

Abstract
The thermotropic phase behavior and organization of aqueous dispersions of the quadruple-chained, anionic phospholipid tetramyristoyl diphosphatidylglycerol or tetramyristoyl cardiolipin (TMCL) was studied by differential scanning calorimetry, x-ray diffraction, 31P NMR, and Fourier-transform infrared (FTIR) spectroscopy. At physiological pH and ionic strength, our calorimetric studies indicate that fully equilibrated aqueous dispersions of TMCL exhibit two thermotropic phase transitions upon heating. The lower temperature transition is much less cooperative but of relatively high enthalpy and exhibits marked cooling hysteresis, whereas the higher temperature transition is much more cooperative and also exhibits a relatively high enthalpy but with no appreciable cooling hysteresis. Also, the properties of these two-phase transitions are sensitive to the ionic strength of the dispersing buffer. Our spectroscopic and x-ray diffraction data indicate that the lower temperature transition corresponds to a lamellar subgel (Lc′) to gel (Lβ) phase transition and the higher temperature endotherm to a Lβ to lamellar liquid-crystalline (Lα) phase transition. At the Lc′/Lβ phase transition, there is a fivefold increase of the thickness of the interlamellar aqueous space from ∼11 Å to ∼50 Å, and this value decreases slightly at the Lβ/Lα phase transition. The bilayer thickness (i.e., the mean phosphate-phosphate distance across the bilayer) increases from 42.8 Å to 43.5 Å at the Lc′/Lβ phase transition, consistent with the loss of the hydrocarbon chain tilt of ∼12°, and decreases to 37.8 Å at the Lβ/Lα phase transition. The calculated cross-sectional areas of the TMCL molecules are ∼79 Å2 and ∼83 Å2 in the Lc′ and Lβ phases, respectively, and we estimate a value of ∼100 Å2 in the Lα phase. The combination of x-ray and FTIR spectroscopic data indicate that in the Lc′ phase, TMCL molecules possess tilted all-trans hydrocarbon chains packed into an orthorhombic subcell in which the zig-zag planes of the chains are parallel, while in the Lβ phase the untilted, all-trans hydrocarbon chains possess rotational mobility and are packed into a hexagonal subcell, as are the conformationally disordered hydrocarbon chains in the Lα phase. Our FTIR spectroscopic results demonstrate that the four carbonyl groups of the TMCL molecule become progressively more hydrated as one proceeds from the Lc′ to the Lβ and then to the Lα phase, while the two phosphate moieties of the polar headgroup are comparably well hydrated in all three phases. Our 31P-NMR results indicate that although the polar headgroup retains some mobility in the Lc′ phase, its motion is much more restricted in the Lβ and especially in the Lα phase than that of other phospholipids. We can explain most of our experimental results on the basis of the relatively small size of the polar headgroup of TMCL relative to other phospholipids and the covalent attachment of the two phosphate moieties to a single glycerol moiety, which results in a partially immobilized polar headgroup that is more exposed to the solvent than in other glycerophospholipids. Finally, we discuss the biological relevance of the unique properties of TMCL to the structure and function of cardiolipin-containing biological membranes.
INTRODUCTION
Cardiolipin (CL) or diphosphatidylglycerol is an anionic phospholipid occurring in the plasma membranes of many types of Gram-negative and Gram-positive bacteria and in the mitochondrial and chloroplast inner membranes of eukaryotes (1–5). CL is usually a relatively small component (≤10 mol %) of the total membrane lipid in such membranes and its primary role appears to be supporting the function of key membrane proteins (5). Specifically, CL plays an important role in the structural stabilization and activation of many mitochondrial enzymes, especially those involved in ATP synthesis and energy transduction (6–12), and in maintaining the structure and function of the Type II photoreaction center of photosynthetic bacteria and plants (13). In higher eukaryotes, CL is an effector of the cytochrome P-450-dependent cholesterol side-chain cleavage enzyme and activates cytochrome c oxidase and the mitochondrial phosphate carrier protein (14–16). It has been suggested that the capacity for structurally specific interactions with membrane CL may actually be highly conserved in such proteins (17). CL may also be found in substantially higher quantities (∼55–60 mol %) in the membranes of some microbial organisms, wherein elevated levels of CL and its derivatives appear to enhance the tolerance of these organisms to halophilic and resource-depletion stress (5,18–21). However, it is not clear whether elevated CL levels reflect a requirement for the enhancement of the structural integrity of the cell membrane, for the support of stress-related increases in energy procurement and transduction, or both (12,18).
Unlike other naturally occurring glycerophospholipids, CL is a quadruple-chained amphiphile composed of two phosphatidate moieties esterified to the 1- and 3-hydroxyl groups of a single glycerol molecule (referred to here as the headgroup glycerol). Consequently, one would expect the CL polar headgroup to be a relatively rigid, mobility-restricted entity, the cross-sectional area of which would be small relative to the combined cross-sectional areas of the four hydrocarbon chains. Thus, CL should be more prone to form inverted nonlamellar phases than phospholipids with larger headgroups such as phosphatidylcholine (PC) and phosphatidylglycerol (PG), although the formation of such inverted nonlamellar phases would be expected to be inhibited by the high surface charge of the CL bilayer (see (22)). Indeed, the Lα phase of CL membranes only converts to the inverted hexagonal phase when the negatively charged phosphate groups at the surface of such bilayers are protonated at low pH or screened by dispersal in solutions of high ionic strength, or if divalent metal cations are added (23–26). It has been suggested that these properties of CL may be an important aspect of the capacity of certain microorganisms to regulate the lamellar/nonlamellar phase propensity of their membrane lipids (27,28).
