

Abstract
Background
Mitochondrial DNA (mtDNA) mutations are of increasing interest due to their involvement in aging, disease, fertility, and their role in the evolution of the mitochondrial genome. The presence of reactive oxygen species and the near lack of repair mechanisms cause mtDNA to mutate at a faster rate than nuclear DNA, and mtDNA deletions are not uncommon in the tissues of individuals, although germ-line mtDNA is largely lesion-free. Large-scale deletions in mtDNA may disrupt multiple genes, and curiously, some large-scale deletions persist over many generations in a heteroplasmic state. Here we examine the phenotypic effects of one such deletion, uaDf5, in Caenorhabditis elegans (C. elegans). Our study investigates the phenotypic effects of this 3 kbp deletion.
Results
The proportion of uaDf5 chromosomes in worms was highly heritable, although uaDf5 content varied from worm to worm and within tissues of individual worms. We also found an impact of the uaDf5 deletion on metabolism. The deletion significantly reduced egg laying rate, defecation rate, and lifespan. Examination of sperm bearing the uaDf5 deletion revealed that sperm crawled more slowly, both in vitro and in vivo.
Conclusion
Worms harboring uaDf5 are at a selective disadvantage compared to worms with wild-type mtDNA. These effects should lead to the rapid extinction of the deleted chromosome, but it persists indefinitely. We discuss both the implications of this phenomenon and the possible causes of a shortened lifespan for uaDf5 mutant worms.
Background
Mitochondria have relatively tiny genomes (< 16,000 bp) that support the mitochondrial respiratory chain. These genomes are highly susceptible to mutation [1-3], because mitochondria are the primary source of damaging reactive oxygen species (ROS), and because mitochondria do not have the complete DNA repair repertoire of the nuclear genome [4,5]. Therefore, mitochondrial DNA (mtDNA) experiences mutation at a greater rate than does nuclear DNA, and the asexual nature of mitochondria exposes mtDNA to the mutational meltdown known as Muller's Ratchet [3]. Surprisingly though, most individuals begin life with a near pristine set of mitochondrial chromosomes, due to a unique set of events that conspire to protect and select the "best" mtDNA in the female germ line across generations [6-8]. It is only after the onset of development that mtDNAs begin to accumulate mutations [9,10]. Mutations in the mtDNA generally vary at the level of the tissue, the cell, and even of the mitochondrion. These mtDNA mutations are implicated in the overall metabolic slowdown that accompanies aging as the mutations disrupt the mitochondrial respiratory chain.
When mtDNA mutations slip past the vigilant oocyte screening machinery and are inherited, nearly every mitochondrion in the organism bears the mutation. The effects of these inherited mtDNA mutations are more severe, and in humans, may result in metabolic disorders that affect metabolically active tissues such as muscle and neural tissue, causing defects in cognition, vision, hearing, and muscle function [11-13]. Perhaps less appreciated are the effects of mtDNA mutations on sperm function. In humans and other mammals, mtDNA mutations result in abnormal sperm and loss of fertility [14-16]. In species with female multiple mating and sperm competition, mtDNA mutations may reduce sperm competitiveness to the point that many males fail to reproduce, thereby reducing the effective population size [17]. These effects appear so deleterious that mtDNA mutations should be removed rapidly by natural selection. However, this is not always the case for some of the most severe mutations, such as deletions that remove large segments of mtDNA. Some deletions may persist for many generations in populations [18,19]. While it is not entirely clear why such deleterious deletions persist, an attractive hypothesis is that they result in a replication advantage to the smaller chromosome [20,21].
Here, we examine an experimentally induced deletion in the mtDNA in a strain of the nematode C. elegans. This deletion, uaDf5, removes nearly 25% of the chromosome (3,054 bp), deleting some or all of four protein-coding and seven tRNA genes [22,23]. Tsang and Lemire [22] showed that this deletion persists indefinitely in populations, having remained for several hundred generations in their laboratory. Affected individuals are heteroplasmic: approximately 60% of their mitochondrial chromosomes harbor the deletion, and mutant and wild-type chromosomes most likely exist together in the same mitochondrion [22]. In their description of this heteroplasmic deletion, Tsang and Lemire (2002) showed that homoplasmic individuals never appeared due to forces preventing the elimination of either mtDNA species. They also could not detect any obvious phenotype associated with the uaDf5 deletion. We wondered whether the uaDf5 deletion produced a subtle phenotype, and the experiments we report here reveal a significant impact of this deletion on metabolic rate, reproduction, life span, and sperm function.
Results
PCR deletion assay
In order to examine the effects of the uaDf5 deletion on worm fitness, we developed an assay to estimate the proportion of uaDf5 chromosomes in a given worm or tissue. Our PCR assay involved three primers (Fig. 1): two outside the deletion, which result in amplification only on deleted chromosomes and a third primer, internal to the deletion, which gives amplification only on wild-type chromosomes. Figure 2A shows PCR products of the correct size amplified from single uaDf5; him-8(e1498)IV worms (henceforth, uaDf5; him-8; N.B. him-8 virgin hermaphrodites produce male progeny that we used in subsequent experiments). Worms taken from the same culture differed in the intensities of the deleted and wild-type products. We were able to determine the proportion of uaDf5 DNA in the PCR products by measuring the relative densities of the uaDf5 and wild-type bands in digitized images of gels using ImageJ software [24].
Figure 1.

The C. elegans mitochondrial chromosome showing the uaDf5 deletion (after reference [22]). The coding genes and rRNAs are shown as large arrows. The tRNA genes are designating by the one-letter amino acid code. The short arrows on the interior represent PCR primers: 1 – Beavis; 2 – U1; 3 – Cemt4555; 4 – Cemt5012; 5 – Cemt1A; 6 – U2.
Figure 2.
