

Abstract
The effects of the linker arm rigidity and size on melanocortin receptor selectivity were explored in a series of compounds using cyclic lactam α-melanocyte-stimulating hormone template. A variety of dicarboxylic acid linkers introduced between the α-amino group of His6 and the ɛ-amino group of Lys10 lead to high-affinity, selective human melanocortin receptor-1 and -5 (hMC1R and hMC5R) antagonists. The incorporation of hydrophilic functions into the linker arm was found to be unfavorable for both binding potency and receptor selectivity. Analogs 8 and 9 containing highly conformationally constrained hydrophobic linkers (m- and p-phthalic acids) were found to be selective nanomolar range hMC1R antagonists (IC50 = 7 and 4 nM, respectively), whereas the employment of a small conformationally constrained linker (maleic acid) resulted in a high-affinity (IC50 = 19 nM) and selective hMC5R antagonist (analog 12). These newly developed melanotropins will serve as critical biochemical tools for elucidating the full spectrum of functions performed by the physiologically important melanocortin-1 and -5 receptors.
Keywords: α-melanocyte-stimulating hormone, antagonist, human melanocortin-1 receptor, human melanocortin-5 receptor, macrocyclic, melanocortin, peptide
Abbreviations: All, allyl; Alloc, allyloxycarbonyl; Boc, tert-butyloxycarbonyl; Fmoc, fluorenylmethoxycarbonyl; CH3CN, acetonitrile; DCM, dichloromethane; DIPEA, diisopropylethylamine; DMF, N,N-dimethylformamide; DIC, diisopropyl carbodiimide; HBTU, 2-(1H-benzotriazole-1-yl)-1, 1, 3, 3-tetramethyluronium hexafluorophosphate; HOBt, N-hydroxybenzotriazole; hMCR, human melanocortin receptor; MSH, melanocyte-stimulating hormone; Nal(2′), 2′-naphthylalanine; Pbf, 2, 2, 4, 6, 7-pentamethyldihydrobenzofuran-5-sulfonyl; PyBOP, benzotriazol-1-yloxy-tris-pyrrolidinophosphonium hexafluorophosphate; TFA, trifluoroacetic acid; Trt, trityl; SPPS, solid-phase peptide synthesis; RP-HPLC, reverse-phase high-performance liquid chromatography; hMC1R, human melanocortin-1 receptor; α-MSH, Ac-Ser-Tyr-Ser-Met-Glu-His-Phe-Arg-Trp-Gly-Lys-Pro-Val-NH2; NDP-α-MSH, Ac-Ser-Tyr-Ser-Nle-Glu-His-D-Phe-Arg-Trp-Gly-Lys-Pro-Val-NH2
The natural melanocortin agonists, α-, β-, γ-melanocyte-stimulating hormones (MSHs), and adrenocorticotropin have been receiving great attention in the recent years because of their involvement in a large number of multifaceted biological actions, including skin pigmentation (1–3), control of the immune system (1–4), erectile function (5–9), blood pressure and heart rate (10), control of feeding behavior and energy homeostasis (11–17), modulation of aggressive/defensive behavior (18), and mediation of pain (19). These endogenous neuropeptides are ligands for the five known subtypes of human melanocortin receptors, which are expressed in various tissues, including skin [human melanocortin-1 receptor (hMC1R)] (1–4), adrenal cortex (hMC2R) (20,21), and throughout the central nervous system (hMC3R, hMC4R, and hMC5R) (22).
Previous reports from our laboratories (23–28) had focused on finding potent and selective agonists and antagonists for the hMC3R and hMC4R, which have been implicated to play complementary roles in weight control (11–17). Recently, Kavarana et al. (24,25) and Bednarek et al. (29) have reported that replacing the Ac-Nle4-Asp5 part of the non-selective superagonist MT-II (Ac-Nle4-c[Asp5, D-Phe7, Lys10]α-MSH(4–10)-NH2) (30) with a variety of dicarboxylic acid linkers yielded several potent and selective hMC3R and hMC4R agonists and antagonists. Extending these studies, a series of novel cyclic α-MSH analogs possessing a variety of flexible aliphatic and constrained aromatic and heterocyclic dicarboxylic acid linkers (c[CO-R-CO-His-D-Phe/D-Nal(2′)-Arg-Trp-Lys]-NH2) have been designed and synthesized by solid-phase methods to further investigate the effects of macrocycle size and flexibility on melanocortin receptor selectivity.
Peptide Design
MT-II, a superpotent but non-selective human melanocortin receptor agonist (30), along with potent non-selective hMC3R/hMC4R antagonist SHU-9119 (Ac-Nle4-c[Asp5, D-Nal(2′)7, Lys10]α-MSH(4–10)-NH2) (31) provided an excellent template for design of the more selective melanocortin ligands. The MT-II template in this study was modified to include the following variety of dicarboxylic acid linkers between the α-amino group of histidine and the ɛ-amino group of lysine: constrained aromatic acids (isophthalic and terephthalic acids), constrained heterocyclic acids (2,6-pyridinedicarboxylic acid and 2,3-pyrazinedicarboxylic acid), constrained unsaturated acid (maleic acid), and flexible aliphatic acids (pimelic and adipic acids). In addition, the effects of linker hydrophilicity were investigated on the analogs containing glutamic acid as the linker (Table 1).
Table 1.