Most of the studies that have been performed on CL model membranes have concentrated on characterizing its role in supporting the function of enzymes such as cytochrome c oxidase (6–10), and on a characterization of its pH, divalent metal, and salt-induced lamellar-nonlamellar phase behavior (23–26). As a result, relatively little is known about the overall thermotropic phase behavior, or about the detailed structure and organization, of CL bilayer membranes. However, some differential scanning calorimetry (DSC) studies have shown that the Lβ/Lα phase transition temperatures of CL bilayers are higher than those of the corresponding diacyl PGs (24,29,30), and are more sensitive to the nature and amounts of mono- and divalent cations present in the dispersal media (29,30). Also, Fourier transform infrared (FTIR) spectroscopic studies suggest that the headgroup glycerol of CL may be involved in strong intramolecular interactions, presumably with the phosphate entities of the polar headgroup (31). In this study, we present a rigorous and systematic DSC, x-ray diffraction, and FTIR and 31P-NMR spectroscopic characterization of the overall thermotropic phase behavior of tetramyristoyl cardiolipin (TMCL), and of the structure and organization of the various lamellar phases that TMCL can form at physiological pH and ionic strength.
MATERIALS AND METHODS
TMCL was obtained from Avanti Polar Lipids (Alabaster, AL) and was used without further purification. Samples were prepared for DSC under the following set of conditions. In one set of measurements, 4–5 mg of TMCL were dispersed in 600 μl of a buffer composed of 50 mM Tris, 150 mM NaCl, 5 mM EDTA, and 1 mM NaN3, pH 7.4, by vigorous agitation at temperatures near 60°C, and a 500 μl aliquot was introduced into the Hastelloy capsule of a Calorimetry Sciences (Spanish Fork, UT) Multi-Cell DSC. Before initial data acquisition, the sample was preconditioned by heating to 50°C followed by cooling to −7°C at scanning rates near 60°C h−1. Data acquisition scans were then performed at scanning rates near 10°C h−1. Typically, the process first involves three cycles of heating and cooling between −7°C and 60°C. Subsequently, the samples were cooled to −30°C and incubated at that temperature for 12 h, then heated to 8°C and incubated at that temperature to ensure that all ice had completely melted. This low-temperature incubation protocol was repeated twice and the sample was then cooled to −7°C and a further set of data-acquisition runs, consisting of two cycles of heating and cooling between −7°C and 60°C, were performed. In another set of DSC measurements, ∼1 mg of lipid was dispersed in 1 ml of buffer composed of 100 mM sodium phosphate, 150 mM NaCl, and 5mM EDTA, pH 7.4, by methods similar to those described above. Subsequently, the sample was incubated at −20°C overnight and then allowed to thaw slowly at temperatures near 4°C. Subsequently, 0.5 ml of sample was loaded into a precooled sample cell of a Microcal (Northampton, MA) VP-DSC instrument and data were acquired between 1 and 55°C at heating and cooling rates near 10°C h−1 and 60°C h−1. The phosphate buffer was utilized to facilitate a direct comparison of our DSC and x-ray diffraction results with previous studies, but this buffer is not suitable for FTIR and 31P-NMR spectroscopic studies, because the presence of large amounts of phosphate in the buffer swamps the lipid phosphate signals, so the Tris buffer was utilized instead. Moreover, the use of two different buffers allowed us to investigate the effect of buffer composition and ionic strength on TMCL phase behavior.
FTIR spectroscopy was performed on samples containing 2–3 mg of TMCL. Samples were dispersed as described above in 100 μl of a D2O-based buffer containing 100 mM sodium phosphate and 150 mM NaCl, pD 7.4. The dispersion was squeezed between the CaF2 windows of a heatable, demountable liquid cell (NSG Precision Cells, Farmingdale, NY) equipped with a 25 μM Teflon spacer. Once mounted in the sample holder of the spectrometer, the sample could be heated between −20°C and 90°C by an external, computer-controlled water bath. Before initial data acquisition, samples were subject to the same low-temperature incubation protocol described above for the DSC samples. Infrared spectra were then acquired as a function of temperature with a Digilab FTS-40 Fourier-transform Spectrometer (Bio-Rad, Digilab Division, Cambridge, MA) using data acquisition parameters similar to those described by Mantsch et al. (32). The experiment involved a sequential series of 2°C temperature ramps with a 20 min interramp delay for thermal equilibration, and was equivalent to an effective scanning rate of 4°C per hour. The data obtained were analyzed using computer programs obtained from the instrument manufacturer and from the National Research Council of Canada. In cases where absorption bands appeared to be a summation of components, a combination of Fourier deconvolution and curve-fitting procedures were used to obtain estimates of the position of the component bands and to reconstruct the contours of the original band envelope.