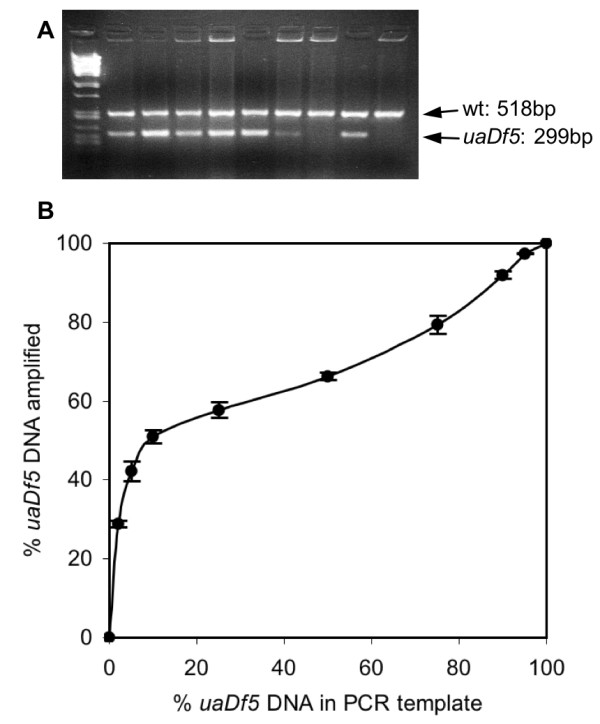
Results of PCR assay for uaDf5 and wild-type mtDNA. (A) Each lane contains the PCR assay products from an individual worm taken arbitrarily from our laboratory stock, except the leftmost lane, which is a molecular size marker (lambda phage genome digested with Pst I). (B) The relationship between the percent of mtDNA molecules with the uaDf5 deletion in the template and the resulting percent of uaDf5 in the amplified products. The percentages of uaDf5 in the template reactions were 0, 2, 5, 10, 25, 50, 75, 90, 95, and 100. Three replicate reactions for each concentration were run. The plotted points represent the mean for the three reactions with the standard deviations shown as error bars.
To determine the proportion of uaDf5 DNA in the template, we performed the PCR deletion assay on known ratios of uaDf5 and wild-type DNA and then compared the template ratios to the product ratios. The assay was very reproducible, but it was especially sensitive to the deleted template molecules, over-estimating their representation when the templates contained small proportions of deleted templates (Fig. 2B). The relationship in Figure 2 was used to calibrate our PCR assay reactions in order to determine the template DNA ratios from our worms. We obtained equations for lines connecting each pair of neighboring points in Fig. 2 and used those equations to estimate the percent of deleted DNA in the template. All of the assay results we report are calibrated and therefore reflect the molar ratios of deleted to wild-type molecules present in the source tissues.
uaDf5 compromises egg laying, defecation, and longevity
We found significant negative correlations between uaDf5 content and the rates of both egg laying (r = -0.450, P = 0.002, N = 44) and defecation (r = -0.387, P = 0.008, N = 46; Fig. 3). When compared with wild-type strain N2, worms harboring the uaDf5 deletion defecated at a slower rate (N2: 1.32 defs/min.; uaDf5: 1.14 defs/min.; F(1,54) = 5.273, P = 0.026) and laid eggs at a slower rate (N2: 9.6 eggs/hour; uaDf5: 6.2 eggs/hour; F(1,54) = 15.239, P = 0.011).
Figure 3.

The rates of egg laying (A) and defecation (B) as a function of uaDf5 content. Age synchronized uaDf5; him-8 worms were assayed for both variables before being subjected to PCR deletion assay.
Longevity was also affected by the uaDf5 mitochondrial deletion. uaDf5; him-8 worms lived significantly shorter lives than did him-8 worms bearing wild-type mitochondrial chromosomes (Fig. 4). Mean lifespan for uaDf5; him-8 worms was 11.5 ± 0.2 (SEM) days (N = 127) compared to 13.9 ± 0.2 days (N = 181) for him-8 (t = 6.45, P < 0.001). Moreover, within the uaDf5 worms, there was a strong negative correlation between survivorship and uaDf5 content (r = -0.868, P < 0.001, N = 45; Fig. 5). It is interesting that the average uaDf5 content of the worms in the longevity experiment (81.2%) was greater than that measured for our worms in our other experiments. Perhaps this is related to age, since these worms were assayed for their uaDf5 content posthumously, compared to worms in our egg-laying and defecation experiments, which were assayed early in adulthood.
Figure 4.

Survivorship of uaDf5; him-8 and him-8 worms at 20°C. Age synchronized uaDf5; him-8 worms (N = 127) and him-8 worms (N = 181) were grown from egg in groups of 5–10 per plate and were transferred to new plates daily during the fertile period to separate them from their offspring. Worms were considered dead when they showed no movement, pharyngeal pumping or response to touch.
Figure 5.

The relationship between uaDf5 content and lifespan for uaDf5; him-8 worms. The dead worms from Figure 4 underwent the PCR deletion assay for uaDf5 content.
uaDf5 reduces sperm crawling rate
Sperm harboring the mitochondrial deletion crawled slower on average than did sperm with wild-type mtDNA (Fig. 6). In vitro, some sperm attach to the substrate by their pseudopodia and crawl; for other sperm, the pseudopodia never attach even though pseudopodial projections treadmill from the tip of the pseudopod to the base. Our estimates of sperm crawling rate included both direct measures of crawling sperm and, when the sperm's pseudopod did not attach, indirect measures using pseudopodial treadmilling speed (averaged from the velocity of two pseudopodial projections for each sperm), the rate of which is known to be equivalent to crawling rate [25,26]. Sperm from him-8 worms crawled at 19.0 ± 0.7 μm/minute (mean ± SEM, N = 61), which is significantly faster than sperm from uaDf5; him-8 at 10.4 ± 0.7 μm/minute (N = 54; F1,114 = 67.79; P < 0.001). Overall, sperm size (cell body diameter) was a significant covariate of sperm crawling rate (F1,114 = 7.289; P = 0.008), but when examined separately, sperm size was a significant predictor of crawling rate for him-8 sperm (r = 0.541; P < 0.001), but not for uaDf5 sperm (r = 0.071; n.s.).
