cell lysates were then separated into mitochondrial and cytosolic fractions by differential centrifugation, and the proteins were analyzed by Western blot with a monoclonal antibody specific to cytochrome c (1 μg/ml) following separation by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) on a 10% gel.
Co-immunoprecipitation and Western blot analysis
For detection of protein complexes, cells were either mock-infected or infected with MHV and mock-treated or treated with the Fas/Fc chimera protein. At indicated time points p.i., cells were lysed with the lysis buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 0.5% sodium deoxycholate, 1% nonidet P-40 [NP-40]) containing protease inhibitor cocktail tablets (Roche, Mannheim, Germany). The cell lysates were clarified from cell debris by centrifugation. The protein concentration was measured by using Bio-Rad protein assay kit (Bio-Rad). Cellular proteins with equivalent number of cells for each sample were precipitated with an antibody specific to caspase-8 (for detecting Fas, FADD and TRADD) or to FADD (for detecting caspase-8) for 4 h at 4 °C on a rocking platform. The immunocomplexes were precipitated with protein A-agarose beads, denatured and separated by SDS–PAGE and analyzed by Western blot as described previously (Liu et al., 2006). The primary antibodies used in this study include a rabbit polyclonal antibody specific to Fas (Cat.# M-20) (200 ng/ml), a goat polyclonal antibody specific to FADD (Cat.# S-18) (200 ng/ml), a rabbit polyclonal antibody against TRADD (Cat.# H-278, 100 ng/ml) and a mouse monoclonal antibody specific to caspase-8 p20 (Cat.# D-8) (100 ng/ml). These antibodies were purchased from Santa Cruz Biotechnology, Inc. A monoclonal antibody specific to β-actin as an internal control (1:2,500) was purchased from Sigma. Presence of the specific proteins was visualized with an Enhanced Chemiluminescence (ECL) system using peracid as a substrate (Amersham Pharmacia Biotech) followed by autoradiography with exposure times ranging from 30 s to 5 min.
Statistical analysis
The results are expressed as mean ± standard deviation (SD) and the mean values were compared using Student t-test. Values of p < 0.05, p < 0.01 and p < 0.001 were considered statistically significant.
Acknowledgments
We thank Dr. Kay Holmes (University of Colorado at Denver) for the MHV receptor monoclonal antibody CC1, Dr. John Fleming (University of Wisconsin at Madison) for the monoclonal antibodies to MHV spike protein and Kelli Halcom (Zhang's laboratory) for proofreading the manuscript. This work was supported by a grant (NS047499) and in part by a grant (P30-NS047546) from the National Institutes of Health.
References
- Barac-Latas V., Suchanek G., Breitschopf H., Stuehler A., Wege H., Lassmann H. Patterns of oligodendrocyte pathology in coronavirus-induced subacute demyelinating encephalomyelitis in the Lewis rat. Glia. 1997;19:1–12. doi: 10.1002/(sici)1098-1136(199701)19:1<1::aid-glia1>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Cheng J., Zhou T., Liu C., Shapiro J.P., Brauer M.J., Kiefer M.C., Barr P.J., Mountz J.D. Protection from Fas-mediated apoptosis by a soluble form of the Fas molecule. Science. 1994;263:1759–1762. doi: 10.1126/science.7510905. [DOI] [PubMed] [Google Scholar]
- Cohen G.M. Caspases: the executioners of apoptosis. Biochem. J. 1997;326:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dandekar A.A., Perlman S. Virus-induced demyelination in nude mice is mediated by gamma delta T cells. Am. J. Pathol. 2002;161:1255–1263. doi: 10.1016/s0002-9440(10)64402-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dveksler G.S., Pensiero M.N., Cardellichio C.B., Williams R.K., Jiang G.S., Holmes K.V., Dieffenbach C.W. Cloning of the mouse hepatitis virus (MHV) receptor: expression in human and hamster cell lines confers susceptibility to MHV. J. Virol. 1991;65:6881–6891. doi: 10.1128/jvi.65.12.6881-6891.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming J.O., Stohlman S.A., Harmon R.C., Lai M.M.C., Frelinger J.A., Weiner L.P. Antigenic relationships of murine coronaviruses: analysis using monoclonal antibodies to JHM (MHV-4) virus. Virology. 1983;131:296–307. doi: 10.1016/0042-6822(83)90498-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming J.O., Wang F.I., Trousdale M.D., Hinton D.R., Stohlman S.A. Interaction of immune and central nervous systems: contribution of anti-viral Thy-1+ cells to demyelination induced by coronavirus JHM. Reg. Immunol. 1993;5:37–43. [PubMed] [Google Scholar]