For the 31P-NMR spectroscopic measurements, 10–15 mg of TMCL were dispersed in 0.6 ml of a buffer composed of 100 mM Tris, 150 mM NaCl, 5 mM EDTA, and 1 mM NaN3, pH 7.4. Before initial data acquisition, samples were subjected to the same low-temperature incubation protocol described above for the DSC samples. Spectra were acquired as a function of temperature with a Varian Unity-300 spectrometer (Varian, Palo Alto, CA) operating at 121.41 MHz for 31P. Spectra were acquired using previously published single-pulse data acquisition techniques and other data acquisition parameters (33).
Samples for x-ray diffraction measurements were also prepared by dispersing 5 mg of TMCL in 0.1 ml of both buffers (100 mM sodium phosphate, 150 mM NaCl, and 5 mM EDTA, pH 7.4; and 50 mM Tris, 150 mM NaCl, 5 mM EDTA, and 1 mM NaN3, pH 7.4, respectively), by vigorous vortex mixing at temperatures near 60°C. Before initial data acquisition, samples were subjected to the same low-temperature incubation protocol described above for the DSC samples. X-ray diffractograms were recorded with a SWAX-camera (HECUS X-Ray Systems, Graz, Austria), which was mounted on a sealed-tube Seifert generator (Ahrensburg, Germany) operating at 2 kW (50 kV and 40 mA). The x-ray beam was filtered for CuKα-radiation (λ = 1.54 Å) using an Ni foil and a pulse-height discriminator, built into the detection system. The scattered intensity was recorded with two linear position-sensitive detectors (HECUS X-Ray Systems) simultaneously in the small- and wide-angle regimes of 10−3 Å−1 < q < 1 Å−1 (SAXS) and 1.2 Å−1 < q < 2.7 Å−1 (WAXS), where q = 4 π sin(θ)/λ is the modulus of the scattering wave vector. Calibration in the small-angle region was performed with silver stearate and in the wide-angle region with a p-bromo-benzoic acid standards, respectively. Samples were loaded into thin-walled quartz capillaries that were thermally equilibrated for 10 min before initiating data acquisition. Temperature was controlled with an accuracy of 0.1°C with a programmable Peltier unit. Diffraction patterns were recorded at each temperature for 2700 s.
Background-corrected SAXS patterns were analyzed in terms of a global analysis technique described previously (34–36). Briefly, the scattered intensity of randomly oriented bilayers
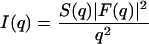 |
is separated into the contributions from the lamellar lattice, given by the structure factor S(q) and the modulation of the electron density across the lipid bilayer, expressed by the form factor F(q). The electron density profile, ρ(z), is modeled as a simple summation of Gaussian peaks, where two Gaussians are located at the position of the phosphate group on each side of the bilayer and a third Gaussian of negative amplitude at the center of the bilayer. F(q) is the Fourier transform of ρ(z) and contains an adjustable parameter, dPP, as a measure for the membrane thickness. From dPP, the phosphate-phosphate distance across the bilayer, we further calculate the hydrocarbon chain length by the equation
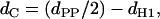 |
where dH1 is the distance from the phosphate group to the polar/apolar interface (37). With PC bilayers, Nagle and co-workers have typically reported values of ∼4.95 Å for dH1 (38). However, Pabst et al. (37) have shown that a dH1 value of ∼4 Å gives a good overall agreement between the dC values obtained by Nagle's group and the values derived from the global data analysis technique. This is because the global analysis technique models the lipid headgroup with a single peak, whereas Nagle and co-workers model the PC headgroup with two contributions, one for the phosphate group and one for the choline group. The latter reports slightly larger dPP values than those derived by the global analysis technique used here, which is compensated for by a smaller dH1 value. The dH1 value used for these studies (4 Å) assumes that the distance between the phosphate group and the polar/apolar interface of TMCL bilayers is comparable to those of PC bilayers, which seems to be justified by our results (see below). The hydrocarbon-chain tilt angle (θt) was estimated from the dC values by the equation
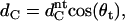 |
where the 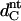 refers to nontilted hydrocarbon chain length, as approximated by the product of the distance between two C-bonds (1.27 Å, (39)) and the number of hydrocarbons per chain, which yields a value of ∼17.8 Å. The projected lateral area (A) per TMCL molecule was estimated from the hydrocarbon-chain tilt angle and the per chain area (AC) by the following equation:
refers to nontilted hydrocarbon chain length, as approximated by the product of the distance between two C-bonds (1.27 Å, (39)) and the number of hydrocarbons per chain, which yields a value of ∼17.8 Å. The projected lateral area (A) per TMCL molecule was estimated from the hydrocarbon-chain tilt angle and the per chain area (AC) by the following equation:
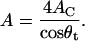 |
The structure factor, S(q), is obtained within the frameworks of the modified Caillé theory (40) or the paracrystalline theory (41). The modified Caillé theory takes into account the decrease in positional correlations due to bending fluctuations, whereas paracrystalline theory considers stacking disorder in perfectly flat lamellae. Hence, it is reasonable to apply the modified Caillé theory in the Lα phase, which exhibits considerable bending fluctuations, and paracrystalline theory in the lamellar gel phases below the Tm, where fluctuations are negligible due to the much larger bending rigidity (∼1 order of magnitude). The most relevant fit parameters of S(q) are the lamellar repeat distance, d, and a fluctuation parameter, which is the dimensionless quantity η (often referred to as the Caillé parameter), which depends inversely on the bending rigidity and the bulk modulus of bilayer interactions. The fluctuation parameter σPT of the paracrystalline theory has the units of length and gives the standard deviation of an assumed Gaussian distribution of bilayer separations. Both fluctuation parameters affect the diffuse decay of the Bragg peak intensity, but in a different way. It should be noted that geometric smearing imposed by the rulerlike shape of the applied x-ray beam leads to Bragg peaks that exhibit a slow intensity decay toward lower q-values and a sharp falloff in the opposite direction. This imposes some limitations on the determination of the fluctuation parameters on an absolute scale (35). However, mainly because the model considers the full q-range, the relative changes of η or σPT (for example, as a function of temperature) can be obtained reliably.