Sequences and the physicochemical properties of the cyclic α-MSH analogs
|
m/z (M + 1)
|
TLC Rfb |
||||||
|---|---|---|---|---|---|---|---|
| No. | Sequence | Calculated | Observed (ESI) | HPLC retention time (min)a | 1 | 2 | 3 |
| 1 | c[CO-m-C6H4-CO-His-D-Phe-Arg-Trp-Lys]-NH2 | 902.4426 | 902.4440 | 14.12 | 0.56 | 0.10 | 0.68 |
| 2 | c[CO-p-C6H4-CO-His-D-Phe-Arg-Trp-Lys]-NH2 | 902.4426 | 902.4518 | 15.26 | 0.53 | 0.04 | 0.61 |
| 3 | c[CO-2,6-pyridine-CO-His-D-Phe-Arg-Trp-Lys]-NH2 | 903.4378 | 903.4237 | 14.37 | 0.64 | 0.11 | 0.67 |
| 4 | c[CO-2,3-pyrazine-CO-His-D-Phe-Arg-Trp-Lys]-NH2 | 904.4331 | 904.4195 | 14.38 | 0.58 | 0.04 | 0.60 |
| 5 | c[CO-cis-CH=CH-CO-His-D-Phe-Arg-Trp-Lys]-NH2 | 852.4269 | 852.4312 | 12.87 | 0.63 | 0.05 | 0.66 |
| 6 | c[CO-(CH2)4-CO-His-D-Phe-Arg-Trp-Lys]-NH2 | 882.4739 | 882.4685 | 12.39 | 0.61 | 0.05 | 0.63 |
| 7 | c[CO-(CH2)5-CO-His-D-Phe-Arg-Trp-Lys]-NH2 | 896.4895 | 896.4908 | 12.46 | 0.65 | 0.06 | 0.65 |
| 8 | c[CO-m-C6H4-CO-His-D-Nal(2′)-Arg-Trp-Lys]-NH2 | 952.4582 | 952.4563 | 16.50 | 0.71 | 0.11 | 0.70 |
| 9 | c[CO-p-C6H4-CO-His-D-Nal(2′)-Arg-Trp-Lys]-NH2 | 952.4582 | 952.4618 | 16.98 | 0.62 | 0.06 | 0.69 |
| 10 | c[CO-2,6-pyridine-CO-His-D-Nal(2′)-Arg-Trp-Lys]-NH2 | 953.4535 | 953.4491 | 16.52 | 0.71 | 0.14 | 0.69 |
| 11 | c[CO-2,3-pyrazine-CO-His-D-Nal(2′)-Arg-Trp-Lys]-NH2 | 954.4487 | 954.4502 | 15.21 | 0.41 | 0.07 | 0.69 |
| 12 | c[CO-cis-CH=CH-CO-His-D-Nal(2′)-Arg-Trp-Lys]-NH2 | 902.4426 | 902.4474 | 14.63 | 0.66 | 0.06 | 0.69 |
| 13 | c[CO-(CH2)4-CO-His-D-Nal(2′)-Arg-Trp-Lys]-NH2 | 932.4895 | 932.4916 | 14.42 | 0.64 | 0.05 | 0.66 |
| 14 | c[CO-(CH2)5-CO-His-D-Nal(2′)-Arg-Trp-Lys]-NH2 | 946.5052 | 946.4997 | 14.23 | 0.69 | 0.11 | 0.68 |
| 15 | H-c[Glu-His-D-Nal(2′)-Arg-Trp-Lys]-NH2 | 933.4848 | 933.4884 | 13.33 | 0.55 | 0.03 | 0.59 |
| 16 | Ac-c[Glu-His-D-Nal(2′)-Arg-Trp-Lys]-NH2 | 975.4952 | 975.4921 | 15.51 | 0.63 | 0.06 | 0.67 |
α-MSH, α-melanocyte-stimulating hormone; HPLC, high-performance liquid chromatography; TLC, thin-layer chromatography; ESI, electrospray ionization.
HPLC column: Vydac 218TP104, 250 × 4.6 mm, 10 μm, 300 Å; HPLC solvent A, 0.1% trifluoroacetic acid in water; solvent B, acetonitrile; gradient: 10–90% B in A over 40 min, flow rate 1.0 mL/min.
TLC system 1: ethyl acetate/acetic acid/water/pyridine (5:1:3:5); TLC system 2: n-propanol/acetic acid/water (10:1:1); TLC system 3: n-butanol/acetic acid/water/pyridine (5:1:4:5).
Methods and Materials
Nα-fluorenylmethoxycarbonyl (Fmoc)-amino acids, peptide coupling reagents and Rink amide AM resin were obtained from Novabiochem (San Diego, CA, USA), except Nα-Fmoc-Glu(O-All)-OH and Nα-Fmoc-Lys(Alloc)-OH, which were purchased from Neo-MPS (San Diego, CA, USA). The following side-chain protecting groups were used: Glu(Oγ-All), Trp(Nin-Boc), Arg(Nɛ-Pbf), His(Nim-Trt), and Lys(Nɛ-Alloc). ACS grade organic solvents were purchased from VWR Scientific (West Chester, PA, USA), and other reagents were obtained from Sigma-Aldrich (St Louis, MO, USA) and used as commercially available. The polypropylene reaction vessels (syringes with frits) (32) were purchased from Torviq (Niles, MI, USA). The purity of the peptides was checked by analytical reverse-phase high-performance liquid chromatography (RP-HPLC) using a Vydac C18 218TP104 column (Western Analytical Products, Murrieta, CA, USA) monitored at 230 and 254 nm, and by thin-layer chromatography (TLC), which was performed using three different solvent systems. Analytical TLC was carried out on 0.25-mm glass-backed silica gel 60 F254 plates (EM Science 5715, VWR Scientific). The TLC chromatograms were visualized by ultraviolet light, and by dipping in potassium permanganate solution followed by heating (hot plate).