Figure 6.

In vitro motility of uaDf5; him-8 and him-8 sperm as a function of the diameter of the sperm cell body. Sperm motility and its diameter were determined using time-lapse videos and OpenLab™ cell imagimg software.
The uaDf5 deletion also had an impact on sperm crawling in vivo. Spermless fer-1(hc13ts) hermaphrodites were mated to either uaDf5; him-8 males or him-8 males and were examined immediately thereafter. We tallied sperm in two regions of their reproductive tracts: the vulval region, which we defined as within 50 μm of the vulva, and the spermathecal region, which we defined as distal to the vulval region. Each worm had two spermathecal regions because the reproductive tract has two lobes, but only one vulva (Fig. 7). We found a significantly greater proportion of the sperm in the vulval region of worms mated to uaDf5; him-8 males compared to those mated to him-8 males (Fig. 7; t = 5.05; P < 0.001; N = 30). Normal sperm activate immediately after insemination and leave the uterus as they crawl to the spermatheca. At this time after insemination, sperm of uaDf5; him-8 males found near the vulva likely cannot crawl at a normal pace.
Figure 7.

Sperm location in the hermaphrodite reproductive tract. The top portion shows an inverted epifluorescent image of a fer-1 hermaphrodite that was mated to uaDf5; him-8 males. Only one lobe of the reproductive tract is shown, extending from the vulva to the spermathecal region. The lobe extending on the other side of the vulva was also examined. The compact spots representing the sperm nuclei (S) were assigned a location: those within 50 μm of the vulva were assigned to the vulval region, whereas those sperm outside the vulval region were assigned to the spermathecal region. The data in the bottom half of the figure represent the numbers (and percent) of sperm found in the two regions for hermaphrodites mated to either him-8 males (N = 15) or uaDf5; him-8 males (N = 15).
The uaDf5 deletion is highly heritable
Even though the uaDf5 mitochondrial deletion has numerous deleterious effects on worm fitness, we found that it persisted in our laboratory populations indefinitely. Consistent with the findings of Tsang and Lemire [22], our laboratory populations have been maintained for several years without the loss of the uaDf5 deletion, although individual worms do vary in their proportions of uaDf5 chromosomes (Fig. 2A). In an effort to understand the transmission of the deleted chromosomes we assessed heritability by comparing individual hermaphrodites with six of their progeny. Figure 8 shows that the uaDf5 content is highly heritable. Estimating heritability, h2, in the narrow sense by regressing the mean uaDf5 content of the progeny with that of the parent gives an estimate of 0.98 [27]. The variation among full sibs was small: the average standard deviation among each progeny cohort equaled only 6.0%.
Figure 8.

Comparison of the uaDf5 content (%) in hermaphrodites and six of their progeny. uaDf5 inheritance was determined by assaying 10 individual hermaphrodites and six of their progeny.
Heteroplasmic worms are genetic mosaics
We examined the uaDf5 content of the gut, testis, and body remains of male uaDf5; him-8 worms (Fig. 9). The testis and the gut differed in their uaDf5 content, and both differed from the dissected body remains, although the uaDf5 contents of the tissues were correlated. The best correlation occurred between the body remains and the testis (r = 0.720, N = 15, P = 0.002; Fig. 9B), whereas the gut correlated less well with both the testis (r = 0.628, N = 12, P = 0.029; Fig. 9C) and the body remains (r = 0.503, N = 14, P = 0.067; Fig. 9D). The sample sizes varied among the comparisons because the PCR reactions sometimes failed on either the gut or the testis. These results indicate that during development the uaDf5 mitochondrial deletion is not segregated equally among tissues. In fact, the testis of one male apparently lost most if not all wild-type mitochondrial chromosomes.
Figure 9.
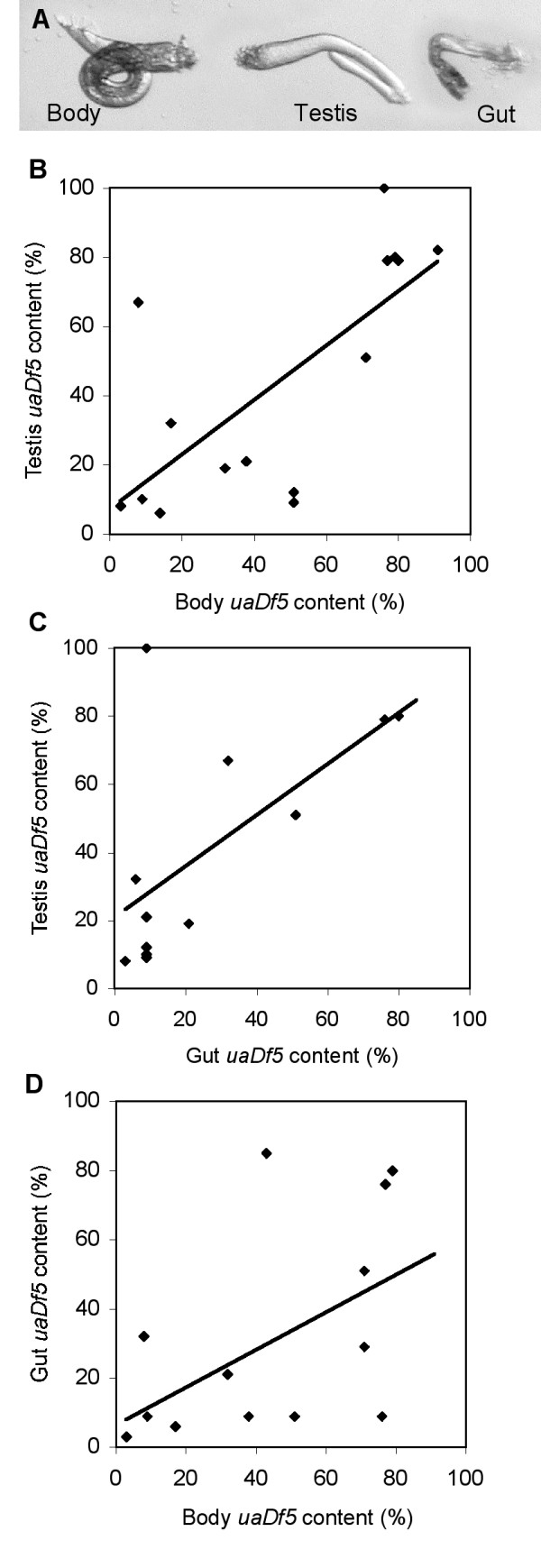
Tissue content of uaDf5 from individual worms. (A) Micrographs of the tissues of a dissected male that were subjected to our PCR assay. (B-C) The relationships between the uaDf5 content of the testis, gut, and body remains.