RESULTS
Differential scanning calorimetry
Illustrated in Figs. 1 and 2 are heating and cooling DSC thermograms that typify the thermotropic phase behavior of dispersions of TMCL in the two buffer systems used. Once fully equilibrated at low temperatures, aqueous dispersions of TMCL exhibit two major heating endothermic transitions. The lower-temperature transition is less cooperative than the higher temperature transition but the energetics of the two processes are comparable in magnitude. However, the thermodynamic properties of these two transitions differ in the two buffer systems and with the instrument used. Thus, for example, at a scanning rate of 10°C h−1, the temperatures, enthalpies, and widths (at half-height) of the lower-temperature transition obtained using the Tris buffer in the Multi-Cell DSC are 24.9°C, 11.5 kcal/mol, and 2.5°C, whereas with phosphate buffer in the VP-DSC the corresponding values are 29.2°C, 14.8 kcal/mol, and 1.2°C. In the case of the higher-temperature transition, the temperatures, enthalpies, and widths recorded with the Tris buffer and Multi-Cell DSC are 38.9°C, 12.9 kcal/mol, and 1.5°C, whereas the corresponding values obtained with phosphate buffer in the VP-DSC are 41.2°C, 13.5 kcal/mol, and 0.3°C, respectively. Nevertheless, when examined in the high-sensitivity power-compensation instrument (i.e., the Microcal VP-DSC), the properties of the latter transition are relatively insensitive to the scanning rate in the range 10°C–60°C h−1 (data not shown). Using data obtained in our x-ray-diffraction and spectroscopic studies (see below), we have assigned the lower-temperature event to a subgel-to-gel (Lc′/Lβ) transition and the higher-temperature event to a gel-to-liquid-crystalline (Lβ/Lα) phase transition involving the melting of lipid hydrocarbon chains.
FIGURE 1.

DSC thermograms illustrating the polymorphic phase behavior exhibited by dispersions of TMCL in Tris buffer (see Materials and Methods). The heating scan A was obtained after low-temperature equilibration of the sample as described in Materials and Methods. Cooling scans B and D were obtained upon cooling from high temperature to temperatures near −7°C and 18°C, respectively. Heating scan C was obtained by immediately reheating the sample after cooling to temperatures near −7°C, whereas heating scan E was obtained by immediate reheating the sample after cooling to temperatures near 18°C. These data were acquired with the Multi-Cell DSC at a scanning rate of 10°C h−1.
FIGURE 2.

DSC thermograms illustrating the polymorphic phase behavior exhibited by dispersions of TMCL in sodium phosphate buffer (see Materials and Methods). Heating scan A represents the pattern of behavior observed upon initial heating of samples that were previously extensively incubated at low temperatures. The other thermograms are typical of those obtained upon subsequent cooling (B) and reheating (C) between 0°C and 55°C. These data were acquired with the Microcal DSC at a scanning rate of 60°C h−1.
Figs. 1 and 2 also show that upon cooling, the conversion of the Lα phase of TMCL to the Lβ phase may be associated with either one (phosphate buffer) or two (Tris buffer) cooling exotherms, depending on the nature of the dispersal buffer. These exothermic events are observed at temperatures only slightly below those observed upon heating, and have been identified as interconversions between the Lα and Lβ phases of this lipid on the basis of our FTIR spectroscopic and x-ray diffraction results (data not shown). Multiple cooling exotherms at the Lα/Lβ phase transitions have been observed with some mixed-chain PCs (42) and cationic phospholipids (43). This phenomenon usually arises from heterogeneity in the domain sizes from which the Lβ is nucleated (42,44). Thus, the differences between the cooling exotherms observed in Tris and phosphate buffer is probably reflecting ionic strength-induced variations in the sizes of the domains formed in the La phase. Unlike the Lα/Lβ phase transition of TMCL, conversion of the Lβ phase to the Lc′ phase exhibits a pronounced cooling hysteresis, being initiated at temperatures some 10°C below the onset of the heating endotherm (Figs. 1 B and 2 B) and, as indicated by the considerably smaller enthalpy change observed (∼2.5 kcal/mol), this phase transition is not completed during the cooling scan. Interestingly, when aqueous dispersions of TMCL are reheated within a few hours after cooling to temperatures near 0°C, one usually observes two low-temperature endotherms centered near 14–18°C and 25–30°C (see Figs. 1 C and 2 C). Moreover, the combined enthalpy of these two peaks (∼6–8 kcal/mol) is usually smaller than that of the single Lc′/Lβ single phase transition observed upon heating the fully equilibrated samples. Moreover, the size of the peak near 25–30°C seems to increase at the expense of the lower-temperature peak upon incubation at temperatures near 0°C. The latter observation suggests that the formation of the stable Lc′ phase formed using the low-temperature incubation protocols described in the Materials and Methods could be proceeding via the formation of an intermediate subgel phase. However, the results of our time-resolved FTIR spectroscopic studies (not shown here) showed no evidence for the formation of such an intermediate Lc′ phase.