Peptide Synthesis
All peptides in this study were synthesized manually by the Nα-Fmoc solid-phase methodology (25,26). Rink amide AM resin [4-(2′,4′-dimethoxyphenyl-Fmoc-aminomethyl)phenoxy resin, 4.0 g, 0.637 mmol/g] was placed into a 50-mL polypropylene syringe with the frit on the bottom and swollen in N,N-dimethylformamide (DMF; 20 mL) for 1 h. The Fmoc protecting group on the Rink linker was removed by 25% piperidine in DMF (1 × 5 and 1 × 15 min). The resin was washed with DMF (6 × 15 mL), and the first Nα-Fmoc amino acid was coupled using preactivated ester [3 equiv. of Nα-Fmoc amino acid, 3 equiv. of N-hydroxybenzotriazole (HOBt), and 3 equiv. of 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU)] in DMF solution containing 6 equiv. of diisopropylethylamine (DIPEA). The coupling mixture was transferred into the syringe with the resin and shaken for 60 min. The resin was washed with DMF (3 × 15 mL) and thrice with dichloromethane (DCM) (3 × 15 mL), and the unreacted amino groups were capped using acetic anhydride (2 mL) and pyridine (2 mL) in DCM (15 mL) for 30 min, and the resin was once again washed with DCM (6 × 15 mL). The next four amino acids, Trp, Arg, D-Phe or D-Nal(2′), and His were consecutively coupled using the procedure described earlier, using the Kaiser ninhydrin test (33) to check the extent of coupling. In the case of a positive Kaiser test, the coupling was repeated until a negative Kaiser test was obtained. The resulting batch of the resin-bound protected pentapeptide Nα-Fmoc-His-D-Phe/D-Nal(2′)-Arg-Trp-Lys was carefully washed with DMF (5 × 15 mL), DCM (5 × 15 mL), methanol (5 × 15 mL), and diethyl ether (5 × 15 mL), and dried under reduced pressure (16 h). The dry resin was split in eight equal portions, which were placed in separate 10-mL fritted polypropylene syringes, and swollen with DMF as described earlier. The same coupling procedure was followed to append the dicarboxylic acid linkers. Pyrazinedicarboxylic and maleic acids were converted into their corresponding monoallyl esters prior to appending to the peptides to minimize competing formation of cyclic imides, by stirring the corresponding anhydrides with allyl alcohol (1 equiv.) in DCM in the presence of a catalytic (5 mol%) amount of DIPEA until homogeneous solutions were obtained. The resulting solutions were concentrated under reduced pressure and used for peptide synthesis without any further purification. The other dicarboxylic acids were used as commercially available. The orthogonal allylic protection for the side-chain of Lys11 and the linker (if applicable) was removed with 0.1 equiv. Pd(PPh3)4/20 equiv. PhSiH3 in DCM (2 × 30 min) prior to the peptide cyclization (34). The deprotected resin-bound peptide was washed with DCM (6 × 5 mL) and DMF (3 × 5 mL). The peptide cyclizations were accomplished with 6 equiv. benzotriazol-1-yloxy-tris-pyrrolidinophosphonium hexafluorophosphate (PyBOP) (35), 6 equiv. HOBt, and 6 equiv. DIPEA in DMF (36 h), and were monitored by Kaiser ninhydrin test (33). The PyBOP treatment was repeated until a negative Kaiser test was obtained. This procedure was found to be vastly superior to the previously described cyclization methodology that employed HBTU/HOBt/DIPEA (25,26), as it was not accompanied with competing guanylation of the primary amine function of Lys10, commonly associated with acyluronium reagents, such as HBTU (36). The new procedure also resulted in improved overall yield of the target peptides. Upon completion of cyclization, the resin was treated with 5% solution of sodium diethyldithiocarbamate trihydrate in DMF (20 min) to remove any remaining traces of the Pd catalyst, then washed with DMF (5 × 15 mL), DCM (3 × 15 mL), methanol (5 × 15 mL), and diethyl ether (5 × 15 mL), and dried under reduced pressure (16 h). The cyclized peptides were cleaved off the solid support with 82.5% v/v trifluoroacetic acid (TFA), 5% water, 5% thioanisol, 2.5% 1,2-ethanedithiol, and 5% phenol (5 mL, 3 h), and the crude peptides were precipitated out by the addition of a chilled 3:1 mixture of diethyl ether and petroleum ether (50 mL) to give white precipitates. The resulting peptide suspensions were centrifuged for 10 min at 6500 r.p.m., (300 g) and the liquid was decanted. The crude peptides were washed with diethyl ether (4 × 50 mL), and after the final centrifugation, the peptides were dried under vacuum (2 h). The resulting white residues were dissolved in 2 M acetic acid, and the insoluble impurities were removed by passing the solutions through Gelman Laboratory Acrodisc 13-mm syringe filters with 0.45-μM polytetrafluoroethylene (PTFE) membranes (Pall Corporation, East Hills, NY, USA). The clear filtrates were lyophilized, the obtained white powders (50–80 mg) were redissolved in glacial acetic acid (1 mL), the resulting solutions were diluted with water (4 mL) to a peptide concentration of about 1 mg/mL, and passed through a Sephadex G-15 column (520 × 30 mm) using 1 M aqueous acetic acid as the eluent. Fractions containing the target peptides, as determined by TLC, were combined and lyophilized. Final purification was accomplished by preparative RP-HPLC on a C18-bonded silica column (Vydac 218TP152022, 250 × 22 mm, 15–20 μM, 300 Å) using a Shimadzu SCL-10A HPLC system (Shimadzu Scientific Instruments, Pleasanton, CA, USA). The peptides were eluted with a linear gradient of 20–80% acetonitrile in 0.1% aqueous TFA solution over 50 min with 10-mL/min flow rate. The purified peptides were isolated in 25–30% overall yield. The structures of the pure peptides were confirmed by high-resolution electrospray ionization (ESI) mass spectrometry using an IonSpec Fourier-transform mass spectrometer with a HiRes ESI source.
Biological Activity Assays
Receptor binding assay
Competition binding experiments were carried out as described earlier (37) using whole HEK-293 (Human embryonic kidney) cells stably expressing hMC1R, hMC3R, hMC4R, and hMC5R. Briefly, HEK-293 cells transfected with human melanocortin receptors (38–40) were seeded on 96-well plates 48 h before assay (100 000 cells/well). For the assay, the cell culture medium was aspirated and cells were washed twice with a freshly prepared binding buffer containing 100% minimum essential medium (MEM) with Earle's salt (Gibco Invitrogen, Carlsbad, CA, USA), 25 mM HEPES (pH 7.4), 0.2% bovine serum albumin, 1 mM 1,10-phenanthrolone, 0.5 mg/L leupeptin, 200 mg/L bacitracin. Next, cells were incubated with different concentrations of unlabeled peptide and labeled [125I]-[Nle4, D-Phe7]-α-MSH (Perkin-Elmer Life Science, Wellesley, MA, USA, 100 000 c.p.m./well, 0.1386 nM) for 40 min at 37 °C. The assay medium was subsequently removed and each well was washed twice with the binding buffer. The cells were then lysed by the addition of 250 μL of 0.1 M NaOH and 250 μL of 1% Triton-X-100. The lysed cells were transferred to 12 × 75 mm glass tubes and the radioactivity was measured by a Wallac 1470 WIZARD gamma-counter (Perkin-Elmer).
