

Abstract
The central glycolytic genes repressor (CggR) controls the transcription of the gapA operon encoding five key glycolytic enzymes in Bacillus subtilis. CggR recognizes a unique DNA target sequence comprising two direct repeats and fructose-1,6-bisphosphate (FBP) is the inducer that negatively controls this interaction. We present here analytical ultracentrifugation and fluorescence anisotropy experiments that demonstrate that CggR binds as a tetramer to the full-length operator DNA in a highly cooperative manner. We also show that CggR binds as a dimer to each direct repeat, the affinity being ∼100-fold higher for the 3′ repeat. In addition, our studies reveal a bimodal effect of FBP on the repressor/operator interaction. At micromolar concentrations, FBP leads to a change in the conformational dynamics of the complex. In the millimolar range, without altering the stoichiometry, FBP leads to a drastic reduction in the affinity and cooperativity of the complex. This bimodal response suggests the existence of two sugar-binding sites in the repressor, a high affinity site at which FBP acts as a structural co-factor and a low affinity site underlying the molecular mechanism of gapA induction.
INTRODUCTION
In bacteria, gene expression is primarily controlled at the level of transcription through complex interactions between regulatory proteins and operator DNA sequences, which are further modulated by a number of effector molecules. Understanding the mechanisms of transcriptional regulation requires a thorough physicochemical analysis of the parameters governing these DNA/protein/ligand linked equilibria. Recent bacterial genomic studies have lead to the discovery of a plethora of putative transcriptional regulators, defining new families and subfamilies of regulatory proteins for which nothing or very little is known. A major challenge consists now in characterizing prototype members of these families and elucidating the molecular basis of these new regulation systems.
The central glycolytic genes repressor (CggR) is one of the transcriptional repressors that have been recently identified by functional analysis of the Bacillus subtilis genome (1,2). CggR belongs to a family of proteins that control the expression of genes and operons involved in the metabolism of sugar substrates. These proteins present a variable helix-turn-helix motif at their N-terminus, probably involved in DNA binding, followed by a putative sugar-binding domain modulating the repressor activity. SorC from Klebsiella pneumoniae, that regulates the expression of sorbose assimilation operons, is the prototype of this family. So far, besides CggR, only DeoR from Bacillus subtilis (not related to the well-characterized DeoR from Escherichia coli) has been purified and characterized in vitro (3). SorC/bsDeoR on one hand and CggR on the other define two subfamilies of repressors that can be distinguished on the bases of their amino sequences (4).
CggR plays a central role in the regulation of the carbon flux through the main carbohydrate catabolic pathways, glycolysis (5). The central part of glycolysis consists of five steps leading to the conversion of dihydroxyacetone-phosphate into phosphoenolpyruvate. In B. subtilis, these reactions are catalyzed by enzymes whose genes are physically linked in the CggR-regulated gapA operon. (1,6). The most strongly regulated gene of this operon is gapA, encoding a glyceraldehyde-3-phosphate dehydrogenase, which is active only in the catabolic direction, the corresponding anabolic reaction being catalyzed by a different enzyme encoded by the gapB gene (2). CggR is encoded by the first gene of the hexacistronic gapA operon and it exerts its transcriptional regulation activity through conditional binding to a DNA operator in the gapA leader region (4). Recently, the sequence of the CggR binding site located in between the promoter of the cggR-gapA genes and the start of the coding sequence was determined (4). The DNA fragment protected from nuclease attack upon CggR binding spans over 45 basepairs (bp) comprising two long direct repeats. A short palindrome is present in each direct repeat, resulting in four regularly spaced elements separated by 10 nucleotides (Fig. 1). However, there is so far no evidence that these DNA elements constitute a common recognition motif for CggR oligomers. Fructose-1,6-bisphosphate (FBP) has been identified as the effector sugar, which modulates the interaction of CggR with its target DNA, inducing gapA transcription when cells are grown in the presence of glucose or other carbohydrates that are catabolized via glycolysis or the pentose phosphate pathways (6). FBP is also a key metabolic signal for carbon catabolite repression mediated by the pleiotropic regulator CcpA, which controls indirectly CggR activity (7). Indeed, FBP is a well-suited modulating molecule, since its concentration in B. subtilis varies considerably, depending on the nature of the carbon sources available. It has been shown, for instance, that its concentration is 10 times higher in the bacterial cells grown in presence of glucose than in presence of succinate (8).
FIGURE 1.

The B. subtilis gapA operator region protected upon CggR binding, as reported in Doan and Aymerich (4). The sequence of the full-length operator DNA (OLR), and the two half-site operator DNAs (OL and OR) used in this study are indicated by dotted lines. The tandem direct repeat and the regularly spaced inverted repeats of GAC triplets are also indicated (black arrows). Aligned sequences of the gapA operator region from different Bacillus species are shown (Bsub, B. subtilis; Bhal, B. halodurans; Bste, B. stearothermophilus; Bant, B anthracis; Bmeg, B megaterium).
In the present work, using a combination of fluorescence-based techniques and analytical ultracentrifugation, we have assessed the stoichiometry, energetics, and dynamics of CggR binding to its operator DNA and the mechanism of modulation of this interaction by the sugar effector, FBP. We have determined the stoichiometry and affinity of the final and intermediate CggR complexes, free or liganded by FBP, with the full operator DNA. Using target DNAs containing each of the tandem repetitions separately, we have been able to determine the affinity of CggR for each operator half site and thereby evaluate the intrinsic cooperativity of DNA binding in absence and in presence of the modulator sugar. Our results suggest that induction of gapA transcription is linked to a bi-modal effect of the inducer sugar on the cooperative binding to DNA of the tetrameric repressor. Given the high level of homology between CggR and other members of the SorC family, as well as the sequence similarity of the corresponding operators in different Gram positive bacteria, the present results are likely to be relevant to the mechanisms of gene expression regulation by other members of this family of newly identified regulatory proteins.
MATERIALS AND METHODS
Protein expression and purification
All experiments were conducted using an N-terminal His-tagged CggR construct, which has been shown to behave as the wild-type repressor in vivo (4). Purification was performed by affinity chromatography, essentially as described in Doan and Aymerich (4), followed by size exclusion chromatography for the protein preparations used in fluorescent anisotropy or ultracentrifugation experiments. An E. coli M15-pREP4 strain transformed with a pQE30 (Quiagene, Hilden, Germany) derivative encoding His6-CggR was grown overnight in LB medium at 37°C in presence of ampicillin and kanamycin. A 20-fold dilution of the initial culture was grown at 37°C until an OD600 of 0.8 was reached and after addition of 0.25 mM IPTG, the culture was resumed for 4 h at 28–30°C to allow CggR overproduction. Cells were harvested by 15 min centrifugation at 8000 rpm (12,000 g), washed with 10 mM Tris-HCl pH 7.5, 100 mM NaCl and frozen at −80°C until use. The cell pellet was thawed and resuspended in 50 mM Tris-HCl, 100 mM NaCl, 10 mM imidazole, 3 mM β-mercaptoethanol, 1 mM benzamidine, pH 8 buffer (HT buffer) containing 5 mM benzamidine and 0.3% v/v of antiprotease cocktail (Roche, Nutley, NJ). Cells were lysed by sonication after 30 min incubation on ice with 0.2 mg/mL of lysozyme together with 2 μg/mL of DNase 1 and 60 mM Na2HPO4. After centrifugation at 18,000 rpm (40,000 g) for 20 min at 4°C, the cell supernatant was loaded onto a Ni2+-NTA agarose resin (Qiagen, Germantown, MD) and incubated for ∼30 min at 4°C with gentle shaking. The resin was then washed extensively with HT buffer supplemented with, successively, 0.125% glycerol and 50 mM Na2HPO4, 1 M NaCl, 0.4 M KCl, and 20 mM imidazole, before eluting the protein with a 70–300 mM gradient of imidazole. The most concentrated fractions were loaded onto a Superdex 200 (Pharmacia, Peapack, NJ) AK16/70 column for gel filtration in 10 mM Tris-HCl, 100 mM NaCl, 2 mM DTT, and 2 mM EDTA buffer, pH 8. The pure and most concentrated fractions were pooled and protein aliquots were stored at −80°C. Protein concentration was determined by three independent methods—Bradford assay, BCA test and by the UV absorbance at 280 nm using a theoretical extinction coefficient of 14,770 M−1 cm−1.