Discussion and conclusion
The uaDf5 mtDNA deletion has deleterious effects. Its presence reduces the rates of egg-laying, defecation and sperm motility. In fact, the greater the uaDf5 content, the more severe the effect on egg-laying and defecation rates. We infer from these results that the uaDf5 deletion disrupts the function of the mitochondrial respiratory chain, resulting in a metabolic defect. While Tsang and Lemire reported that the uaDf5 deletion had no phenotype [22], the defects we identified are not obvious in laboratory culture, given the inherent capacity for growth exhibited by populations of C. elegans. Indeed, under routine laboratory culture, our uaDf5; him-8 plates appear to grow at rates similar to wild-type. By itself, the effect of the deletion might be greater, but Tsang and Lemire [22] have shown that hermaphrodite uaDf5; him-8 worms have double the normal mtDNA copy number, perhaps as a mechanism to compensate for the deletion. Under such conditions however, the ratios of the respiratory chain components would be altered, a condition that affects respiratory function [28]. Even though the uaDf5 phenotype appears subtle in the laboratory, such phenotypes may lead to severe selective disadvantage. For example, Hodgkin and Barnes [29] showed that a slight increase in generation time (due to a lengthened period of spermatogenesis) led to a competitive disadvantage. In addition, working with the lifespan gene age-1, Walker et al. [30] showed that the conditional allele age-1(hx546), which has no apparent phenotype at 20°C, was at a competitive disadvantage to the wild-type allele under starvation stress. Therefore, even a slight fitness cost will lead to a competitive disadvantage, and our results suggest that uaDf5 should disappear rapidly in nature. Rather than disappear, the uaDf5 deletion persists indefinitely in laboratory populations.
The persistence of the uaDf5 deletion is a puzzling phenomenon, which might be the result of laboratory culture. The worms are kept in the laboratory under near ideal conditions where they have abundant food, optimal hydration, pH, solute availability, etc. Under such conditions, metabolically compromised worms might not suffer dire consequences. However, even under ideal conditions, competition does not cease. Worms that reproduce efficiently should hold an advantage over the less efficient, as Hodgkin and Barnes [29] and Walker et al. [30] found with their laboratory studies. Gemmell et al. [31] theorize that mtDNA mutations that affect only sperm may persist due to the strict maternal inheritance of mitochondria. However, our studies show that hermaphrodites suffer deleterious effects as well. Thus, there appears to be a factor(s) that maintains this deletion in the laboratory populations.
Tsang and Lemire [22] suggested that the "wild-type" mtDNA molecules in heteroplasmic uaDf5 worms may harbor a deleterious mutation. Such a mutation would prevent the appearance of homoplasmic wild-type individuals from heteroplasmic lines. While this is certainly possible, it is also possible that the deleted molecules are maintained in populations due to a replication advantage. Smaller mitochondrial chromosomes are thought to replicate faster, giving them a competitive advantage over larger chromosomes [20,32].
Although resolution of this issue awaits experimentation, it might provide clues to the evolution of the mitochondrial genome. If mitochondrial deletions ever persist in nature like uaDf5 does in laboratory populations, then one mechanism to resolve the deleterious effects is nuclear expression of the deleted genes. This hypothetical mechanism may have driven a large proportion of the ancestral bacterial genes of the mitochondria to the nucleus [32], a process that began perhaps as early as two billion years ago [1]. In support of this hypothesis, the nuclear genome is frequently invaded by mitochondrial DNA. In Arabidopsis thaliana for instance, a complete copy of the mtDNA is duplicated on chromosome II [33]. If some of these insertions pick up mitochondrial insertion sequences [34], the stage would be set for nuclear transfer.
Multiple copies of the mitochondrial chromosomes are bound together with protein forming a nucleoid within each mitochondrion [35,36]. If each chromosome replicates once, new nucleoids should duplicate the "genotype" of the parent nucleoid. Our high estimate of the heritability of the proportion of uaDf5 within worms suggests that faithful duplication of the parental nucleoid genotype occurs with only little deviation in the female germline. Somatically, we found somewhat greater deviation, where the variation of uaDf5 within the tissues of males was more pronounced. Perhaps mtDNA replication in the germline is under more strict control than that in the soma. In fact, our data from the longevity experiment suggest the deletion may increase in frequency with age, since our dead worms had a greater percentage of deleted chromosomes than did our younger worms. Tsang and Lemire [22] showed that uaDf5 content remained constant from the first larval stage through the early adult stage, but they did not look at older worms. It is not unreasonable that the deletion should increase in frequency in older worms, because aging is associated with proliferation of mtDNA deletions in C. elegans [37].