Adenylate cyclase assay
HEK-293 cells transfected with human melanocortin receptors (38) were grown to confluence in MEM medium (Gibco Life Technologies) containing 10% fetal bovine serum, 100 units/mL penicillin and streptomycin, and 1 mM sodium pyruvate. The cells were seeded on 96-well plates 48 h before assay (100 000 cells/well). For the assay, the cell culture medium was removed and the cells were rinsed with 1 mL of MEM buffer (Gibco Life Technologies) or with Earle's balanced salt solution (EBSS; Gibco Invitrogen). An aliquot (0.4 mL) of the EBSS was placed in each well along with 5 μL of 0.5 mM isobutylmethylxanthine (IBMX) for 1 min at 37 °C. Next, aliquots of melanotropin peptide solutions with varying concentrations (0.1 mL) were added, and the cells were incubated for 3 min at 37 °C. The reaction was stopped by aspirating the assay buffer and adding 0.15-mL ice-cold Tris/ethylenediaminetetraacetic acid buffer to each well. After dislodging the cells with the help of trypsin, the cells were transferred to polypropylene micro-centrifuge tubes and placed in a boiling water bath for 15 min. The cell lysate was then centrifuged for 2 min at 14,000 g (max 16000 g), and 50 μL of the supernatant was aliquoted into an Eppendorf tube. The total cAMP content was measured by competitive binding assay according to the TRK 432 assay kit instructions (Amersham Corp., Piscataway, NJ, USA). IC50 and EC50 values represent the mean of two experiments performed in triplicate. IC50 and EC50 estimates and their associated standard errors were determined by fitting the data using a non-linear least-squares analysis, with the help of GraphPad Prism 4 (GraphPad Software, San Diego, CA, USA). The maximal cAMP produced at 10 μM concentration of each ligand was compared with the amount of cAMP produced at 10 μM concentration of the standard agonist MT-II, and is expressed in per cent (as % max effect) in Table 2.
Table 2.
Binding affinities and cAMP activities of cyclic α-MSH analogs at hMCRs
| hMC1R
|
hMC3R
|
hMC4R
|
hMC5R
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | Code | IC50 (nM) | EC50 (nM) | % max effect | IC50 (nM) | EC50 (nM) | % max effect | IC50 (nM) | EC50 (nM) | % max effect | IC50 (nM) | EC50 (nM) | % max effect |
| 1 | c[CO-m-C6H4-CO-His-D-Phe-Arg-Trp-Lys]-NH2 | >1000 | >1000 | 0 | 550 ± 70 | >1000 | 0 | >10 000 | >10 000 | 0 | 260 ± 30 | >1000 | 44 |
| 2 | c[CO-p-C6H4-CO-His-D-Phe-Arg-Trp-Lys]-NH2 | >1000 | 11 ± 2 | 4 | 390 ± 40 | >1000 | 0 | >1000 | >1000 | 0 | >2000 | 600 ± 60 | 17 |
| 3 | c[CO-2,6-pyridine-CO-His-D-Phe-Arg-Trp-Lys]-NH2 | >1000 | >1000 | 8 | 420 ± 60 | 560 ± 60 | 4 | >3500 | >5000 | 0 | >1500 | >1000 | 13 |
| 4 | c[CO-2,3-pyrazine-CO-His-D-Phe-Arg-Trp-Lys]-NH2 | >10 000 | 670 ± 89 | 10 | 4000 | >10 000 | 0 | >1000 | >1000 | 0 | >10 000 | >3000 | 61 |
| 5 | c[CO-cis-CH=CH-CO-His-D-Phe-Arg-Trp-Lys]-NH2 | 330 ± 30 | >1100 | 0 | 110 ± 10 | 94 ± 10 | 69 | 60 ± 6 | 110 ± 11 | 80 | 69 ± 7 | 55 ± 6 | 39 |
| 6 | c[CO-(CH2)4-CO-His-D-Phe-Arg-Trp-Lys]-NH2 | 140 ± 15 | 38 ± 4 | 70 | 72 ± 8 | 3 ± 0.3 | 87 | 110 ± 11 | 60 ± 7 | 90 | 85 ± 8 | 6 ± 0.6 | 46 |
| 7 | c[CO-(CH2)5-CO-His-D-Phe-Arg-Trp-Lys]-NH2 | 1000 | >1000 | 0 | 140 ± 15 | 80 ± 8 | 100 | 160 ± 14 | 250 ± 30 | 80 | 150 ± 16 | 200 ± 23 | 67 |
| 8 | c[CO-m-C6H4-CO-His-D-Nal(2′)-Arg-Trp-Lys]-NH2 | 7.0 ± 1 | >10 000 | 0 | 160 ± 23 | >1000 | 17 | 140 ± 15 | >10 000 | 0 | 550 ± 60 | 110 ± 10 | 63 |
| 9 | c[CO-p-C6H4-CO-His-D-Nal(2′)-Arg-Trp-Lys]-NH2 | 4.0 ± 0.8 | >10 000 | 0 | 120 ± 13 | >1000 | 25 | 110 ± 11 | >5000 | 0 | 670 ± 70 | 210 ± 21 | 55 |
| 10 | c[CO-2,6-pyridine-CO-His-D-Nal(2′)-Arg-Trp-Lys]-NH2 | 100 ± 10 | >1000 | 0 | 110 ± 20 | >10 000 | 0 | 540 ± 60 | >10 000 | 0 | >1000 | 610 ± 70 | 64 |
| 11 | c[CO-2,3-pyrazine-CO-His-D-Nal(2′)-Arg-Trp-Lys]-NH2 | 100 ± 11 | 400 ± 50 | 25 | 280 ± 20 | >1000 | 0 | 420 ± 45 | 99 | 36 | 660 ± 60 | >1000 | 93 |
| 12 | c[CO-cis-CH=CH-CO-His-D-Nal(2′)-Arg-Trp-Lys]-NH2 | 250 ± 22 | 96 ± 10 | 28 | 56 ± 6 | 310 ± 40 | 15 | 7 ± 8 | >1000 | 11 | 19 ± 2 | >10 000 | 0 |
| 13 | c[CO-(CH2)4-CO-His-D-Nal(2′)-Arg-Trp-Lys]-NH2 | >2800 | >3000 | 0 | 15 ± 2 | 300 ± 30 | 23 | 16 ± 2 | >1000 | 20 | 10 ± 1.2 | 59 ± 6 | 5 |
| 14 | c[CO-(CH2)5-CO-His-D-Nal(2′)-Arg-Trp-Lys]-NH2 | 1000 | 930 ± 88 | 0 | 64 ± 8 | 940 ± 11 | 13 | 60 ± 8 | 120 ± 13 | 19 | 44 ± 5 | 11 ± 2 | 4 |
| 15 | H-c[Glu-His-D-Nal(2′)-Arg-Trp-Lys]-NH2 | >1000 | 42 ± 6 | 56 | 27 ± 3 | 18 ± 2 | 15 | 900 ± 100 | >1000 | 28 | 470 ± 50 | 62 ± 4 | 34 |
| 16 | Ac-c[Glu-His-D-Nal(2′)-Arg-Trp-Lys]-NH2 | 500 ± 100 | 400 ± 122 | 95 | 42 ± 4 | 370 ± 40 | 75 | 42 ± 4 | 36 ± 4 | 23 | 77 ± 7 | 32 ± 3 | 198 |
| 17 | Ac-Nle-c[Asp-His-D-Phe-Arg-Trp-Lys]-NH2 | 0.2 ± 0.01 | 0.3 ± 0.04 | 100 | 1.25 ± 0.2 | 1.85 ± 0.2 | 100 | 1.07 ± 0.3 | 2.87 ± 0.52 | 100 | 7.47 ± 0.23 | 3.3 ± 0.7 | 100 |
hMC1R, -3R, -4R, and -5R, human melanocortin receptor-1, -3, -4, and -5; IC50, concentration of peptide at 50% specific binding (n = 4); EC50, effective concentration of peptide that was able to generate 50% maximal intracellular cAMP accumulation (n = 4); % max effect, % of cAMP produced at 10 μM ligand concentration, in relation to MT-II. The peptides were tested at a range of concentration from 10−10 to 10−5 nM.
Results and Discussion