Fluorescent labeling of DNA oligonucleotides
The oligonucleotides containing the full CggR binding site (Fig. 1) were purchased from GENSET (Paris, France) in HPLC purified form. The sense oligonucleotide was already labeled on its 5′ end with a fluorescein phosphoramidate bearing a six-carbon linker. The labeling ratio, estimated using extinction coefficients of 73,000 and 452,800 M−1 cm−1 for fluorescein at 495 nm and for the DNA at 260 nm, respectively, was ∼70%. The oligonucleotides containing the half-sites, OL and OR operator (Fig. 1), were purchased from Sigma-Genosys (Heverhill, United Kingdom) in HPLC purified form and derivatized with an aminolink-2, which provides a primary amino group separated from the 5′ end by a six-carbon alkyl chain. Sense oligonucleotides were covalently labeled with Alexa Fluor 488 succinimidyl ester (Molecular Probes, Invitrogen, Eugene, OR). For the labeling, the DNA oligonucleotides were further purified by chloroform extraction followed by ethanol precipitation and mixed with a 20-fold excess of the fluorophore in 0.1 M sodium borate buffer, pH 9. The labeling reaction was allowed to proceed overnight at room temperature with continuous shaking. Most of the unreacted dye was eliminated by ethanol precipitation and the DNA was dissolved in 0.05 M TEAA buffer and loaded onto a C 18 column to separate labeled from unlabeled DNA by reverse phase chromatography. The degree of labeling was determined by using extinction coefficients of 71,000, 219,940, and 229,494 M−1 cm−1 for the fluorophore at 495 nm, for the OL and OR operator oligonucleotides at 260 nm, respectively, yielding a labeling ratio for the OL operator of 20% and for the OR DNA of 50%. The extinction coefficients for the DNA were given by the supplier. The sense and anti-sense strands were hybridized by heating a mixture containing a 10% excess of the nonlabeled strand to 90–95 C for 10 min and slowly cooling down to room temperature using a thermocycler.
Analytical ultracentrifugation
Analytical ultracentrifugation experiments were performed using an Optima XL-A analytical ultracentrifuge (Beckman-Coulter, Palo Alto, CA) equipped with an UV-visible absorbance detection system. A Ti60 eight-hole rotor and standard (12-mm optical path) double sector (sedimentation velocity) or six-channel (sedimentation equilibrium) centerpieces of Epon-charcoal were used. Absorbance scans of samples containing fluorescein or Alexa 488 labeled DNA were carried out at 495 nm; in the case of nonlabeled DNA, the detection wavelength was 260 nm. The buffer was 50 mM Tris-HCl, 150 mM NaCl, 0.05 mg/mL BSA, 2 mM EDTA, 2 mM TCEP, pH 8. When detected at 260 nm, BSA was removed from the buffer. In the sedimentation velocity experiments (which were carried out at 10°C) the samples were centrifuged at 40,000 rpm. Short-column (70 μl) sedimentation equilibrium was performed at 5°C at two consecutive speeds (8000 and 13,000 rpm) with a final centrifugation at high speed (50,000 rpm) to determine the baseline offset.
The sedimentation coefficient distributions were determined by direct linear least-squares boundary fitting of the sedimentation velocity profiles using the software SEDFIT (9). All the s-values reported were corrected to standard conditions (20°C and water) using the software SEDNTERP (retrieved from the RASMB server (10)). The same software was used to estimate the partial specific volume of CggR from its amino-acid sequence and the buffer viscosity and density at different temperatures. Whole-cell weight average buoyant molecular weights (bMW values) were obtained by fitting the equation describing the radial concentration distribution of an ideal solute at sedimentation equilibrium to the experimental data using the program EQASSOC (11). Because the partial specific volumes of protein (0.74 ml/g) and DNA (0.54 ml/g) are very different, the analysis to obtain the molecular weight of the macromolecular mixtures from the corresponding buoyant masses is not straightforward. Therefore the sedimentation equilibrium data of the macromolecular mixtures were analyzed assuming the linear approximation for the buoyant masses,
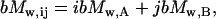 |
(1) |
where ij refers to the complex AiBj, and bMw,A and bMw,B are the bMW values of pure A and pure B, respectively (12). In addition, the sedimentation equilibrium gradients were analyzed using a model of mixtures of independently sedimenting solutes with the program MULTMX (kindly provided by Dr. Allen Minton, National Institutes of Health, Bethesda, MD).
Fluorescence anisotropy binding assays
Steady-state fluorescence anisotropy experiments were performed using a Beacon 2000 Fluorescence Polarization System (Panvera, Madison, WI) equipped with automatic temperature control. The buffer for the measurements was 50 mM Tris-HCl, 150 mM NaCl, 1% glycerol, 0.05 mg/mL BSA, 2 mM EDTA, 2 mM DTT, pH 8, unless otherwise stated and the temperature was always 21°C. DNA titrations were performed by adding increasing concentrations of purified protein to independent tubes containing the labeled DNA with or without FBP. The final concentration of OLR, OL, and OR operator in the titrations was 0.9, 5.1, and 1.7 nM, respectively. Anisotropy values plotted represent the average of 5–7 values measured by the instrument after equilibration. Each curve is the average of 3–5 independent experiments.
Analysis of the binding curves was performed by using Bioeqs software (13), which makes use of a numerical solver engine that permits to determine the free energies of formation of the postulated complexes from the individual elements. The input parameters for the analysis using this software are the values for the total concentration of all the individual elements at each concentration point in the titration and those of the free energies of formation of all the postulated complexes from these elements. Given those initial values, the program solves the set of nonlinear free energy equations for the species concentration vector using a constrained optimization algorithm and incorporating the total elemental concentration constraints as Lagrange multipliers (13). Using the values for the anisotropy of each of the proposed species, BIOEQS then calculates the anisotropy curve for each titration and then uses a Marquardt-Levenberg algorithm to adjust all of the parameter values (anisotropies and free energies) to best fit the data. The error in the determined free energies is then estimated using the rigorous confidence limit testing at the 67% confidence level, in which the uncertainties arising from parameter correlations were taken into account.
Time-resolved fluorescence
Time-resolved fluorescence measurements were carried out using an ISS KOALA sample chamber and ISS analog frequency domain electronics (ISS, Champaign IL). Excitation was performed at 450 nm by frequency doubling of the pulse picked emission at 900 nm of a Spectra Physics Tsunami tunable Ti:sapphire infra-red mode-locked picosecond laser pumped by a 10 watt Millennia diode laser (Newport-Spectraphysics, Mountainview, CA). Emission was observed via a monochromator at 520 nm. The reference lifetime compound was fluorescein in a buffered solution, pH 9.0. Phase and modulation data (lifetime = 4.0 ns) were collected until errors were <0.3 and 0.005° of phase and modulation units, respectively. Excitation was vertically polarized and emission was set at magic-angle for lifetime measurements. In the samples, fluorescein-labeled full DNA operator concentration was 3 μM, CggR concentration was 10-times larger, and FBP concentration was 2 mM. The buffer and the temperature (21°C) were the same as for the anisotropy binding titrations.