X-ray diffraction
Illustrated in Fig. 3 A are the SAXS profiles exhibited by TMCL in the sodium phosphate buffer system at temperatures bracketing the calorimetrically-resolved Lc′/Lβ and Lβ/Lα phase transitions. The bilayer properties calculated from these x-ray diffraction data are summarized in Table 1 and Fig. 4. At temperatures near 2°C, TMCL exhibits small-angle diffractograms containing a sharp, resolution-limited, first-order Bragg peak. Aside from the fourth-order peak, the higher-order diffraction peaks are barely visible, which is analogous to diffraction patterns displayed by phosphatidylethanolamines (PEs) (45). The global data analysis gives a good overall fit to these data except for the fourth-order peak. This can be attributed to simplifications involved in polycrystalline theory (36), but has no significant affect on the structural parameters obtained. As reported in Table 1, we find that at 2°C, TMCL forms a multibilayer system with a lamellar repeat distance (d) of 54.2 Å, where the individual layers of thickness dPP ∼ 42.8 Å are separated by a small water layer dW ≡ d – dPP ∼ 11 Å, again very much like the PEs (45). At temperatures near 35°C, TMCL exhibits two SAXS peaks that can be indexed as the first and second lamellar diffraction order. The peaks appear to be considerably broader than at 2°C, which is due to an increase of the stacking fluctuations from σPT (2°C) = 3.7 Å to σPT (35°) = 10.3 Å. The central peak width given by the scattering domain size remains unaffected. The approximately threefold increase of σPT manifests itself also in a larger d of ∼93.3 Å. The observed increase of dPP of ∼43.5 Å is much less pronounced, and hence the increase of d is largely due to a swelling of the water layer by ∼40 Å. Finally, at temperatures above the higher-temperature phase transition, aqueous TMCL exhibits Bragg peaks that index on a lamellar lattice with a d of ∼83 Å at 55°C. The membrane thickness (dPP ∼ 37.8 Å) is now significantly smaller than observed at lower temperatures, as expected for the melting of the hydrocarbon chains. The bilayer separation has decreased slightly to ∼45 Å, but is still significantly larger than in uncharged lipid/water systems, which is also reflected in the fluctuation parameter η (Table 1), which is approximately three times the value exhibited in phosphatidycholine (PC) dispersions (36). Comparable x-ray diffraction experiments were also performed with TMCL in the lower ionic strength Tris buffer (not shown). Our analyses of the data obtained in those experiments indicate that the structural parameters such as bilayer thickness and hydrocarbon chain packing were the same as those obtained in phosphate buffer. However, the positional correlation of the bilayers decreased in the Tris buffer, resulting in broader Bragg peaks and an increase in the thickness of the water layer between the bilayers (i.e., larger d-values). The latter is consistent with the expected effects of increased electrostatic repulsion under those experimental conditions.
FIGURE 3.

Scattered x-ray intensity of TMCL in small-angle (A) and wide-angle regime (B) at three temperatures in the Lc′ (2°C), Lβ (35°C), and Lα (55°C) phase. Data shown were acquired during heating using samples that were initially fully equilibrated at low temperatures. Arrows in panel A indicate the position of the first-order Bragg peak except for the pattern at 2°C, which has an additional arrow marking the fourth diffraction order. Solid lines show the best fits to the SAXS data applying a global analysis technique (see Materials and Methods). Bracketed numbers in panel B give the Miller indices of the corresponding Bragg rods.
TABLE 1.
Bilayer properties calculated for the three polymorphic phases exhibited by aqueous dispersions of TMCL
| T2 (°C) | d (Å) | dpp (Å) | dC (Å) | dW (Å) | σPT (Å) | η | Ac (Å)* | A (Å)* |
|---|---|---|---|---|---|---|---|---|
| 2 | 54.2 | 42.8 | 17.4 | 11.4 | 3.7 | — | 19.3 | 78.9 |
| 35 | 93.3 | 43.5 | 17.8 | 49.8 | 10.3 | — | 20.8 | 83.2 |
| 55 | 83 | 37.8 | 14.9 | 45.2 | — | 0.305 | — | — |
TMCL: 100 mM sodium phosphate, 150 mM NaCl, and 5 mM EDTA, pH 7.4.
For abbreviations, see text.
FIGURE 4.

Low-resolution, one-dimensional electron density profiles along the bilayer normal calculated from the small-angle scattering diffraction patterns exhibited by aqueous dispersions of TMCL. Electron density profiles were calculated for (A) Lc′ phase at 2°C; (B) Lβ phase at 35°C; and (C) Lα phase at 55°C.