Analog 1 was found to be a very weak antagonist at the hMC1R and the hMC3R, entirely inactive at the hMC4R, and a weak partial agonist at the hMC5R (maximum stimulation 44%). Replacement of the isophthalic acid linker with terephthalic acid (analog 2) resulted in no change in the biological profile of the cyclic peptide. Similar results were obtained for the peptides with 2,6-pyridinedicarboxylic acid (analog 3) and 2,3-pyrazinedicarboxylic acid linkers (analog 4). This suggests that the macrocycle expansion and introduction of aromatic heterocyclic linkers into the MT-II template do not produce favorable peptide topography for melanocortin activity. The introduction of the small constrained maleic acid linker yielded analog 5, which exhibited weak antagonist binding at the hMC1R and nanomolar range partial agonist activities at the hMC3R (IC50 = 110 nM), hMC4R (IC50 = 60 nM), and hMC5R (IC50 = 69 nM), with negligible receptor selectivity. This result contrasts sharply with our earlier findings that short aliphatic linkers, such as succinic acid, placed into the MT-II template produced potent hMC4R selective agonist VJH-085 (c[CO-(CH2)2-CO-His-D-Phe-Arg-Trp-Lys]-NH2) (25). It seems plausible that the differences in the biological activities of these peptides stem from the unfavorable dihedral constraint introduced by the maleic acid linker, which is locked in the cis-configuration, whereas the nuclear magnetic resonance structure of VJH-085 reported by our laboratories (41) indicates the opposite anti-conformation of the succinic acid linker. Analogs 6 and 7 were obtained by further expansion of the lactam macrocycle with flexible adipic acid and pimelic acid linkers, respectively. Analog 6 demonstrated nanomolar range partial agonist activities at all four receptor subtypes, while analog 7 displayed no interaction with the hMC1R, full agonist activity at the hMC3R and modest partial agonist activities at the hMC4R and hMC5R. Overall, these aliphatic linkers showed little tendency to produce selective ligands, which may be due to higher flexibility of the corresponding macrocycles.
Analogs 8–14 were produced by replacing D-Phe in position 7 of peptides 1–7 with D-Nal(2′) and the biological activities of these two subsets were compared. Analogs 8 and 9 exhibited very similar biological profiles, where both peptides were found to be antagonists at the hMC1R with high binding affinity (IC50 = 7 and 4 nM, respectively), weak partial agonists at the hMC3R, weak antagonists at the hMC4R and weak partial agonists at the hMC5R. These compounds show a good selectivity for the hMC1R (up to 30-fold versus the hMC3R, 25-fold versus the hMC4R, and up to 170-fold versus the hMC5R), which is a particularly important finding as reports of potent hMC1R antagonists are scarce in the literature (42–45). The meta- and para-stereochemistry of the phthalic acid linker clearly has very little effect on the biological activities of these two compounds. Interestingly, the ortho-isomeric peptide (c[CO-o-C6H4-CO-His-D-Nal(2′)-Arg-Trp-Lys]-NH2), reported earlier from our laboratories (25), showed similar biological properties at the hMC3–5R, albeit with a somewhat higher binding potencies, which was likely due to the smaller size of the lactam macrocycle. In contrast, replacement of the phthalic acid linker with more polar heterocyclic linkers produced peptides (analogs 10 and 11) with drastically different biological profiles. Thus, analog 10 exhibited weak antagonist affinities at the hMC1–4R, and a weak partial agonist activity at the hMC5R, while analog 11 was found to be a weak partial agonist at the hMC1R (25% cAMP stimulation), a weak antagonist at the hMC3R, a partial agonist at the hMC4R (36% cAMP stimulation) and a weak agonist at the hMC5R. Analog 12, with maleic acid linker, showed nanomolar range binding affinities at all four receptor subtypes, and partial agonist activities at the hMC1–4R (28%, 15%, and 11% maximum cAMP stimulation, respectively). At the same time, analog 12 was found to be a high-affinity hMC5R antagonist (IC50 = 19 nM). It is interesting to compare the biological activities of this peptide with those of MK-7 (c[CO-o-C6H4-CO-His-D-Nal(2′)-Arg-Trp-Lys]-NH2) (25). While maleic acid linker offers the same type of dihedral constraint for the peptide chain as o-phthalic acid linker used in MK-7, it lacks the aromaticity and the steric bulk of the latter. This structural difference may account for the observed conversion of the hMC5R agonist MK-7 to antagonist (analog 12). Furthermore, analogs 13 and 14, featuring longer aliphatic linkers, were determined to be weak antagonists at the hMC1R, partial agonists at the hMC3R (23% and 13% maximum cAMP stimulation, respectively) and hMC4R (20% and 19% maximum cAMP stimulation, respectively), and antagonists at the hMC5R, while exhibiting high binding affinities to this receptor sub-type (IC50 = 10 and 44 nM, respectively). Interestingly, the increase in the size of the flexible lactam macrocycle results in loss of binding affinities of the respective peptides (analogs 13 and 14) toward the hMC1R (IC50 = 2.8 and 1.0 μM, respectively), while favoring binding to the other receptor subtypes (hMC3–5R). This is in contrast to the observed binding affinities of the peptides containing highly conformationally constrained linkers (analogs 8 and 9) to the hMC1R (IC50 = 7 and 4 nM, respectively). This finding offers evidence for the effects of the macrocycle rigidity on the hMC1R receptor selectivity.