Our longevity experiment also showed that worms harboring the uaDf5 deletion not only died sooner than those with normal mtDNA, but there was a strong negative relationship between uaDf5 content and lifespan. Therefore, the metabolic deficit associated with the uaDf5 deletion reduces lifespan. While many mutations in metabolic genes extend lifespan [38,39], others reduce longevity [40]. For example, mev-1 encodes a subunit of cytochrome b560 [41], a member of the mitochondrial respiratory chain, and the mutation mev-1(kn1) reduces lifespan [42]. The mev-1(kn1) defect is hypothesized to increase ROS production. uaDf5 differs from other metabolic mutations in C. elegans because it is mitochondrial and, rather than introducing a defective protein, it alters the ratio of components of the mitochondrial respiratory chain. This imbalance may produce a respiratory chain bottleneck that not only slows metabolism but also increases production of ROS [28].
Sperm performance was also affected by the uaDf5 deletion. Those sperm bearing the uaDf5 deletion crawled much more slowly than wild-type, both in vitro and in vivo. Deletions in mtDNA have been associated with reduced fertility in mammals including humans [14,15], but the fertility of our worms was not affected by the uaDf5 mutation, even though the sperm crawl slowly. We suspect that in the absence of competition, even slow sperm fertilize eggs efficiently. It may be that a uaDf5 sperm performance phenotype becomes apparent when the sperm are in competition to fertilize oocytes [17], a phenomenon that we will be investigating in future research.
Methods
Worm strains and culture conditions
Growth and handling of C. elegans strains were as described previously [43,44]. The strains N2 and him-8(e1498)IV were provided by the Caenorhabditis Genetics Center (USA). N2 is the designated wild-type strain, and hermaphrodites of the him-8(e1498)IV strain produce male progeny even when unmated. The fer-1(hc13ts)I strain was supplied by S. Ward (University of Arizona), and fer-1 mutant hermaphrodites are sterile due to a sperm defect at 25°C. The uaDf5; him-8(e1498)IV strain was kindly provided by B. Lemire (University of Alberta), who originally isolated the uaDf5 mitochondrial deletion.
Single-worm PCR deletion assay
After extracting DNA from single worms [45], PCR was performed with three primers (see Fig. 1 for locations on the mtDNA): two designed to sites outside the deletion (forward primer U1: 5'-CCATCCGTGCTAGAAGACAA-3', and reverse primer Cemt1A: 5'-CTTCTACAGTGCATTGACCTAGTC-3'), and a third primer internal to the deletion (Cemt5012: 5'-TTGGTGTTACAGGGGCAACA-3'). The two outer primers amplify a 298 bp product only from deleted chromosomes. The internal primer binds only to wild-type chromosomes and amplifies with Cemt1A to produce a 518 bp wild-type product. PCR products were separated on 2% agarose gels in a TBE buffer system and visualized by ethidium bromide staining. Images of the gels were digitized and analyzed for DNA content using ImageJ [24].
To check the accuracy of our PCR assay, we performed the assay on DNA templates that contained known ratios of both deleted and wild-type template molecules. These template molecules were first amplified with primers designed to sites outside the positions of our assay primers. Deleted template was amplified with primers Beavis (5'-AAAATCGTCTAGGGCCCACCAA-3') and U2 (5'-CTCTCCCAACTCGTGTACTG-3'). Wild-type template was amplified with Cemt4555 (5'-GGGATGTTGGTGACATTGCCA-3') and U2. We then cloned the template PCR products using the AccepTor™ vector system and NovaBlue Singles™ competent cells (Novagen). Wild-type and uaDf5 mutant plasmids were isolated from overnight bacterial cultures (37°C). After quantifying the concentration of each plasmid spectrophotometrically, we mixed various molar ratios of the wild-type and uaDf5 mutant template molecules, holding total template concentration at 5 pM. We then ran our PCR assay on the template mixtures.
Egg laying and defecation rate
We assayed the egg-laying and defecation rates for age-synchronized uaDf5; him-8 worms that had been adults for two days. These worms were isolated for two hours on 35 mm plates seeded with OP50 and then removed. We counted the eggs that were laid and calculated the egg-laying rate. Either immediately preceding or just after the egg-laying assay, the defecation rate was measured for each worm. We recorded the interval between the obvious posterior body contractions that load the rectum for defecation [46]. The defecation interval was measured three times for each worm, and after the defecation and egg-laying assays, the worms were subjected to the PCR deletion assay. The relationships between uaDf5 content and egg-laying and defecation rates were examined using correlation analysis, and worm strains were compared with ANOVA.
Lifespan
We assessed the lifespan of another set of worms as described by Van Voorhies and Ward [39]. Age-synchronized cohorts of worms were grown from egg at 20°C. After two days, L4 hermaphrodites were picked to new plates (5–10 worms per plate on 35 mm plates). These fertile hermaphrodites were transferred to new plates daily during the fertile period to distinguish them from their offspring and to prevent them from mating with their male progeny, which might reduce hermaphrodite life span [47]. Worms were recorded as dead when they had no pharyngeal pumping, no movement and no response to touch. Individual uaDf5 worm corpses were subjected to the PCR deletion assay described above. Differences in longevity between worm strains were examined using t-Tests, and we used correlation analysis to determine the relationship between longevity and uaDf5 content.
Sperm crawling performance
We evaluated the crawling performance of sperm from uaDf5; him-8 males in two experiments. In the first, we measured crawling rate for these amoeboid sperm cells in vitro. Two days after they were isolated as L4 larvae (e.g. two-day-old virgin), males were dissected on poly-L-lysine coated slides under SM1 buffer [48] containing 200 μg/ml Pronase, a protease mixture that causes the released spermatids to undergo spermiogenesis and begin to crawl [49]. Time-lapse videos of sperm motility were captured, and sperm crawling rate was measured with OpenLab™ cell imaging software. In addition, the diameter of the cell body was measured and used as an indication of sperm cell size [50]. The crawling rate of him-8 sperm was measured for comparison.