Data analysis was performed using Globals software (Laboratory of Fluorescence Dynamics, Urbana, IL). This program uses a nonlinear least-squares procedure to minimize the squared deviations between the observed and expected phase and modulation values for multiple experimental data sets. Correlated error analysis (i.e., one parameter is varied near the minimum while the other parameters are all free) were performed on the rotational correlation times, and the rigorous 67% confidence limits are reported for each parameter. For lifetime analysis, a model involving a single exponential fluorescence intensity decay, I(t), was found to adequately describe the experimental data,
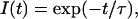 |
(2) |
where τ is the fluorescence lifetime. In contrast, when analyzing the anisotropy decays, r(t), a double-exponential model was found to best fit the experimentally recovered decays,
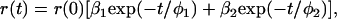 |
(3) |
where r(0) is the time-zero anisotropy, φi are the rotational correlation times and βi the fractional amplitude of each rotational correlation time (β1 + β2 = 1). Anisotropy decays determined for the fluorescein-labeled operator alone, in presence of CggR and with added FBP were analyzed globally and the time-zero anisotropy was a linked parameter in the fittings.
RESULTS
CggR binds to the full operator DNA as a tetramer
As a first step toward a thorough biophysical characterization of CggR interactions with its operator DNA, analytical ultracentrifugation (AUC) experiments were conducted to establish the stoichiometry of the CggR/operator complexes. Sedimentation velocity of a DNA fragment comprising the gapA operator region (OLR, Fig. 1) was first carried out using a 3 μM solution of the fluorescein-labeled oligonucleotide under the appropriate buffer conditions used for specific binding (see below). The profiles were analyzed in terms of a distribution of sedimentation coefficients, allowing an evaluation of solute homogeneity (Fig. 2). The sedimentation velocity profile of the OLR operator DNA alone is best described by a single sedimentation coefficient distribution with an s-value of 3.6 S. To determine the size of the solute species, sedimentation equilibrium was carried out in parallel. Fig. 3 A shows the corresponding sedimentation equilibrium gradient of the OLR operator DNA. It can be well fit by a single sedimenting species with a bMW of 12,700, compatible with the double-stranded oligonucleotide. These data demonstrated that the OLR operator DNA used in this study was present as a homogeneous species, and hence that the DNA preparation contained no detectable amount of single-stranded labeled DNA or DNA aggregates. Simulation of the DNA shape and size using the HYDROSUB (14) software showed that the sedimentation coefficient of 3.6 S determined for OLR was compatible with a hydrated DNA molecule of ∼16 and 1.8 nm in length and width, respectively, in good agreement with the length of a 45-bp oligonucleotide and with the 2 nm diameter reported for B-DNA (15).
FIGURE 2.

Sedimentation coefficient distributions of the fluorescein-labeled full-length gapA operator alone and in complex with CggR, in absence and in presence of FBP. The profiles correspond to free OLR DNA (dot-dashed line), OLR in complex with CggR (solid lines), and the CggR/OLR complex in 2 mM FBP solution (dotted lines). The concentration of DNA was 3 μM and the molar ratio of CggR (in monomer units) to DNA was 10:1 (thick lines) and 5:1 (thin lines). The distributions shown here were corrected to standard conditions (20°C).
FIGURE 3.

Sedimentation equilibrium gradients of CggR in complex with the fluorescein-labeled full operator DNA. Sedimentation gradients correspond to the free OLR DNA (A), the OLR/CggR complex alone (B) and in presence of 2 mM FBP (C). In the graphs, solid lines correspond to the best fitting of the experimental data. Theoretical gradients corresponding to complexes integrated by a CggR dimer (dashed lines) or tetramer (dotted line) bound to the full operator are shown for comparison. DNA concentration was 3 μM and the repressor was in a 10-fold molar excess (in monomer units) with respect to DNA.
Addition of CggR to the labeled OLR DNA solutions resulted in different sedimentation coefficient distributions depending on the CggR/DNA ratio in the sample. In solutions containing a fivefold molar excess of CggR over the operator DNA (in monomer units) (Fig. 2, thin solid lines), two peaks could be distinguished, one centered around 3.6 S corresponding to the free DNA, and another one at ∼9.3 S corresponding to the repressor/DNA complex. However, when CggR was added in 10-fold molar excess (Fig. 2, thick solid lines), most of the labeled DNA was detected in a single symmetric peak at 9.3 S, suggesting that the repressor/DNA complex behaved essentially as a single species. In contrast, the repressor/DNA solutions supplemented with 2 mM FBP remained polydisperse even at a 10-fold molar excess of CggR over DNA (Fig. 2, thick dashed line). Although the free DNA peak at 3.6 S almost completely disappeared, the sedimentation peak of the bound DNA appeared as a broad, highly asymmetric distribution at lower s-values than in absence of FBP. Moreover, the s-value increased upon increasing the repressor concentration.
Sedimentation equilibrium experiments in the absence and presence of FBP were performed with samples containing a 10-fold molar excess of CggR over the labeled DNA (Fig. 3, B and C). Analysis of the equilibrium data of the CggR/DNA solution in the absence of FBP, assuming a single solute, yielded an estimated bMW of 50,300. This value is close to that calculated from the addition of the buoyant masses of four CggR molecules (39,500) and one double-stranded oligonucleotide (12,700). It can therefore be concluded that in absence of inducer, CggR binds as a tetramer to the gapA operator DNA.
The analysis of the sedimentation equilibrium gradients of the CggR/OLR solution supplemented with 2 mM FBP using a single-species model yielded an estimated bMW of 45,300 that is compatible with a complex constituted by 3.2 CggR units and 1 molecule of DNA. However, given the lack of monodispersity of the sedimentation coefficient profiles observed for the CggR/DNA complex in the presence of FBP (Fig. 2), analysis of the sedimentation equilibrium gradients was carried out assuming a mixture of macromolecular species at sedimentation equilibrium. The equilibrium gradients were found to be most compatible with a mixture of two protein/DNA species in equal molar fractions within experimental uncertainty containing one DNA molecule and either two or four CggR monomers. Together with the results of the sedimentation velocity experiments, this analysis suggested that in the presence of FBP, the amount of repressor used in the AUC experiments is not sufficient to fully saturate the OLR operator DNA with four CggR monomers because the activity of the CggR preparations was never 100%. Nevertheless, if saturation were reached, the final protein/DNA complex would be made of four CggR units per molecule of DNA; that is, CggR would saturate the tandem operator sites as a tetramer, in the absence as well as in the presence of FBP.
FBP alters CggR binding to DNA
Anisotropy-based binding assays for the CggR/operator interaction were carried out using fluorescently labeled oligonucleotide targets. This approach can provide fairly precise estimations of thermodynamic parameters of complex binding reactions (16). Changes in the rotational properties of the labeled DNA were monitored by measuring the steady-state anisotropy of the fluorescein-labeled OLR oligonucleotide upon increasing the concentration of CggR. A series of control experiments using specific or nonspecific DNAs at different ionic strengths enabled to determine optimal conditions for anisotropy binding titrations (data not shown). Specific binding was observed at salt concentrations above 100 mM NaCl (but not at 50 mM) and this specificity was maintained in the presence of FBP. Further titrations were therefore carried out at 150 mM NaCl.
Anisotropy-based titrations of the OLR operator DNA were carried out in the absence and in the presence of increasing concentrations of FBP. We note that the curves obtained at the highest three concentrations of FBP (2, 3, and 5 mM) were identical (data not shown), indicating a saturation of the effect by 2 mM FBP. It can be seen in Fig. 4 A that FBP had a profound effect on the binding profiles. First, a considerable shift to higher repressor concentration of the midpoint of binding was observed with increasing FBP. This indicated that the apparent affinity of CggR for its specific DNA target is reduced at high FBP concentrations. Secondly, whereas in the absence of inducer, the binding event takes place essentially between 1 and 10 nM, the titration curve in presence of saturating FBP spans a protein concentration range of over two orders of magnitude (>1–100 nM). This suggests that CggR interacts with its DNA target in a cooperative manner and that FBP decreases the cooperativity of this interaction. Finally, the presence of inducer lowers the asymptotic anisotropy value for the final protein/DNA complex. Under the conditions of the anisotropy titrations, in which the target DNA concentration is extremely low (0.9 nM) to ensure equilibrium binding, the concentration of CggR is largely saturating. Based on the AUC experiments, the complexes are tetrameric at saturation both in absence and in presence of FBP. Therefore, the lower anisotropy value observed for the plateau in presence of FBP is due to a difference in the dynamic properties, and not in the final stoichiometry of the complex.