The analogs 15 and 16 were derived from the hMC3R selective antagonist MK-9 (c[CO-(CH2)3-CO-His-D-Nal(2′)-Arg-Trp-Lys]-NH2) (25), by introducing a hydrophilic function to the linker via replacement of the glutaric acid linker with L-glutamic acid and N-acetyl-L-glutamic acid. Analog 15 was found to be a weak partial agonist at the hMC1R, whereas analog 16 showed a very weak agonist activity at this receptor. Similar to MK-9, analog 15 demonstrated moderate hMC3R binding selectivity, but in contrast to MK-9, it remained a potent partial agonist at this receptor (EC50 = 18 nM, 15% maximum stimulation). Analog 15 also showed weak partial agonist activities at the hMC4R (EC50 > 1.0 μM, 28% maximum stimulation) and the hMC5R (EC50 = 62 nM, 34% maximum stimulation). Acetylation of the N-terminus (analog 16) resulted in weakened partial agonist activity at the hMC3R (EC50 = 374 nM, 75% maximum stimulation), but improved binding affinity and the partial agonist activity at the hMC4R (EC50 = 36 nM, 23% maximum stimulation), and produced a potent hMC5R agonist (EC50 = 32 nM).
Conclusions
In summary, introduction of a hydrophilic function into the linker arm was found to be unfavorable in terms of both potency and receptor selectivity. On the contrary, increasing the overall rigidity of the cyclic D-Nal(2′)7-α-MSH scaffold by introducing highly conformationally constrained hydrophobic linkers (m- and p-phthalic acids) between the α-amino group of His6 and the ɛ-amino group of Lys10 leads to nanomolar range selective hMC1R antagonists (analogs 8 and 9), which suggests the important role of hydrophobic aromatic interactions between the linker arm and the hMC1R binding pocket for hMC1R selectivity. Conversely, more polar aromatic heterocycles used as linker arms (analogs 10 and 11) weakened the peptide binding potencies towards the hMC1R. These findings represent a significant advancement toward development of more potent and highly selective hMC1R antagonists, unprecedented in the current literature. In addition, replacement of o-phthalic acid linker in an hMC5R agonist c[CO-o-C6H4-CO-His-D-Nal(2′)-Arg-Trp-Lys]-NH2 with a small conformationally constrained maleic acid linker resulted in a high-affinity and selective hMC5R antagonist (analog 12), which exemplifies an interesting and potentially useful new way of conversion of an hMC5R agonist into an antagonist. These newly developed melanotropin peptides may find application as biochemical and pharmacological tools for determining the complex physiological roles played by the hMC1R and the hMC5R.
Figure 1.
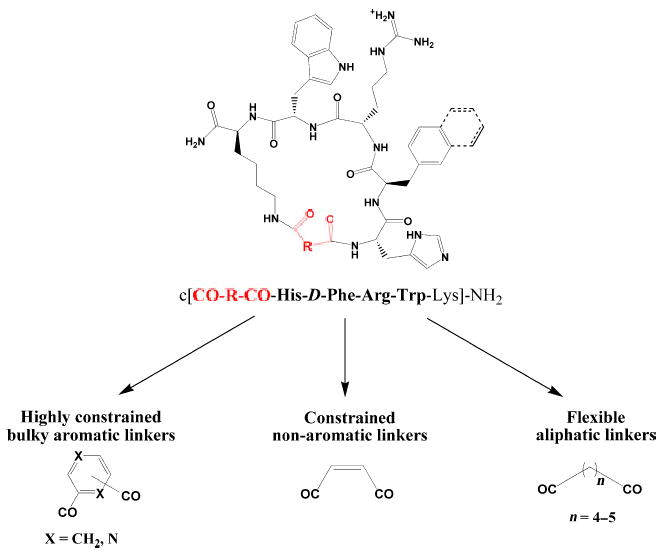
Design of the α-MSH-derived cyclic lactam scaffold.
Acknowledgments
This research was supported by grants from the US Public Health Service, National Institutes of Health DK-17420 and DA-06284. The opinions expressed are those of the authors and not necessarily those of the USPHS.