- Gallagher T.M., Escarmis C., Buchmeier M.J. Alteration of the pH dependence of coronavirus-induced cell fusion: effect of mutations in the spike glycoprotein. J. Virol. 1991;65:1916–1928. doi: 10.1128/jvi.65.4.1916-1928.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano N., Fujiwara K., Hino S., Matumoto M. Replication and plaque formation of mouse hepatitis virus (MHV-2) in mouse cell line DBT culture. Arch. Gesamte Virusforsch. 1974;44:298–302. doi: 10.1007/BF01240618. [DOI] [PubMed] [Google Scholar]
- Houtman J.J., Fleming J.O. Pathogenesis of mouse hepatitis virus-induced demyelination. J. NeuroVirol. 1996;2:361–376. doi: 10.3109/13550289609146902. [DOI] [PubMed] [Google Scholar]
- Hsu H., Shu H.B., Pan M.G., Goeddel D.V. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996;84:299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- Liu Y., Zhang X. Expression of cellular oncogene Bcl-xL prevents coronavirus-induced cell death and converts acute infection to persistent infection in progenitor rat oligodendrocytes. J. Virol. 2005;79:47–56. doi: 10.1128/JVI.79.1.47-56.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Cai Y., Zhang X. Induction of caspase-dependent apoptosis in cultured rat oligodendrocytes by murine coronavirus is mediated during cell entry and does not require virus replication. J. Virol. 2003;77:11952–11963. doi: 10.1128/JVI.77.22.11952-11963.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Pu Y., Zhang X. Role of the mitochondrial signaling pathway in murine coronavirus-induced oligodendrocyte apoptosis. J. Virol. 2006;80:395–403. doi: 10.1128/JVI.80.1.395-403.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNulty S., Crouch M., Smart D., Rumsby M. Differentiation of bipolar CG-4 line oligodendrocytes is associated with regulation of CREB, MAP kinase and PKC signalling pathways. Neurosci. Res. 2001;41:217–226. doi: 10.1016/s0168-0102(01)00280-2. [DOI] [PubMed] [Google Scholar]
- Nijhawan D., Honarpour N., Wang X. Apoptosis in neural development and disease. Annu. Rev. Neurosci. 2000;23:73–87. doi: 10.1146/annurev.neuro.23.1.73. [DOI] [PubMed] [Google Scholar]
- Ramakrishna C., Bergmann C.C., Atkinson R., Stohlman S.A. Control of central Nervous system viral persistence by neutralizing antibody. J. Virol. 2003;77:4670–4678. doi: 10.1128/JVI.77.8.4670-4678.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roulston A., Marcellus R.C., Branton P.E. Viruses and apoptosis. Annu. Rev. Microbiol. 1999;53:577–628. doi: 10.1146/annurev.micro.53.1.577. [DOI] [PubMed] [Google Scholar]
- Schwartz T., Fu L., Lavi E. Differential induction of apoptosis in demyelinating and nondemyelinating infection by mouse hepatitis virus. J. NeuroVirol. 2002;8:392–399. doi: 10.1080/13550280260422695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stohlman S.A., Bergmann C.C., Perlman S. Mouse hepatitis virus. In: Ahmed R., Chen I., editors. Persistent Viral Infections. Wiley; New York, NY: 1999. pp. 537–557. [Google Scholar]
- Strasser A., O'Connor L., Dixit V.M. Apoptosis signaling. Annu. Rev. Biochem. 2000;69:217–245. doi: 10.1146/annurev.biochem.69.1.217. [DOI] [PubMed] [Google Scholar]
- Susin S.A., Zamzami N., Castedo M., Daugas E., Wang H.G., Geley S., Fassy F., Reed J.C., Kroemer G. The central executioner of apoptosis: multiple connections between protease activation and mitochondria in Fas/APO-1/CD95- and ceramide-induced apoptosis. J. Exp. Med. 1997;186:25–37. doi: 10.1084/jem.186.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaux D.L., Cory S., Adams J.M. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- Wang F.I., Stohlman S.A., Fleming J.O. Demyelination induced by murine hepatitis virus JHM strain (MHV-4) is immunologically mediated. J. Neuroimmunol. 1990;30:31–41. doi: 10.1016/0165-5728(90)90050-W. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G.F., Perlman S. Macrophage infiltration, but not apoptosis, is correlated with immune-mediated demyelination following murine infection with a neurotropic coronavirus. J. Virol. 1999;73:8771–8780. doi: 10.1128/jvi.73.10.8771-8780.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G.F., Dandekar A.A., Pewe L., Perlman S. CD4 and CD8 T cells have redundant but not identical roles in virus-induced demyelination. J. Immunol. 2000;165:2278–2286. doi: 10.4049/jimmunol.165.4.2278. [DOI] [PubMed] [Google Scholar]
- Wyllie A.H. Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature. 1980;284:555–556. doi: 10.1038/284555a0. [DOI] [PubMed] [Google Scholar]
- Yang X., Khosravi-Far R., Chang H.W., Baltimore D. Daxx, a novel Fas-binding protein that activates JNK and apoptosis. Cell. 1997;89:1067–1076. doi: 10.1016/s0092-8674(00)80294-9. [DOI] [PMC free article] [PubMed] [Google Scholar]