FIGURE 4.

Fluorescence-based studies of the interaction of the fluorescein-labeled full-length target operator DNA with CggR. (A) Anisotropy binding titrations of the fluorescein-labeled full DNA operator (OLR) at 0.9 nM with CggR at increasing concentration in absence (▪) or in presence of FBP at 10 μM (▵), 100 μM (•) or 2 mM (⋄); solid line curves correspond to data fitting using the model proposed in Fig. 6 (see the main text). The straight dotted lines indicate the protein concentrations used in Fig. 4 B. (B) FBP titration of CggR in complex with the fluorescein-labeled OLR operator (0.9 nM) at protein concentration of 7.7 nM (♦) and 258 nM (▵) in dimer units. Fittings of the data to models involving one (dashed lines) and two (solid lines) binding sites are shown for comparison. The term mA.U. stands for milli-anisotropy units.
Bi-modal effect of FBP on the CggR/operator complex
To explore in more detail the effects of FBP on the CggR/DNA complex, solutions containing a fixed concentration of OLR operator (0.9 nM) and either 7.7 nM or 258 nM of the CggR dimer, were titrated with FBP (Fig. 4 B). In the titrations performed at 7.7 nM CggR, a biphasic transition was detected, with apparent midpoints around 1 and 100 μM effector sugar. The single transition observed for the curve obtained at 258 nM CggR exhibited a midpoint near 10 μM. The shift from 1 to 10 μM for the first FBP binding event upon increasing the CggR concentration arises from the fact that the free repressor, which is unlabeled and present at ∼256 nM, competes for FBP binding. Isothermal titration calorimetry, as well as fluorescence correlation spectroscopy (FCS) experiments performed with free repressor confirmed that a first binding event took place between 1 and 10 μM FBP (results not shown). For the 7.7 nM CggR solution, increasing FBP concentration above 100 μM not only accentuated the anisotropy drop, but also resulted in a significant reduction of CggR affinity for the operator DNA. Indeed the presence of saturating FBP leads to repressor release from the operator DNA (Fig. 4 A, dotted line). The second ligand binding event did not produce any detectable transition in the titrations carried out at 258 nM dimeric CggR, since at this protein concentration virtually all the operator DNA is bound by a CggR tetramer according to the AUC results above, even at saturating FBP concentration, and thus, no protein release would be expected upon FBP binding (Fig. 4 A, solid line). Hence, two distinct binding events occurred upon addition of increasing concentrations of FBP to the CggR/DNA solutions, one in the mid-micromolar range that modifies the dynamics of the complex, and a second above 10 μM that decreases the affinity.
CggR dimers bind to each direct repeat with different affinity
The full-length ORL operator site consists of a tandemly repeated DNA sequence, each containing a short palindrome (Fig. 1). Since the sequence of the left (OL) and right (OR) repeats differ by several basepairs, analysis of the binding of CggR to each half-site operator and any effects of the inducer sugar on the stoichiometry and affinity of these interactions were investigated.
The stoichiometry of the repressor/DNA complexes involving half-site operators was investigated in the presence/absence of FBP. AUC experiments were performed on samples containing 400 nM of nonlabeled OR or OL operator and a 10-fold molar excess of CggR in monomer units. Sedimentation coefficient distributions obtained from the velocity profiles were similar in the presence of FBP, yielding an s-value of 5.2 S for both target DNAs (Fig. 5 A). The analysis of the corresponding sedimentation equilibrium gradients yielded average bMW values of 24,600 and 25,800 for the complexes involving the OL and OR operator, respectively, compatible with a stoichiometry of one CggR dimer per oligonucleotide (data not shown). Although the fitting of the equilibrium gradients with this simple model was not optimum, and the presence of larger complexes was suspected, the CggR dimer-bound DNA complex was clearly the major species for both half-site targets. In the absence of FBP, the heterogeneous distribution of the sedimentation coefficients precluded further characterization of the complexes. For both targets, a broad distribution was observed, predominantly above 10 S but also around the peak corresponding to the dimer/DNA complex, indicating that both in the absence and in the presence of FBP, CggR interacts primarily as a dimer with each of the half-operator sites. The high molecular weight species observed in the absence of FBP might correspond to repressor/DNA complexes with additional CggR dimers bound to the initial complex through protein/protein interactions favored by the high concentrations used in the ultracentrifugation experiments. Note that AUC (both equilibrium and sedimentation velocity) experiments on the free repressor at 23 μM revealed that CggR forms dimers and higher order species under these conditions, while FCS experiments in the submicromolar concentration range (which is relevant for the DNA titrations) yielded a diffusion coefficient that is consistent with dimer below 200 nM of free CggR (unpublished).
FIGURE 5.

Stoichiometry and affinity of CggR interaction with the two half-operator DNA sequences. (A) Sedimentation coefficient distribution of CggR complexes with nonlabeled double-stranded oligonucleotides corresponding to the right (OR, thick lines) and left (OL, thin lines) half-site operator DNAs, corrected to standard conditions (20°C). Solid and dashed lines correspond to experiments in presence and in absence of FBP, respectively. The concentration of DNA used was 400 nM and CggR was in 10-fold molar excess (in monomer units) with respect to DNA. (B) Anisotropy binding titrations of the fluorescein-labeled full operator (ORL, ▴) and of the Alexa-488 labeled half-site DNAs (OR, •; OL, ▪) with CggR in absence of FBP or in presence of 2 mM FBP (OR, ○; OL, □). The concentration of OLR, OR and OL operator double-stranded DNA was 0.9 nM, 1.7 nM, and 5.1 nM, respectively. The fittings shown in this figure correspond to the most suitable models discussed for each interaction (see the main text). The term mA.U. stands for milli-anisotropy units.
The intrinsic affinity of the CggR dimer for the half-sites was compared by anisotropy binding titrations with 5′-Alexa labeled OL or OR (Fig. 5 B). Titration curves in the absence of FBP showed drastic differences in the interaction of CggR with each target DNA. In the case of the right half-operator, a first binding event occurred in the subnanomolar range of repressor concentration, in the same range as the binding observed for the full-length target. However, the corresponding anisotropy increase was only half as large as that observed on the full-length target. This first binding event on the right half-site was followed by a second binding event at significantly higher protein concentrations that exhibited a final plateau value close to that observed for the full-length target. In contrast, the binding profile observed for the OL target DNA appeared as a single event that took place at much higher repressor concentrations compared to the OR or OLR titration, indicating that the affinity of CggR for the left repeat is considerably lower than for the right repeat alone or the tandem repeats. When performed in the presence of 2 mM FBP, CggR binding to each half target DNA occurs with somewhat lower affinity than in the absence of inducing sugar, and with a severe reduction in the anisotropy of the final protein/DNA complex. In the case of the OR half-site, the second binding event no longer took place upon addition of FBP, the anisotropy reaching a plateau value similar to that of the first binding event observed in absence of FBP.
All together, the stoichiometry and affinity data on CggR interaction with the half-operators suggest that the repressor binds primarily as a dimer to each direct repeat, with much higher affinity for the right repeat, and that the protein-protein interactions responsible for the subsequent binding event are of much lower affinity when inducer is bound.
Energetics of CggR binding to DNA
Based on a stoichiometry of one dimer per half-site operator for the CggR/DNA complexes determined by AUC, quantitative analysis of the anisotropy binding profiles presented in Fig. 5 B was performed (Table 1). In the model used for the analysis of the binding curves, the formation of a single complex (DO) was postulated, consisting of one CggR dimer (D) and one molecule of the half-site operator DNA (O) arising from the initial elements, the CggR dimer and the OL or OR operator. Results of studies of the behavior of the free protein using FCS and AUC (manuscript in preparation) indicate that below 150 nM the protein exhibits translational diffusion behavior consistent with dimer, while above that concentration it self-associates into a number of different higher order species in a protein concentration-dependent manner. This higher order protein association behavior is unlikely to be artifactual (i.e., due to the His-tag, for example) since it is modulated by FBP. Moreover, the in vivo phenotype in strains expressing the His-tagged protein was identical to that observed for the wild type.