Sperm crawling performance was also analyzed in vivo. One-day-old virgin uaDf5; him-8 or him-8 males were paired for two hours on 35 mm Petri dishes with fer-1(hc13ts) adult hermaphrodites at a 1:3 ratio. The fer-1(hc13ts) mutation disrupts spermatogenesis, producing self-sterile hermaphrodites at 25°C [51]. Therefore, any sperm present in the fer-1 hermaphrodites came from their male partners. Immediately after the mating interval, the fer-1 hermaphrodites were fixed in ethanol on microscope slides and stained with the DNA label DAPI (4',6-diamidino-2-phenylindole; 20 μg/ml in PBS) to visualize the nuclei [50]. The specimens were flattened under a coverslip until all the nuclei were within one plane of focus under epifluorescence, and the characteristically compact sperm nuclei were counted on captured images of the specimens. Specifically, we counted the number of sperm that were within 50 μm of the vulva, an indication that those sperm had not crawled very far into the hermaphrodite reproductive tract. We calculated the proportion of these "slow" sperm compared with the sperm that had moved farther into the reproductive tract, and we evaluated the difference between the uaDf5; him-8 and him-8 strains by t-Test after arcsine transformation of the proportions.
uaDf5 tissue content
In order to compare the proportion of deleted mitochondrial chromosomes within the tissues of single worms, we dissected males under M9 buffer [44]. The males were cut with a micro-scalpel just behind the pharynx. After a short time, the exposed gut and testis were dissected away from the remains of the body. The gut, testis, and body remains were each subjected individually to our PCR deletion assay and the uaDf5 content compared among tissues by correlation.
Inheritance and persistence of uaDf5
We determined the inheritance of the uaDf5 deletion by individually assaying hermaphrodites and six of their progeny. The parents were allowed to lay eggs for one day before undergoing DNA extraction. Six of their hermaphrodite progeny were arbitrarily chosen and subjected to DNA extraction. The PCR deletion assay was run on each "family" at once, and the proportion of uaDf5 DNA of the parents was compared to that of the offspring.
Authors' contributions
WSL and AGS contributed equally, conducting the lifespan study, taking part in the egg-laying rate, defecation rate, and sperm crawling rate studies, and contributing to manuscript preparation. CD conducted the heritability and mosaicism studies, and she read and approved the manuscript. KC performed the egg-laying and defecation rate studies and contributed to manuscript preparation. CWL designed the PCR assay and its calibration, conducted the in vitro sperm crawling rate study, and contributed to manuscript preparation. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
We thank Oussama Bouban and Derek Rosenzweig for technical assistance. Several worm strains were kindly provided by the Caenorhabditis Genetics Center, which is funded by the NIH National Center for Research Resources. Bernard Lemire graciously supplied the strain uaDf5; him-8. This research was funded by a grant from the U.S. National Institutes of Health (Award 5 S06 GM053933-10 to CWL).
Contributor Information
Wei-Siang Liau, Email: wliau@csupomona.edu.
Aidyl S Gonzalez-Serricchio, Email: aidyl@csupomona.edu.
Cleonique Deshommes, Email: nique07@yahoo.com.
Kara Chin, Email: karachin20@hotmail.com.
Craig W LaMunyon, Email: cwlamunyon@csupomona.edu.
References
- Andersson SGE, Kurland CG. Reductive evolution of resident genomes. Trends Microbiol. 1998;6:263–268. doi: 10.1016/S0966-842X(98)01312-2. [DOI] [PubMed] [Google Scholar]
- Berg OG, Kurland CG. Why mitochondrial genes are most often found in nuclei. Mol Biol Evol. 2000;17:951–961. doi: 10.1093/oxfordjournals.molbev.a026376. [DOI] [PubMed] [Google Scholar]
- Lynch M. Mutation accumulation in transfer RNAs: molecular evidence for Muller's ratchet in mitochondrial genomes. Mol Biol Evol. 1996;13:209–220. doi: 10.1093/oxfordjournals.molbev.a025557. [DOI] [PubMed] [Google Scholar]
- Bohr VA. Repair of oxidative DNA damage in nuclear and mitochondrial DNA, and some changes with aging in mammalian cells. Free Radic Biol Med. 2002;32:804–812. doi: 10.1016/S0891-5849(02)00787-6. [DOI] [PubMed] [Google Scholar]
- Croteau DL, Stierum RH, Bohr VA. Mitochondrial DNA repair pathways. Mutat Res. 1999;434:137–148. doi: 10.1016/s0921-8777(99)00025-7. [DOI] [PubMed] [Google Scholar]
- Jansen RP, de Boer K. The bottleneck: mitochondrial imperatives in oogenesis and ovarian follicular fate. Mol Cell Endocrinol. 1998;145:81–88. doi: 10.1016/S0303-7207(98)00173-7. [DOI] [PubMed] [Google Scholar]
- Cummins JM. The role of mitochondria in the establishment of oocyte functional competence. Eur J Obstet Gynecol Reprod Biol. 2004;115 Suppl 1:S23–9. doi: 10.1016/j.ejogrb.2004.01.011. [DOI] [PubMed] [Google Scholar]
- Hsieh RH, Tsai NM, Au HK, Chang SJ, Wei YH, Tzeng CR. Multiple rearrangements of mitochondrial DNA in unfertilized human oocytes. Fertil Steril. 2002;77:1012–1017. doi: 10.1016/S0015-0282(02)02994-1. [DOI] [PubMed] [Google Scholar]