Illustrated in Fig. 3 B are the patterns of WAXS reflections exhibited by the three polymorphic phases of TMCL. The pattern of diffraction peaks observed in this region arises from the two-dimensional packing of the lipid fatty acyl chains. At temperatures near 2°C, the WAXS diffractogram contains a relatively sharp Bragg peak centered near 4.6 Å and a broader Bragg reflection centered near 3.8 Å, suggesting that the latter arises from an off-equatorial reflection typical for hydrocarbon chains being tilted with respect to the bilayer plane (46). These peaks were assigned to the (2 0) and (1 1) spacings of an orthorhombic subcell with lattice parameters a = 9.2 Å, b = 4.2 Å, and a per-chain area of 19.3 Å2 (see Table 1). From the SAXS data we estimate a hydrocarbon-chain tilt angle of some 12°, from which we obtain a projected lateral area per TMCL molecule of some 80 Å2 (Table 1). At temperatures near 35°C, the WAXS pattern of TMCL exhibits a single sharp and symmetric reflection centered at 4.27 Å. This is consistent with the assembly of untilted hydrocarbon chains into two-dimensional hexagonal subcells with lattice constants of a = 8.5 Å and b = 4.9 Å, and a mean per-chain area of 20.8 Å2 (46,47). For this phase the projected lateral area (∼83.2 Å) is simply four times the Ac value and is only slightly larger than obtained for the Lc′ phase at 2°C (Table 1). We also note that the dC value obtained from the analysis of the SAXS data (∼17.8 Å) is in excellent agreement with our estimates of the untilted length of TMCL all-trans hydrocarbon chains, consistent with the conclusion that TMCL hydrocarbon chains are untilted at temperatures between the two phase transitions. The increase of the membrane thickness across the lower-temperature transition can thus be entirely explained by the removal of the chain tilt. Finally, at temperatures above the higher-temperature transition, TMCL exhibits a broad diffuse WAXS peak, consistent with the low degree of lateral packing order, which occurs in highly mobile melted hydrocarbon chains (i.e., the Lα phase). However, in the absence of dilatometric measurements we were unable to determine the lateral area per molecule in the Lα phase. Nevertheless, if the area expansion across the hydrocarbon chain-melting phase transition of TMCL is of comparable magnitude to that observed with PCs (∼25%, see (48)), the projected lateral area should be near 100 Å2.
Fourier-transform infrared spectroscopy
Illustrated in Fig. 5 are the regions of the C-H stretching, C=O stretching, CH2 scissoring, and asymmetric phosphate O-P-O stretching bands of the FTIR spectra exhibited by aqueous dispersions of TMCL at temperatures bracketing the two thermotropic phase transitions reported by DSC. These regions of the spectra encode information about hydrocarbon chain conformation, bilayer interfacial conformation and hydration, hydrocarbon chain packing interactions, and the polarity of the phosphate polar headgroups, respectively. It is clear from even a cursory examination of these spectra that the two phase transitions detected by DSC are structurally distinct thermotropic events. Thus, for example, in the C-H stretching region (2800–3000 cm−1), the changes accompanying the lower-temperature transition centered near 25–30°C involve relatively small decreases in the intensities of all absorption bands, whereas the changes accompanying the higher-temperature transition near 39–42°C are considerably larger and involve significant increases in both band frequency and bandwidth (Fig. 5 A), indicating that the major changes in hydrocarbon chain conformation coincide with the higher-temperature event. In contrast, the C=O stretching, CH2 scissoring, and asymmetric phosphate stretching regions of the FTIR spectrum exhibit major changes in band frequency, band shape, and band width at the lower-temperature transition and considerably smaller changes at the higher-temperature transition (Fig. 5, B–D), indicating that the major changes in interfacial conformation and hydration, hydrocarbon chain packing, and polar headgroup polarity occur at the lower-temperature transition. A more detailed examination and structural interpretation of the spectroscopic changes associated with these two phase transitions is presented below.
FIGURE 5.

FTIR spectra observed upon heating fully equilibrated aqueous dispersions of TMCL. The absorbance spectra shown were acquired at the temperatures indicated and typify the C-H stretching (A), the C=O stretching (B), the CH2 scissoring (C), and the asymmetric phosphate stretching (D) regions of the IR spectra of this compound at temperatures bracketing the two calorimetrically resolved thermotropic phase transitions. The vertical dashed line (A) marks the position of the maxima of CH2 symmetric stretching band observed in the in the three polymorphic forms of this lipid. The dashed lines (B) represent the subcomponents of the observed absorption band. The asterisks (D) mark the positions of the peaks of the CH2 wagging band progression.