TABLE 1.
Free energy parameters for CggR interaction with the DNA operator half-sites
|
D + OL ⇆ DOL
|
D + OR ⇆ DOR
|
2D + OR ⇆ D2OR
|
|||
|---|---|---|---|---|---|
| [FBP] (μM) | ΔG0 (kcal/mol) | Kd (nM) | ΔG0 (kcal/mol) | Kd (nM) | ΔG0 (kcal/mol) |
| 0 | 9.8 ± 0.2 | 52 | 14 (−0.6; +1.5) | 0.03 | 23 (−0.5; +6.5) |
| 2000 | 9.6 ± 0.2 | 73 | 11.9 ± 0.3 | 1.4 | |
D represents a CggR dimer. Confidence limits were determined from rigorous confidence limit testing at the 67% level (mean ± 1 SD). ΔG0 values consigned correspond to the dissociation reaction. Values indicated in italics are approximate, the very low Kd values compared to DNA concentration (1.7 nM) precluding more accurate determination.
For the right operator in absence of FBP, the binding of a second dimer was included in the model to account for the second binding event. In the analysis, the anisotropy values of the free DNA and of the complex were floating parameters. On the OR operator DNA the dimer/DNA complex was found to be of very high affinity (∼30 picomolar) in the absence of FBP, although because the target DNA was present at 1.7 nM, this value was not well-determined. In the presence of 2 mM FBP, the Kd for the OR DNA increased by a factor of ∼50 to 1.4 nM, whereas the second binding event was not observed to occur in presence of inducer. Compared to OR, CggR exhibits a much lower overall affinity for the left half-site, OL, 52 and 73 nM in the absence and presence of FBP, respectively.
The binding curves obtained with the full-length target (Fig. 4 A) were analyzed using the model given in Fig. 6, including the four species, free DNA (O), free CggR dimer (D), the final tetrameric-repressor/operator complex (D2O), and the dimer-bound intermediate complex (DO). The results of the analysis presented in Table 2 and Fig. 6 show that the free energy of formation of the complexes (total free energy from the free elements to the complex in question) is significantly larger in absolute value in the absence than in the presence of FBP. In particular the free energy of formation of the final tetrameric complex (ΔGT) decreased by 3.5 kcal/mol in presence of saturating FBP. This corroborates the qualitative results described above that together demonstrate that FBP binding reduces significantly the affinity of the final repressor/operator complex. The evolution of the free energies of binding to the full-length target as a function of FBP (Table 2) confirms that the major effect of FBP on the operator affinity occurs above 10 μM inducer.
FIGURE 6.

Energetic diagram used to analyze the conditional interaction of CggR with its full operator DNA and illustration of the cooperativity estimation performed. ΔG0 values were taken from Tables 1 and 2 and correspond to the dissociation reaction. The model takes into account the markedly different affinity of CggR for the two half-sites. One CggR dimer first binds to the right half-site of the OLR DNA with a slightly higher affinity in the absence than in the presence of saturating FBP (ΔG1 values taken from Table 2 at 0 and 2 mM FBP). In the absence of cooperativity, binding of a second CggR dimer to the left half-site would occur with the free energies measured with the OL half-operator DNA (ΔG0 values taken from Table 1). The difference between the total free energies calculated by addition of the free energies of the two independent binding events (dashed lines) and the total free energies of the binding reaction determined experimentally (solid lines, ΔGT values taken from Table 2) corresponds to the cooperative free energy (ΔGcoop).
TABLE 2.
Free energy parameters for CggR interaction with the tandem DNA operator sites
| 2D+OLR ⇆ D2OLR
|
2D +OLR ⇆ D2OLR
|
DOLR +D ⇆ D2OLR
|
|||
|---|---|---|---|---|---|
| [FBP] (μM) | ΔG1 (kcal/mol) | Kd (nM) | ΔGT (kcal/mol) | ΔG2 = ΔGT−ΔG1 (kcal/mol) | Kd (nM) |
| 0 | 12.8 (−0.9; +2.5) | 0.3 | 24.9 (−0.6; +1.5) | 12.1 | 1.0 |
| 10 | 12.2 (−1.3; +0.8) | 0.9 | 24.2 ± 0.5 | 12 | 1.2 |
| 100 | 12.1 ± 0.4 | 1.0 | 23.1 ± 0.3 | 11 | 6.6 |
| 2000 | 11.3 ± 0.4 | 4.0 | 21.4 ± 0.3 | 10.1 | 31 |
D refers to CggR dimer. Errors were determined from rigorous confidence limiting testing at the 67%. ΔG values consigned correspond to the dissociation reaction.
To obtain a reliable estimation of the intrinsic cooperativity of the repressor/operator binding reaction one must account for the markedly different affinity of CggR for the two half-sites forming its DNA target sequence determined above. The overall free energy of formation of the tetrameric complex can be considered to be the sum of two free energies, those of binding of the first, and then the second dimer. Since the right half site is that of highest affinity, the intermediate repressor/DNA complex must correspond to CggR binding to the right half-site, while binding to the left-half site gives rise to the final complex. Included in this second free energy is the contribution of the protein-DNA interactions, as well as the cooperative (likely protein-protein) interactions that occur in the tetrameric complex. Following this reasoning, subtraction of the free energy change determined for the binding of a CggR dimer to the OL operator (Table 1) from that obtained from the analysis for the binding of the second CggR dimer to the full operator sequence (ΔG2 in Table 2) provides an estimate of the cooperative free energy. We note that despite the large confidence limits on the value of ΔG1, the covariance curves from the rigorous confidence limit testing reveal that the difference between ΔGT and ΔG1 is well defined (12.1 ± 0.3 kcal/mol), and it is this difference that determines the cooperativity. An illustration of this approach is shown in Fig. 6. It demonstrates that the binding of the repressor to the full-length target in the absence of FBP occurred with a cooperative free energy equal to 2.3 kcal/mol ± 0.5 kcal/mol (taking into account as well the confidence limits, ±0.2 kcal/mol, on the free energy of dimer binding to OL). In presence of saturating inducer sugar, this cooperative free energy was considerably reduced (0.5 ± 0.5 kcal/mol). Thus, assuming that this cooperative free energy arises from protein-protein interactions, we conclude that FBP at millimolar concentrations significantly destabilizes the interactions between CggR dimers in the protein/operator complex.
Conformational dynamics of the labeled DNA/CggR complex
Finally, we wanted to clarify the FBP-induced change in dynamics of the CggR/OLR complexes observed by anisotropy binding titrations (Fig. 4). To this end, time-resolved fluorescence anisotropy experiments in the frequency domain were undertaken on the free DNA target and the CggR/OLR complexes in the presence/absence of inducer (Table 3, Fig. 7). Inducer was present at 2 mM, but despite its dual effect on the affinity and the dynamics of the CggR/operator at this concentration, the high concentrations of protein (30 μM) and DNA (3 μM) ensured that no dissociation of the complex occurred and that any changes in the anisotropy would be due to the dynamics.
TABLE 3.