- Kraytsberg Y, Nekhaeva E, Bodyak NB, Khrapko K. Mutation and intracellular clonal expansion of mitochondrial genomes: two synergistic components of the aging process? Mech Ageing Dev. 2003;124:49–53. doi: 10.1016/S0047-6374(02)00169-0. [DOI] [PubMed] [Google Scholar]
- Dufour E, Larsson NG. Understanding aging: revealing order out of chaos. Biochim Biophys Acta. 2004;1658:122–132. doi: 10.1016/j.bbabio.2004.04.020. [DOI] [PubMed] [Google Scholar]
- Gattermann N, Retzlaff S, Wang YL, Hofhaus G, Heinisch J, Aul C, Schneider W. Heteroplasmic point mutations of mitochondrial DNA affecting subunit I of cytochrome c oxidase in two patients with acquired idiopathic sideroblastic anemia. Blood. 1997;90:4961–4972. [PubMed] [Google Scholar]
- Schon EA, Bonilla E, DiMauro S. Mitochondrial DNA mutations and pathogenesis. J Bioenerg Biomembr. 1997;29:131–149. doi: 10.1023/A:1022685929755. [DOI] [PubMed] [Google Scholar]
- DiMauro S, Andreu AL. Mutations in mtDNA: are we scraping the bottom of the barrel? Brain Pathol. 2000;10:431–441. doi: 10.1111/j.1750-3639.2000.tb00275.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao SH, Chao HT, Wei YH. Multiple deletions of mitochondrial DNA are associated with the decline of motility and fertility of human spermatozoa. Mol Hum Reprod. 1998;4:657–666. doi: 10.1093/molehr/4.7.657. [DOI] [PubMed] [Google Scholar]
- Nayernia K, Adham IM, Burkhardt-Gottges E, Neesen J, Rieche M, Wolf S, Sancken U, Kleene K, Engel W. Asthenozoospermia in mice with targeted deletion of the sperm mitochondrion-associated cysteine-rich protein (Smcp) gene. Mol Cell Biol. 2002;22:3046–3052. doi: 10.1128/MCB.22.9.3046-3052.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St John JC, Jokhi RP, Barratt CL. Men with oligoasthenoteratozoospermia harbour higher numbers of multiple mitochondrial DNA deletions in their spermatozoa, but individual deletions are not indicative of overall aetiology. Mol Hum Reprod. 2001;7:103–111. doi: 10.1093/molehr/7.1.103. [DOI] [PubMed] [Google Scholar]
- Gemmell NJ, Sin FY. Mitochondrial mutations may drive Y chromosome evolution. Bioessays. 2002;24:275–279. doi: 10.1002/bies.10062. [DOI] [PubMed] [Google Scholar]
- Beziat F, Touraille S, Debise R, Morel F, Petit N, Lecher P, Alziari S. Biochemical and molecular consequences of massive mitochondrial gene loss in different tissues of a mutant strain of Drosophila subobscura. J Biol Chem. 1997;272:22583–22590. doi: 10.1074/jbc.272.36.22583. [DOI] [PubMed] [Google Scholar]
- Petit N, Touraille S, Debise R, Morel F, Renoux M, Lecher P, Alziari S. Developmental changes in heteroplasmy level and mitochondrial gene expression in a Drosophila subobscura mitochondrial deletion mutant. Curr Genet. 1998;33:330–339. doi: 10.1007/s002940050344. [DOI] [PubMed] [Google Scholar]
- Diaz F, Bayona-Bafaluy MP, Rana M, Mora M, Hao H, Moraes CT. Human mitochondrial DNA with large deletions repopulates organelles faster than full-length genomes under relaxed copy number control. Nucleic Acids Res. 2002;30:4626–4633. doi: 10.1093/nar/gkf602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selosse MA, Albert BR, Godelle B. Reducing the genome size of organelles favours gene transfer to the nucleus. Trends Ecol Evol. 2001;16:135–141. doi: 10.1016/S0169-5347(00)02084-X. [DOI] [PubMed] [Google Scholar]
- Tsang WY, Lemire BD. Stable heteroplasmy but differential inheritance of a large mitochondrial DNA deletion in nematodes. Biochem Cell Biol. 2002;80:645–654. doi: 10.1139/o02-135. [DOI] [PubMed] [Google Scholar]
- Lemire B. Mitochondrial genetics. In: The C elegans Research Community , editor. WormBook. 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramoff MD, Magelhaes PJ, Ram SJ. Image processing with ImageJ. Biophotonics Int. 2004;11:36–42. [Google Scholar]
- Roberts RM, King KL. Centripetal flow and directed reassembly of the major sperm protein (MSP) cytoskeleton in the amoeboid sperm of the nematode, Ascaris suum. Cell Motil Cytoskel. 1991;20:228–241. doi: 10.1002/cm.970200306. [DOI] [PubMed] [Google Scholar]
- Ward S, Hogan E, Nelson GA. The initiation of spermiogenesis in the nematode Caenorhabditis elegans. Dev Biol. 1983;98:70–79. doi: 10.1016/0012-1606(83)90336-6. [DOI] [PubMed] [Google Scholar]
- Falconer DS, Mackay TFC. Introduction to Quantitative Genetics. 4. Essex , Longman Group, Ltd.; 1996. [Google Scholar]
- Rossignol R, Faustin B, Rocher C, Malgat M, Mazat JP, Letellier T. Mitochondrial threshold effects. Biochem J. 2003;370:751–762. doi: 10.1042/BJ20021594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin J, Barnes TM. More is not better: brood size and population growth in a self-fertilizing nematode. Proc R Soc Lond B. 1991;246:19–24. doi: 10.1098/rspb.1991.0119. [DOI] [PubMed] [Google Scholar]
- Walker DW, McColl G, Jenkins NL, Harris J, Lithgow GJ. Evolution of lifespan in C. elegans. Nature. 2000;405:296–297. doi: 10.1038/35012693. [DOI] [PubMed] [Google Scholar]