In the C-H stretching region of the FTIR spectrum, the dominant features are the absorption bands centered near 2850 cm−1 and 2915 cm−1 (Fig. 5 A). These bands arise from the symmetric and asymmetric stretching vibrations of CH2 groups of the lipid hydrocarbon chains, respectively (49,50). In particular, the frequency of the symmetric stretching band is determined by the trans/gauche rotamer content of the lipid hydrocarbon chains and is therefore an excellent spectroscopic marker for changes in the conformation of these chains (49,50). At temperatures below 20°C, fully equilibrated TMCL samples exhibit sharp symmetric CH2 stretching bands at frequencies centered near 2850 cm−1 (Fig. 1 A), values consistent with the existence of predominantly all-trans polymethylene chains. Upon heating to temperatures near the Lc′/Lβ phase transition of this lipid, this band broadens slightly without any significant change in its peak frequency, indicating that no gauche conformers are induced at the lower-temperature transition. However, at the Lβ/Lα phase transition temperature, we see discontinuous increases in its frequency and width, indicating significant increases in the gauche rotamer content and overall mobility of the lipid hydrocarbon chains. These results clearly indicate that the lower-temperature transition involves a solid phase rearrangement of the TMCL molecules with all-trans lipid hydrocarbon chains, whereas the higher-temperature transition involves the melting of the TMCL hydrocarbon chains.
In the 1400–1500 cm−1 region of the IR spectrum, the dominant feature is the absorption band between 1466 cm−1 and 1473 cm−1, which arises from the scissoring vibrations of the hydrocarbon chain methylene groups (see Fig. 5 C). The properties of this band are sensitive to the lateral packing interactions between hydrocarbon chains (49,50). At low temperatures, the CH2 scissoring band of TMCL appears as a single, sharp absorption peak centered near 1473 cm1, suggesting that the all-trans lipid chains are packed in a subcell with their zigzag planes parallel. When heated to temperatures in between the Lc′/Lβ and Lβ/Lα phase transitions, this CH2 scissoring band broadens and its peak frequency shifts to values near 1468 cm−1 (Fig. 5 C), suggesting that the hydrocarbon chains are more mobile and packed in a hexagonal subcell. Moreover, since the spectroscopic properties exhibited in the C-H stretching region indicate that the hydrocarbon chains are still in the all-trans conformation (see previous paragraph), we can also conclude that the hydrocarbon chains are rotationally disordered, most probably because of an increase in the rates and amplitude of the reorientational fluctuations of the all-trans polymethylene chains at the Lc′/Lβ phase transition. Finally, when heated to temperatures above the Lβ/Lα phase transition, the CH2 scissoring band of TMCL broadens considerably without a significant change in its peak frequency (see Fig. 5 C). Since the contours of the C-H stretching regions indicate that the hydrocarbon chains are melted at these temperatures (see previous paragraph), we can conclude that the liquid-crystalline phase contains hexagonally arranged, conformationally and rotationally disordered hydrocarbon chains.
The spectra shown in Fig. 5 B also indicate that the major changes in the C=O stretching region between 1650 cm−1 and 1800 cm−1 also occur mainly at the Lc′/Lβ phase transition. Since the infrared absorption in this region arises exclusively from the stretching vibrations of the ester carbonyl groups of TMCL, these bands can provide valuable information about the structure and properties of the polar/apolar interfacial regions located near the backbone glycerol moiety of the three polymorphic forms of this lipid (49,50). At temperatures below the onset of the Lc′/Lβ phase transition, the C=O stretching band seems to be a summation of a relatively sharp band centered near 1735 cm−1 and a broader band of lesser intensity centered near 1716 cm−1 (see dashed curves in Fig. 5 B). These bands arise from the stretching vibrations of subpopulations of free and hydrogen-bonded ester carbonyl groups, respectively (51,52). At temperatures between the Lc′/Lβ and Lβ/Lα phase transitions, the C=O ester stretching band is also resolvable into two components (see dashed curves in Fig. 5 B). However, at these temperatures the component bands are of comparable intensity, are broader, and are centered at frequencies near 1743 cm−1 and 1728 cm−1, respectively. These latter two components are also found at temperatures above the Lβ/Lα phase transition of the lipid, but at those temperatures the lower-frequency component is even broader and is the major contributor to the integrated intensity of the observed C=O stretching band (see dashed curves in Fig. 5 B). These results indicate that there is a progressive increase in the degree of hydrogen bonding of the fatty acid carbonyl groups, resulting from an increased penetration of water into the glycerol backbone region of the TMCL molecules, as one proceeds from the Lc′ through the Lβ into the Lα phase.
An examination of the region encompassing the phosphate O-P-O asymmetric stretching band also indicates that fairly drastic changes occur at the Lc′/Lβ phase transition with relatively minor changes occurring at hydrocarbon chain-melting Lβ/Lα phase transition temperature (Fig. 5 D). At low temperatures, the broad contours of the phosphate O-P-O asymmetric stretching band are largely obscured by several sharp, relatively weak absorption bands arising from the CH2 wagging band progression (see marked bands in Fig. 5 D). These bands arise from the coupled CH2 wagging vibrations of methylene groups on all-trans polymethylene chains, and are usually quite sharp when the rates and amplitudes of chain reorientational fluctuations are low (49,50). However, despite the interference from these bands, the presence of the broad underlying absorption of the phosphate O-P-O asymmetric band is obvious and its peak seems to be centered near 1216 cm−1. Upon heating to temperatures in between the Lc′/Lβ and Lβ/Lα phase transitions, the CH2 wagging band progression largely disappears and the peak of the phosphate O-P-O asymmetric stretching is more easily resolved at frequencies again near 1216 cm−1 (Fig. 5 D). Upon heating to temperatures above the lipid Lβ/Lα phase transition, the phosphate O-P-O asymmetric stretching band narrows slightly, but its peak frequency remains near 1216 cm−1. This frequency is near the lower end of the range expected of phosphate O-P-O asymmetric stretching vibrations, consistent with the phosphate groups being well hydrated and/or bonded to hydrogen-bonding donor groups.