Time-resolved rotational parameters of the fluorescein-labeled operator DNA, alone or bound to CggR
| τ (ns) | φ1 (ns) | β1 | φ2 (ns) | β2 | χ2 | r(0) (linked) | |
|---|---|---|---|---|---|---|---|
| OLR | 4.5 | 0.37 ± 0.08 | 0.80 ± 0.03 | 5 (−1; +2) | 0.20 ± 0.02 | 2.5 | 0.33 ± 0.02 |
| OLR (internal) | 4.5 | 0.31 ± 0.03 | 0.64 ± 0.02 | 31 (−3; +5) | 0.36 ± 0.02 | 3.1 | 0.32 ± 0.02 |
| OLR+CggR | 4.5 | 0.45 ± 0.09 | 0.74 ± 0.02 | 14 (−2; +6) | 0.26 ± 0.02 | 8.6 | 0.33 ± 0.02 |
| OLR+CggR+FBP | 4.5 | 0.42 ± 0.07 | 0.76 ± 0.02 | 10 (−1.5; +4.5) | 0.24 ± 0.02 | 6.9 | 0.33 ± 0.02 |
The concentration of fluorescein-labeled OLR was 3 μM, CggR was added in a 10-fold molar excess with respect to DNA, and the concentration of FBP was 2 mM. The values r(0), φi, and βi are the time-zero anisotropy, the rotational correlation times, and their fractional contribution to the anisotropy decay, respectively. Estimated uncertainties were calculated by performing rigorous confidence limit testing of the recovered parameters at the 67%.
FIGURE 7.

Differential frequency response for the full fluorescein-labeled DNA operator alone and in complex with CggR. Differential phase (bottom panel) and modulation (top panel) of the free full DNA operator (•), of the CggR/DNA complex (▪) and of the complex in presence of FBP (▵) as a function of the excitation frequency. DNA, CggR, and FBP concentrations were 3 μM, 30 μM, and 2 mM, respectively. Excitation and emission wavelengths were 450 and 520, respectively.
The fluorescence lifetime of the fluorescein label on the DNA was a single exponential and independent of both protein and sugar concentration. In all cases, the anisotropy decay could be described well by two rotational correlation times (φ1 and φ2), whose amplitudes (β1 and β2) did not change upon addition of either protein or inducer. The very fast rotation, ∼400 ps, corresponds to the local mobility of the dye and was also independent of both protein and sugar concentration. The large amplitude of the local rotational motions of the dye under all conditions (75%) explains why the total change in steady-state anisotropy in the titrations remains relatively modest. The longer rotational correlation time was well resolved for the free DNA, since its value is similar to that of the fluorescence lifetime. At 5 ns, however, this value is quite small compared to the harmonic mean value expected for the global tumbling of the oligonucleotide estimated by modeling it as a rigid cylinder (∼28 ns) using HYDROSUB (17). Even rotation about the long axis of the cylinder is expected to be much longer, 16 ns. A significantly smaller experimental value for the correlation time indicates a significant contribution of conformational flexibility of the probe or the DNA on an intermediate timescale. To further investigate these two possibilities, we measured the anisotropy decay for an oligonucleotide labeled internally near the 5′-end of the anti-sense strand. From these data we recovered a global tumbling time of 31 ns (Table 3), in good agreement with the expected mean value. This observation rules out any major bending of the DNA as a source of the flexibility. Indeed FCS measurements on the free 5′-labeled target yielded a translational diffusion coefficient (59 μm2/s), in reasonable agreement with the expected value (63 μm2/s), and clearly not a faster diffusion that would be observed in the presence of bending. The intermediate timescale flexibility can therefore be ascribed to a local conformational flexibility near the probe.
Compared to the free DNA, time-resolved anisotropy showed a significant increase in the long correlation time for the CggR/operator complex. Nevertheless, its value of ∼14 ns is much smaller than the 75-ns rotational correlation time expected for a tetrameric CggR/DNA complex. Since the long correlation time represents depolarization from a combination of global tumbling (linked to the molecular weight) and conformational flexibility, the observed increase in its value upon protein binding probably arises from a combination of changes in both. In presence of inducer, the value of the long correlation time is smaller than in absence of FBP, in accord with the lower plateau value observed for the steady-state anisotropy. We can rule out an increase in the fast local probe rotations as the source of this change because the amplitude and the value of the fast correlation time do not change. Thus the effect of FBP on the plateau anisotropy value arises from changes in the intermediate timescale dynamics of the dye bound through a six-carbon linker to the 5′-end of the sense strand of the target oligonucleotide. Dyes bound in this manner are known to participate in stacking interactions at the end of the double-stranded oligonucleotide. With appropriate association and dissociation rate constants, such a dynamic equilibrium could account for the intermediate timescale of the anisotropy decay. Moreover, the interaction of a protein or changes in the bound protein conformation induced by sugar binding could affect this dynamic equilibrium.
DISCUSSION
The present study sought to determine the molecular mechanisms governing the interaction of the transcriptional repressor CggR with its operator DNA and its modulation by the glycolytic metabolite, FBP. A combination of approaches was used, including fluorescence anisotropy (steady-state and time-resolved) and analytical ultracentrifugation (sedimentation velocity and equilibrium sedimentation). From AUC experiments performed with CggR/OLR (full-length) operator complexes, the stoichiometry of the final repressor/operator complex was found to be four CggR molecules per operator in absence as well as in presence of FBP. The finding that this repressor binds to its DNA target as a tetramer is consistent with the structure of its unique target sequence. As highlighted in Fig. 1, the gapA operator region is comprised of two long direct repeats (OL and OR), each of which contains a small inverted repeat of three nucleotides (GAC). The four repeated elements being separated one from another by one turn of the double-helix (GACN7GTCN7GACN7GTC), it is likely that each triplet forms the core of the elementary binding site for one subunit of the CggR tetramer. We then showed that a CggR dimer binds to each OL or OR direct repeat but with different affinity. Thus, each half operator site would constitute the binding site for a symmetric dimer, exhibiting much lower affinity for the left (5′) site than for the right (3′) site. Comparison of the gapA operator DNA sequences from different Bacillus species provides a possible explanation for the different affinities. As observed in Fig. 1, the GAC triplets are flanked by conserved residues that could provide additional and specific contacts between the protein and the target DNA. From this analysis, it can be proposed that the extended binding site for monomeric CggR contains the CGGGACA recognition sequence. In the OR half-site of the B. subtilis gapA operator, we noticed that this heptanucleotide motif is found in two inverted copies within a nearly perfect palindrome (GCGGGACATN3ATGTCCAGC). This extended palindrome would therefore constitute an optimized target sequence for the binding of a symmetric CggR dimer. In contrast, only part of the 5′ half (CGGGAC) of this palindrome is present in the OL operator site, the 3′ half being reduced to only four nucleotides (TGTC). This tetranucleotide sequence would interact less optimally with the repressor subunit, explaining why the CggR dimer does not bind tightly to the OL half-site. Other examples of transcriptional regulators exhibiting differential binding to the two half-sites constituting their full binding sequence have been reported (18,19). For example, the LacI repressor protein, a prototype for transcriptional repression (20), has been found to bind to the left half-site of its operator DNA with a higher affinity than to the right one (21). Although a binding sequence made of two repetitions of the highest affinity binding site could in principle optimize protein binding to the operator DNA, it has been shown that some nucleotides in the naturally occurring binding sequences could be selected not only to confer a high binding affinity but also for their capacity to provide a relative rotational orientation to the half-site within the context of the DNA helix, which could be essential for protein recognition (22,23). In the case of CggR, this differential affinity for the half operator sites could also be related to the mechanism of gapA transcription induction by FBP.
From our results it is clear that FBP in the millimolar range reduced both the affinity and cooperativity of CggR binding to the full operator DNA. The first CggR dimer bound to the target DNA more than 10-fold more tightly in absence than in presence of saturating FBP. For the binding of the second CggR dimer, the difference in affinity was even more notable, >30-fold weaker at high FBP concentration. Estimation of the cooperativity taking into account the difference in intrinsic affinity for the two half sites revealed that the binding of CggR to the DNA was highly cooperative in absence of FBP, and this cooperativity was almost completely abolished in presence of millimolar FBP. We interpret this cooperativity as arising from protein-protein interactions between the two dimers, the dimer bound at the high affinity site (OR) stabilizing the dimer bound at the low affinity site (OL). However, we cannot rule out that structural features other than protein-protein interactions could lead to the observed cooperativity (24,25).