References
- 1.Hadley ME, editor. The Melanotropic Peptides, Vols I–III. Boca Raton, FL, USA: CRC Press; 1988. [Google Scholar]
- 2.Cone RD, editor. The melanocortin system. Ann N Y Acad Sci. 2003;994:1–387. [Google Scholar]
- 3.Cone RD, editor. The Melanocortin Receptors. Totowa, NJ, USA: Humana Press; 2000. [Google Scholar]
- 4.Vaudry H, Eberle AN, editors. The melanotropic peptides. Ann N Y Acad Sci. 1993;680:1–687. doi: 10.1111/j.1749-6632.1993.tb19750.x. [DOI] [PubMed] [Google Scholar]
- 5.Wessells H, Gralnek D, Dorr R, Hruby VJ, Hadley ME, Levine N. Effect of an α-melanocyte stimulating hormone analogue on penile erection and sexual desire in men with organic erectile dysfunction. Urology. 2000;56:641–646. doi: 10.1016/s0090-4295(00)00680-4. [DOI] [PubMed] [Google Scholar]
- 6.Wessells H, Fuciarelli K, Hansen J, Hadley ME, Hruby VJ, Dorr R, Levine N. Synthetic melanotropic peptide initiates erections in men with psychogenic erectile dysfunction: double-blind placebo controlled crossover study. J Urol. 1998;160:389–393. [PubMed] [Google Scholar]
- 7.Bertolini A, Vergoni W, Gessa GL, Ferrari W. Induction of sexual excitement by action of adrenocorticotropic hormone in brain. Nature. 1969;221:667–669. doi: 10.1038/221667a0. [DOI] [PubMed] [Google Scholar]
- 8.Bertolini A, Vergoni W, Gessa GL, Ferrari W. Erection and ejaculation: a central effect of ACTH-like peptides in mammals. In: Sandler M, Gessa GL, editors. Sexual Behavior: Pharmacology and Biochemistry. New York: Raven Press; 1975. pp. 247–257. [Google Scholar]
- 9.Van der Ploeg LHT, Martin WJ, Howard AD, Nargund RP, Austin CP, Guan X, Drisko J, et al. A role for the melanocortin 4 receptor in sexual function. Proc Natl Acad Sci U S A. 2002;99:11381–11386. doi: 10.1073/pnas.172378699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li SJ, Varga K, Archer P, Hruby VJ, Sharma SD, Kesterson RA, Cone RD, et al. Melanocortin antagonists define two distinct pathways of cardiovascular control by α- and γ-melanocyte stimulating hormones. J Neurosci. 1996;16:5182–5188. doi: 10.1523/JNEUROSCI.16-16-05182.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Role of the melanocortinergic neurons in feeding and the Agouti obesity syndrome. Nature. 1997;385:165–168. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- 12.Vergoni AV, Poggioloi R, Bertolini A. Corticotropin inhibits food intake in rats. Neuropeptides. 1986;7:153–158. doi: 10.1016/0143-4179(86)90091-0. [DOI] [PubMed] [Google Scholar]
- 13.Vergoni AV, Poggioloi R, Marrama D, Bertolini A. Inhibition of feeding by ACTH-(1–24): behavioral and pharmacological aspects. Eur J Pharmacol. 1990;179:347–355. doi: 10.1016/0014-2999(90)90175-6. [DOI] [PubMed] [Google Scholar]
- 14.Ramos EJB, Meguid MM, Campos ACL, Coelho JCU. Neuropeptide Y, α-melanocyte-stimulating hormone, and monoamines in food intake regulation. Nutrition. 2005;21:269–279. doi: 10.1016/j.nut.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 15.Yang YK, Harmon CM. Recent developments in our understanding of melanocortin system in the regulation of food intake. Obes Rev. 2003;4:239–248. doi: 10.1046/j.1467-789x.2003.00104.x. [DOI] [PubMed] [Google Scholar]
- 16.Ellacott KLJ, Cone RD. The central melanocortin system and the integration of short- and long-term regulators of energy homeostasis. Recent Prog Horm Res. 2004;59:395–408. doi: 10.1210/rp.59.1.395. [DOI] [PubMed] [Google Scholar]
- 17.Zimanyi IA, Pelleymounter MA. The role of melanocortin peptides and receptors in regulation of energy balance. Curr Pharm Des. 2003;9:627–641. doi: 10.2174/1381612033391234. [DOI] [PubMed] [Google Scholar]
- 18.Morgan C, Thomas RE, Cone RD. Melanocortin-5 receptor deficiency promotes defensive behavior in male mice. Horm Behav. 2004;45:58–63. doi: 10.1016/j.yhbeh.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 19.Mogil JS, Wilson SG, Chesler EJ, Rankin AL, Nemmani KVS, Lariviere WR, Groce MK, et al. The melanocortin-1 receptor gene mediates female-specific mechanisms of analgesia in mice and humans. Proc Natl Acad Sci U S A. 2003;100:4867–4872. doi: 10.1073/pnas.0730053100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gispen WH, Isaacson RL. ACTH-induced excessive grooming in the rat. Pharmacol Ther. 1981;12:209–246. doi: 10.1016/0163-7258(81)90081-4. [DOI] [PubMed] [Google Scholar]
- 21.Gantz I, Fong TM. The melanocortin system. Am J Physiol Endocrinol Metab. 2003;284:E468–E474. doi: 10.1152/ajpendo.00434.2002. [DOI] [PubMed] [Google Scholar]
- 22.Chhajlani B. Distribution of cDNA for melanocortin receptor subtypes in human tissues. Biochem Mol Biol Int. 1996;38:73–80. [PubMed] [Google Scholar]
- 23.Grieco P, Balse PM, Weinberg D, MacNeil T, Hruby VJ. D-Amino acid scan of γ-melanocyte-stimulating hormone: importance of Trp8 on human MC3 receptor selectivity. J Med Chem. 2000;43:4998–5002. doi: 10.1021/jm000211e. [DOI] [PubMed] [Google Scholar]
- 24.Kavarana M, Cai M, Trivedi D, Han G, Hruby VJ. The development of a novel highly selective and potent agonist for human melanocortin 4 receptor. In: Lebl M, Houghten R, editors. Peptides: Wave of the Future, 2nd International and the 17th American Peptide Symposium. Dordrecht: Kluwer Academic Publishers; 2001. pp. 708–709. [Google Scholar]