- Gemmell NJ, Metcalf VJ, Allendorf FW. Mother's curse: the effect of mtDNA on individual fitness and population viability. Trends Ecol Evol. 2004;19:238–244. doi: 10.1016/j.tree.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Blanchard JL, Lynch M. Organellar genes - why do they end up in the nucleus? Trends Genet. 2000;16:315–320. doi: 10.1016/S0168-9525(00)02053-9. [DOI] [PubMed] [Google Scholar]
- Lin X, Kaul S, Rounsley S, Shea TP, Benito MI, Town CD, Fujii CY, Mason T, Bowman CL, Barnstead M, Feldblyum TV, Buell CR, Ketchum KA, Lee J, Ronning CM, Koo HL, Moffat KS, Cronin LA, Shen M, Pai G, Van Aken S, Umayam L, Tallon LJ, Gill JE, Adams MD, Carrera AJ, Creasy TH, Goodman HM, Somerville CR, Copenhaver GP, Preuss D, Nierman WC, White O, Eisen JA, Salzberg SL, Fraser CM, Venter JC. Sequence and analysis of chromosome 2 of the plant Arabidopsis thaliana. Nature. 1999;402:761–768. doi: 10.1038/45471. [DOI] [PubMed] [Google Scholar]
- Martin W, Herrmann RG. Gene transfer from organelles to the nucleus: How much, what happens, and why? Plant Physiol. 1998;118:9–17. doi: 10.1104/pp.118.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battersby BJ, Shoubridge EA. Selection of a mtDNA sequence variant in hepatocytes of heteroplasmic mice is not due to differences in respiratory chain function or efficiency of replication. Hum Mol Genet. 2001;10:2469–2479. doi: 10.1093/hmg/10.22.2469. [DOI] [PubMed] [Google Scholar]
- Kaufman BA, Newman SM, Hallberg RL, Slaughter CA, Perlman PS, Butow RA. In organello formaldehyde crosslinking of proteins to mtDNA: identification of bifunctional proteins. Proc Natl Acad Sci USA. 2000;97:7772–7777. doi: 10.1073/pnas.140063197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melov S, Lithgow GJ, Fischer DR, Tedesco PM, Johnson TE. Increased frequency of deletions in the mitochondrial genome with age of Caenorhabditis elegans. Nucleic Acids Res. 1995;23:1419–1425. doi: 10.1093/nar/23.8.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Voorhies WA. Is life span extension in single gene long-lived Caenorhabditis elegans mutants due to hypometabolism? Exp Gerontol. 2003;38:615–618. doi: 10.1016/S0531-5565(03)00070-6. [DOI] [PubMed] [Google Scholar]
- Van Voorhies WA, Ward S. Genetic and environmental conditions that increase longevity in Caenorhabditis elegans decrease metabolic rate. Proc Natl Acad Sci USA. 1999;96:11399–11403. doi: 10.1073/pnas.96.20.11399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braeckman BP, Houthoofd K, Vanfleteren JR. Patterns of metabolic activity during aging of the wild type and longevity mutants of Caenorhabditis elegans. Age. 2000;23:55–73. doi: 10.1007/s11357-000-0007-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii N, Fujii M, Hartman PS, Tsuda M, Yasuda K, Senoo-Matsuda N, Yanase S, Ayusawa D, Suzuki K. A mutation in succinate dehydrogenase cytochrome b causes oxidative stress and ageing in nematodes. Nature. 1998;394:694–697. doi: 10.1038/29331. [DOI] [PubMed] [Google Scholar]
- Ishii N, Takahashi K, Tomita S, Keino T, Honda S, Yoshino K, Suzuki K. A methyl viologen-sensitive mutant of the nematode Caenorhabditis elegans. Mutat Res. 1990;237:165–171. doi: 10.1016/0921-8734(90)90022-j. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulston J, Hodgkin J. Methods. In: Wood WB, editor. The Nematode Caenorhabditis elegans. Cold Spring Harbor, NY USA , Cold Spring Harbor Laboratory Monograph 17; 1988. p. 591. [Google Scholar]
- Williams BD. Genetic mapping with polymorphic sequence-tagged sites. In: Epstein HF, Shakes DC, editor. Caenorhabditis elegans: Modern Biological Analysis of an Organism. Vol. 48. San Diego , Academic Press; 1995. pp. 81–96. (Methods in Cell Biology). Wilson L., Matsudaira P. [DOI] [PubMed] [Google Scholar]
- Walker DS, Gower NJ, Ly S, Bradley GL, Baylis HA. Regulated disruption of inositol 1,4,5-trisphosphate signaling in Caenorhabditis elegans reveals new functions in feeding and embryogenesis. Mol Biol Cell. 2002;13:1329–1337. doi: 10.1091/mbc.01-08-0422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gems D, Riddle DL. Longevity in Caenorhabditis elegans reduced by mating but not gamete production. Nature. 1996;379:723–725. doi: 10.1038/379723a0. [DOI] [PubMed] [Google Scholar]
- Machaca K, DeFelice LJ, L'Hernault SW. A novel chloride channel localizes to Caenorhabditis elegans spermatids and chloride channel blockers induce spermatid differentiation. Dev Biol. 1996;176:1–16. doi: 10.1006/dbio.1996.9999. [DOI] [PubMed] [Google Scholar]
- Shakes DC, Ward S. Initiation of spermiogenesis in C. elegans: a pharmacological and genetic analysis. Dev Biol. 1989;134:189–200. doi: 10.1016/0012-1606(89)90088-2. [DOI] [PubMed] [Google Scholar]
- LaMunyon CW, Ward S. Larger sperm outcompete smaller sperm in the nematode Caenorhabditis elegans. Proc Biol Sci. 1998;265:1997–2002. doi: 10.1098/rspb.1998.0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achanzar WE, Ward S. A nematode gene required for sperm vesicle fusion. J Cell Science. 1997;110:1073–1081. doi: 10.1242/jcs.110.9.1073. [DOI] [PubMed] [Google Scholar]