Interestingly, we observed a bimodal effect of FBP on the repressor/operator complex. Two distinct ligand binding events, occurring at low and high FBP concentrations, were clearly observed under appropriate experimental conditions. While FBP in the millimolar range leads to the significant differences in the free energies of binding discussed above, an unexpected effect of the inducing sugar was also observed in the micromolar range. Although no significant change in the apparent complex affinity or cooperativity was observed at these low concentrations of FBP, a significant decrease in the value of the plateau anisotropy was repeatedly observed. The AUC experiments in presence of FBP unequivocally demonstrated that a change in the stoichiometry of the final CggR/DNA complexes as a result of FBP binding was not the cause of this difference in anisotropy. Rather, it appeared that the decrease in the steady-state anisotropy was associated with an increase in the conformational dynamics of the complex. Since we observed that FBP also protects CggR from proteolytic cleavage and extensive aggregation in vitro (data not shown), it can be proposed that besides its inducer effect in the millimolar range, FBP may also play a conformational role that is effective at concentrations far below those of the induction conditions.
The most plausible explanation for the observed dual effect of FBP on the CggR/DNA interaction is that the repressor possesses two distinct sugar binding sites. Indeed, the biphasic FBP titration curve is best fitted when using a model considering two ligand binding sites with different affinities. Binding of FBP at the high affinity (micromolar) site would change the conformation of the CggR/operator complex but have no effect on the CggR/DNA affinity. At this site, FBP could be seen as a co-factor, necessary for the folding or stability of the repressor subunit, for instance, but not sufficient to promote the induction response. It is only once bound at the second, low affinity (millimolar) site that FBP induces a decrease in overall operator binding. The presence of two classes of FBP-binding sites has already been reported for other regulator proteins from B. subtilis, in particular the HPr Kinase (26), which regulates the expression of several catabolic genes playing a central role in carbon catabolite repression (27). Structural modeling together with mutagenesis approaches also suggest the presence of two sugar binding sites in the CggR regulatory domain (T. Doan, L. Martin, G. Labesse, S. Aymerich and N. Declerck, unpublished). According to this analysis, the CggR C-terminal domain is structurally similar to the E. coli glucosamine-6-phosphate deaminase (NagB) that possesses two binding sites for phosphorylated sugars: one in an internal cavity where the catalytic reaction takes place, and a second on a surface loop responsible for the allosteric activation of the hexameric enzyme and located at the subunit interface (28). Whether these two sugar binding sites are somehow equivalent to the high and low affinity sites for FBP in CggR remains an open question.
How can the present results shed light on the behavior of CggR in vivo? Among the possible mechanisms of transcriptional repression (29), CggR is more likely to preclude the elongation of the leader transcript than to act at the level of initiation, since the CggR operator is located 32 bp downstream from the transcriptional initiation site (4). Previous studies have shown that mutations in any of the four repeats forming the complete operator site alleviate CggR-mediated repression of gapA expression (4), indicating that only tetrameric CggR would be able to prevent the progression of the transcribing RNA polymerase. Given the high affinity (<1 nM) and cooperativity of the CggR/DNA interaction, a limited number of active repressor molecules would be required to saturate the CggR unique DNA target with the tetrameric repressor and thereby maintain gapA expression in the repressed state. Indeed, cggR mRNA transcripts are very unstable in vivo and <230 molecules of CggR are present per cell (30), indicating that the intracellular concentration of CggR dimers might not exceed 10 nM under nonglycolytic conditions. Changes in metabolic regime leading to an increasing concentration of FBP will reduce both the affinity and cooperativity of the repressor/operator interaction, and this could be enough to fully induce gapA transcription, even though FBP does not abolish CggR binding to DNA. As the concentration of CggR increases upon induction (cggR being the first transcribed gene of the gapA operon), the concentration of FBP required for releasing the repressor from the operator site increases as well. Indeed, it has been reported that the induction of gapA is very strong in cells growing on glycerol or glucose (2,6,7), conditions under which the FBP concentration is ∼6 mM or 14 mM (8), respectively, compared to ∼1 mM when succinate is the only carbon source available (8).
Based on our findings we propose that FBP binding disfavors protein-protein interactions between the two CggR dimers. In this case the CggR tetramer would behave as two individual dimers, each bound independently to one of the half-site operators. Transcription does not necessarily require permanent dissociation of the complex, but simply a window of opportunity in which the polymerase may proceed through the operator site during temporary dissociation or by displacing the unstable CggR/DNA complexes. On its way along the DNA, the RNA polymerase would first encounter the low affinity CggR binding site (OL) either free of repressor or weakly bound by a CggR dimer. It would therefore easily read through the first operator site. The overall free energy of formation of a tetramer/operator complex is significantly larger in absolute value than for a dimer/operator complex. Moreover, these binding equilibria are, in all cases of course, dynamic. Hence, the probability for dissociation of the CggR dimer from the high affinity, OR site is much greater than for the tetramer to dissociate from the full operator site. The polymerase would be able to take advantage of the increased probability of dissociation at the high affinity site bound only by dimer, allowing transcription to occur at a level sufficient for the physiological effects of induction. It is also possible, as other authors have already pointed out, that CggR might respond synergistically to FBP and to an anabolic signal such that total induction of gapA would be achieved by the combination of both, while a single metabolite would result only in partial derepression (7).
Can the proposed mechanism of transcriptional regulation be extrapolated to other members of the SorC repressor family to which belongs CggR? Given the high sequence similarity between CggR paralogs in different Gram positive bacteria on one hand and the gapA operator regions on the other hand, it is likely that all CggR-like repressors function on the same molecular basis. Although the strength of the interactions between the different subunits of the tetramer and the four repeated elementary sites of the operator may vary, the presence of a low affinity binding site flanked by high affinity sites would be key for the inducer-regulated cooperative binding process. Besides CggR, the only other member of the SorC family that has been characterized in vitro is DeoR from B. subtilis (bsDeoR) that regulates the dra-nupC-pdp operon involved in deoxyribonucleoside and deoxyribose utilization and whose preferred inducer is ribose-5-P. bsDeoR recognizes an operator sequence that spans the promoter region of the regulated operon, suggesting that, unlike CggR, this repressor prevents the initiation of transcription by the RNA polymerase. Nevertheless, there are striking similarities between the two repression systems. Although bsDeoR-DNA interactions have not been studied in as much detail as in this study, it has been proposed that this repressor also binds as a tetramer and in a highly cooperative manner to its DNA target. Moreover, as for CggR, binding of the repressor protects a fairly long DNA sequence that contains repetitive DNA elements forming one and a half palindrome (with the missing half in between), suggesting to the authors that only three of the four subunits of the tetramer are (tightly) bound to DNA in the bsDeoR-operator complex (31). These highly unusual features, common to the two classes of repressors, suggest that the regulation mechanism proposed for CggR is also valid for the other members of the SorC/DeoR family.
Acknowledgments
We thank Dr. Stefan Arold for his help with the isothermal titration calorimetry experiments and Dr. Mercedes Jiménez for technical assistance.
This work was supported by a Marie Curie intra-European fellowship, grant No. BFU2005-04087-C02-01 from the Spanish Dirección General de Enseñanza Superior e Investigación to G.R., and an instrumentation grant from the Association pour la Recherche contre le Cancer (grant No. 7882) to CAR and INSERM. S.Z. is the recipient of a European International Fellowship (MEIF-CT-2004-007320). A.O. is the recipient of a fellowship from Fundación Cajamurcia.