- 25.Kavarana MJ, Trivedi D, Cai M, Ying J, Hammer M, Cabello C, Grieco P, et al. Novel cyclic templates of α-MSH give highly selective and potent antagonists/agonists for human melanocortin-3/4 receptors. J Med Chem. 2002;45:2644–2650. doi: 10.1021/jm020021z. [DOI] [PubMed] [Google Scholar]
- 26.Grieco P, Lavecchia A, Cai M, Trivedi D, Weinberg D, MacNeil T, Van der Ploeg LHT, et al. Structure–activity studies of the melanocortin peptides: discovery of potent and selective affinity antagonists for the hMC3 and hMC4 receptors. J Med Chem. 2002;45:5287–5294. doi: 10.1021/jm0202526. [DOI] [PubMed] [Google Scholar]
- 27.Cai M, Mayorov AV, Cabello C, Stankova M, Trivedi D, Hruby VJ. Novel 3D pharmacophore of α-MSH/γ-MSH hybrids leads to selective human MC1R and MC3R analogues. J Med Chem. 2005;48:1839–1848. doi: 10.1021/jm049579s. [DOI] [PubMed] [Google Scholar]
- 28.Grieco P, Cai M, Mayorov AV, Trivedi D, Hruby VJ. Structure–activity studies of new melanocortin peptides containing an aromatic amino acid at the N-terminal position. Peptides. 2006;27:472–481. doi: 10.1016/j.peptides.2005.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bednarek MA, MacNeil T, Tang R, Kalyani RN, Van der Ploeg LHT, Weinberg DH. Potent and selective peptide agonists of α-melanotropin action at human melanocortin receptor 4: their synthesis and biological evaluation in vitro. Biochem Biophys Res Commun. 2001;286:641–645. doi: 10.1006/bbrc.2001.5444. [DOI] [PubMed] [Google Scholar]
- 30.Al-Obeidi F, Hadley ME, Pettitt BM, Hruby VJ. Design of a new class of superpotent cyclic α-melanotropins based on quenched dynamic simulations. J Am Chem Soc. 1989;111:3413–3416. [Google Scholar]
- 31.Hruby VJ, Lu D, Sharma SD, Castrucci A, de L, Kesterson RA, Al-Obeidi FA, Hadley ME, et al. Cyclic lactam alpha-melanotropin analogues of Ac-Nle4-cyclo[Asp5, D-Phe7, Lys10] alpha-melanocyte-stimulating hormone-(4–10)-NH2 with bulky aromatic amino acids at position 7 show high antagonist potency and selectivity at specific melanocortin receptors. J Med Chem. 1995;38:3454–3461. doi: 10.1021/jm00018a005. [DOI] [PubMed] [Google Scholar]
- 32.Krchnak V, Vagner J. Color-monitored solid-phase multiple peptide synthesis under low-pressure continuous-flow conditions. Pept Res. 1990;3:182–193. [PubMed] [Google Scholar]
- 33.Kaiser E, Colescott RL, Bossinger CD, Cook PI. Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Anal Biochem. 1970;34:595–598. doi: 10.1016/0003-2697(70)90146-6. [DOI] [PubMed] [Google Scholar]
- 34.Thieriet N, Alsina J, Giralt E, Guibé F, Albericio F. Use of alloc-amino acids in solid-phase peptide synthesis. Tandem deprotection–coupling reactions using neutral conditions. Tetrahedron Lett. 1997;38:7275–7278. [Google Scholar]
- 35.Coste J, Le-Nguyen D, Castro B. PyBOP®: a new peptide coupling reagent devoid of toxic by-product. Tetrahedron Lett. 1990;31:205–208. [Google Scholar]
- 36.Albericio F, Bofill JM, El-Faham A, Kates SA. Use of onium salt-based coupling reagents in peptide synthesis. J Org Chem. 1998;63:9678–9683. [Google Scholar]
- 37.Mayorov AV, Cai M, Chandler KB, Petrov RR, Van Scoy AR, Yu Z, Tanaka DK, et al. Development of cyclic γ-MSH analogues with selective hMC3R agonist and hMC3R/hMC5R antagonist activities. J Med Chem. 2006;49:1946–1952. doi: 10.1021/jm0510326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cai M, Cai C, Mayorov AV, Xiong C, Cabello CM, Soloshonok VA, Swift JR, et al. Biological and conformational study of substituted prolines in MT-II template: steric effects leading to human MC5 receptor selectivity. J Pept Res. 2004;63:116–131. doi: 10.1111/j.1399-3011.2003.00105.x. [DOI] [PubMed] [Google Scholar]
- 39.Haskell-Luevano C, Miwa H, Dickinson C, Hruby VJ, Yamada T, Gantz I. Binding and CAMP studies of melanotropin peptides with the cloned human peripheral melanocortin receptor, hMC1R. Biochem Biophys Res Commun. 1994;204:1137–1142. doi: 10.1006/bbrc.1994.2581. [DOI] [PubMed] [Google Scholar]
- 40.Gantz I, Miwa H, Konda Y, Shimoto Y, Tashiro T, Watson SJ, Delvalle J, et al. Molecular cloning of a novel melanocortin receptor. J Biol Chem. 1993;268:15174–15179. [PubMed] [Google Scholar]
- 41.Ying J, Kçv3r KE, Gu X, Han G, Trivedi DB, Kavarana MJ, Hruby VJ. Solution structures of cyclic melanocortin agonists and antagonists by NMR. Biopolymers. 2003;71:696–716. doi: 10.1002/bip.10596. [DOI] [PubMed] [Google Scholar]
- 42.Han G, Quillan JM, Carlson K, Sadee W, Hruby VJ. Design of novel chimeric melanotropin–deltorphin analogues. Discovery of the first potent human melanocortin 1 receptor antagonist. J Med Chem. 2003;46:810–819. doi: 10.1021/jm020355o. [DOI] [PubMed] [Google Scholar]
- 43.Hruby VJ, Cai M, Grieco P, Han G, Kavarana M, Trivedi D. Exploring the stereostructural requirements of peptide ligands for the melanocortin receptors. Ann N Y Acad Sci. 2003;994:12–20. doi: 10.1111/j.1749-6632.2003.tb03157.x. [DOI] [PubMed] [Google Scholar]
- 44.Cai M, Mayorov AV, Ying J, Stankova M, Trivedi D, Cabello C, Hruby VJ. Design of novel melanotropin agonists and antagonists with high potency and selectivity for human melanocortin receptors. Peptides. 2005;26:1481–1485. doi: 10.1016/j.peptides.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 45.Holder JR, Haskell-Luevano C. Melanocortin ligands: 30 years of structure–activity relationship (SAR) studies. Med Res Rev. 2004;24:325–356. doi: 10.1002/med.10064. [DOI] [PubMed] [Google Scholar]