References
- 1.Kunst, F., N. Ogasawara, I. Moszer, A. M. Albertini, G. Alloni, V. Azevedo, M. G. Bertero, P. Bessieres, A. Bolotin, S. Borchert, R. Borriss, L. Boursier, A. Brans, M. Braun, S. C. Brignell, S. Bron, S. Brouillet, C. V. Bruschi, B. Caldwell, V. Capuano, N. M. Carter, S. K. Choi, J. J. Codani, I. F. Connerton, A. Danchin et al. 1997. The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature. 390:249–256. [DOI] [PubMed] [Google Scholar]
- 2.Fillinger, S., S. Boschi-Muller, S. Azza, E. Dervyn, G. Branlant, and S. Aymerich. 2000. Two glyceraldehyde-3-phosphate dehydrogenases with opposite physiological roles in a nonphotosynthetic bacterium. J. Biol. Chem. 275:14031–14037. [DOI] [PubMed] [Google Scholar]
- 3.Saxild, H. H., L. N. Andersen, and K. Hammer. 1996. Dra-nupC-pdp operon of Bacillus subtilis: nucleotide sequence, induction by deoxyribonucleosides, and transcriptional regulation by the deoR-encoded DeoR repressor protein. J. Bacteriol. 178:424–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doan, T., and S. Aymerich. 2003. Regulation of the central glycolytic genes in Bacillus subtilis: binding of the repressor CggR to its single DNA target sequence is modulated by fructose-1,6-bisphosphate. Mol. Microbiol. 47:1709–1721. [DOI] [PubMed] [Google Scholar]
- 5.Fuhrer, T., E. Fischer, and U. Sauer. 2005. Experimental identification and quantification of glucose metabolism in seven bacterial species. J. Bacteriol. 187:1581–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leyva-Vazquez, M. A., and P. Setlow. 1994. Cloning and nucleotide sequences of the genes encoding triose phosphate isomerase, phosphoglycerate mutase, and enolase from Bacillus subtilis. J. Bacteriol. 176:3903–3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ludwig, H., N. Rebhan, H. M. Blencke, M. Merzbacher, and J. Stulke. 2002. Control of the glycolytic gapA operon by the catabolite control protein A in Bacillus subtilis: a novel mechanism of CcpA-mediated regulation. Mol. Microbiol. 45:543–553. [DOI] [PubMed] [Google Scholar]
- 8.Mijakovic, I., S. Poncet, A. Galinier, V. Monedero, S. Fieulaine, J. Janin, S. Nessler, J. A. Marquez, K. Scheffzek, S. Hasenbein, W. Hengstenberg, and J. Deutscher. 2002. Pyrophosphate-producing protein dephosphorylation by HPr kinase/phosphorylase: a relic of early life? Proc. Natl. Acad. Sci. USA. 99:13442–13447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schuck, P. 2000. Size distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78:1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laue, T. M., B. D. Shah, T. M. Ridgeway, and S. L. Pelletier. 1992. Computer-aided interpretation of analytical sedimentation data for proteins. In Analytical Ultracentrifugation in Biochemistry and Polymer Science. S. E. Harding and A. J. Rowe, editors. The Royal Society of Chemistry, Cambridge, UK.
- 11.Minton, A. P. 1994. Conservation of signal: a new algorithm for the elimination of the reference concentration as an independently variable parameter in the analysis of sedimentation equilibrium. In Modern Analytical Ultracentrifugation. T. M. Schuster and T. M. Laue, editors. Birkhauser, Boston, MA. 81–93.
- 12.Rivas, G., W. Stafford, and A. P. Minton. 1999. Characterization of heterologous protein-protein interactions using analytical ultracentrifugation. Methods. 19:194–212. [DOI] [PubMed] [Google Scholar]
- 13.Royer, C. A., W. R. Smith, and J. M. Beechem. 1990. Analysis of binding in macromolecular complexes: a generalized numerical approach. Anal. Biochem. 191:287–294. [DOI] [PubMed] [Google Scholar]
- 14.Royer, C. A., and J. M. Beechem. 1992. Numerical analysis of binding data: advantages, practical aspects and implications. Methods Enzymol. 210:481–505. [DOI] [PubMed] [Google Scholar]
- 15.Eimer, W., J. R. Williamson, S. G. Boxer, and R. Pecora. 1990. Characterization of the overall and internal dynamics of short oligonucleotides by depolarized dynamic light scattering and NMR relaxation measurements. Biochemistry. 29:799–811. [DOI] [PubMed] [Google Scholar]
- 16.Grillo, A. O., and C. A. Royer. 2000. The basis for the super-repressor phenotypes of the AV77 and EK18 mutants of Trp repressor. J. Mol. Biol. 295:17–28. [DOI] [PubMed] [Google Scholar]
- 17.Garcia de la Torre, J., and B. Carrasco. 2002. Hydrodynamic properties of rigid macromolecules composed of ellipsoidal and cylindrical subunits. Biopolymers. 63:163–167. [DOI] [PubMed] [Google Scholar]
- 18.Liu, Y. C., and K. S. Matthews. 1993. Dependence of Trp repressor-operator affinity, stoichiometry, and apparent cooperativity on DNA sequence and size. J. Biol. Chem. 268:23239–23249. [PubMed] [Google Scholar]
- 19.Baleja, J. D., W. F. Anderson, and B. D. Sykes. 1991. Different interactions of Cro repressor dimer with the left and right halves of OR3 operator DNA. J. Biol. Chem. 266:22115–22124. [PubMed] [Google Scholar]
- 20.Lewis, M. 2005. The Lac repressor. C. R. Biol. 328:521–548. [DOI] [PubMed] [Google Scholar]
- 21.Kalodimos, C. G., A. M. Bonvin, R. K. Salinas, R. Wechselberger, R. Boelens, and R. Kaptein. 2002. Plasticity in protein-DNA recognition: Lac repressor interacts with its natural operator 01 through alternative conformations of its DNA-binding domain. EMBO J. 21:2866–2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koudelka, G. B. 1998. Recognition of DNA structure by 434 repressor. Nucleic Acids Res. 26:669–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lefstin, J. A., and K. R. Yamamoto. 1998. Allosteric effects of DNA on transcriptional regulators. Nature. 392:885–888. [DOI] [PubMed] [Google Scholar]
- 24.Schumacher, M. A., M. C. Miller, S. Grkovic, M. H. Brown, R. A. Skurray, and R. G. Brennan. 2002. Structural basis for cooperative DNA binding by two dimers of the multidrug-binding protein QacR. EMBO J. 21:1210–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.White, A., X. Ding, J. C. Van der Spek, J. R. Murphy, and D. Ringe. 1998. Structure of the metal-ion-activated diphtheria toxin repressor/tox operator complex. Nature. 394:502–506. [DOI] [PubMed] [Google Scholar]
- 26.Jault, J. M., S. Fieulaine, S. Nessler, P. Gonzalo, A. Di Pietro, J. Deutscher, and A. Galinier. 2000. The HPr kinase from Bacillus subtilis is a homo-oligomeric enzyme which exhibits strong positive cooperativity for nucleotide and fructose 1,6-bisphosphate binding. J. Biol. Chem. 275:1773–1780. [DOI] [PubMed] [Google Scholar]
- 27.Saier, M. H., Jr. 1996. Cyclic AMP-independent catabolite repression in bacteria. FEMS Microbiol. Lett. 138:97–103. [DOI] [PubMed] [Google Scholar]
- 28.Rudino-Pinera, E., S. Morales-Arrieta, S. P. Rojas-Trejo, and E. Horjales. 2002. Structural flexibility, an essential component of the allosteric activation in Escherichia coli glucosamine-6-phosphate deaminase. Acta Crystallogr. D Biol. Crystallogr. 58:10–20. [DOI] [PubMed] [Google Scholar]
- 29.Rojo, F. 2001. Mechanisms of transcriptional repression. Curr. Opin. Microbiol. 4:145–151. [DOI] [PubMed] [Google Scholar]
- 30.Meinken, C., H. M. Blencke, H. Ludwig, and J. Stulke. 2003. Expression of the glycolytic gapA operon in Bacillus subtilis: differential syntheses of proteins encoded by the operon. Microbiology. 149:751–761. [DOI] [PubMed] [Google Scholar]
- 31.Zeng, X., H. H. Saxild, and R. L. Switzer. 2000. Purification and characterization of the DeoR repressor of Bacillus subtilis. J. Bacteriol. 182:1916–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]