31P-Nuclear magnetic resonance spectroscopy
Illustrated in Fig. 6 are the 31P-NMR powder patterns acquired at temperatures bracketing the two thermotropic phase transitions observed by DSC upon heating after prolonged sample incubation at low temperatures. At temperatures below the onset of the lower-temperature transition, fully equilibrated samples of aqueous TMCL exhibit broad 31P-NMR powder patterns with basal line widths near 250 ppm (spectrum A), characteristic of the so-called rigid-limit spectra expected of the crystalline forms of phospholipids (53,54). This suggests that TMCL forms an Lc′ phase in which reorientation of the phosphate moieties of the TMCL headgroup is extremely slow on the 31P-NMR timescale. We also note that despite the effective “immobilization” of the phosphate headgroups, 31P-NMR spectra of the Lc′ phase of TMCL were easily acquired using simple single-pulse data acquisition techniques. This situation differs markedly from what occurs when similar techniques are used to record the 31P-NMR spectra of the subgel phases of phospholipids such as dimyristoyl phosphatidylethanolamine and dimyristoyl phosphatidylglycerol, for which the extremely long longitudinal relaxation times of the 31P nucleus makes the recording of their Lc phase spectra impractical (55,56). Given that such slow longitudinal relaxation of the 31P nucleus have been attributed to long-lived H-bonding interactions between the phosphate and appropriate H-bond donor groups (55,56), it appears that the phosphate moieties of the polar headgroup of TMCL may not be involved in long-lived H-bonding interactions with either solvent molecules of other potential H-bonding donors in their Lc′ phase.
FIGURE 6.

Proton-decoupled 31P-NMR powder patterns exhibited by aqueous dispersions of TMCL. The spectra shown were acquired in the heating mode after prolonged sample equilibration at low temperatures and are representative of (A) Lc′ phase at 2°C; (B) Lβ phase at 32°C; and (C) Lα phase at 48°C.
When heated to temperatures between the completion and onset temperatures of its lower-temperature and higher-temperature thermotropic phase transitions, significant narrowing of the 31P-NMR powder patterns of TMCL occurs (Fig. 6, spectrum B). However, the 31P-NMR powder patterns observed in this phase are themselves quite broad (basal line widths ∼140 ppm), and their widths and contours are atypical of those normally exhibited by the Lβ-like lamellar gel phases of other 1,2-diacyl glycerophospholipids (for examples, see (32,33,55,56)). This observation suggests that in the Lβ phase of this lipid, reorientation of its headgroup phosphate moieties is either significantly slower than that of the Lβ phases of most phospholipid bilayers, or that the range and amplitude of reorientational motion accessible to phosphate groups in the TMCL Lβ phase are much more restricted than is typical of phospholipid Lβ phases, or both.
Finally, at temperatures above its Lβ/Lα phase transition temperature, TMCL exhibits 31P-NMR powder patterns that show some of the features normally observed in the Lα phases of most other phospholipids (Fig. 4, spectrum C). However, the apparent chemical shift anisotropy (∼25 ppm) of fluid TMCL dispersions is considerably smaller than that of the Lα phases of phospholipids such as PC, PE, and PG, for which values near 35–40 ppm are commonly observed (see (32,33,55,56)). This observation suggests that even in its Lα phase, the phosphate moieties of TMCL may be subject to motional restrictions that are not normally observed in most other phospholipids.
DISCUSSION
A number of new and important conclusions about the thermotropic phase behavior and organization of TMCL bilayer membranes can be drawn from the calorimetric, x-ray diffraction, and spectroscopic data presented here. For example, we demonstrate that the overall thermotropic behavior of TMCL is strongly influenced by the nature of the dispersal medium, especially the temperature at which its thermotropic phase transitions are observed. Such behavior is commonly observed with anionic phospholipid membranes (see (57–59)) and was thus generally expected. However, the data presented here and elsewhere (see (29,30,60)) indicates that CL membranes are more sensitive to such effects than membranes composed of other anionic phospholipids such as PG and phosphatidylserine. This observation can probably be attributed to the reduced shielding of the negatively charged phosphate groups on the surface of CL membranes by the single headgroup glycerol moiety to which both phosphates are covalently linked. This is because the relative rigidity and restricted mobility of the CL headgroup effectively reduces its capacity for intra- and intermolecular interaction with other CL phosphate groups, and as a result negative charges at the surfaces of CL bilayers will not be as shielded from interaction with the solvent and its dissolved ions, as would be the case with anionic lipids such as PG and phosphatidylserine. In this respect, the properties of CL bilayers may actually be more comparable to those of bilayers composed of the anionic glycerophospholipid phosphatidic acid (PA), which is also very sensitive to the properties of the dispersal medium, presumably because its negatively charged phosphate headgroups are not shielded from interaction with the water and ions.
The calorimetric data presented here is in general agreement with previous calorimetric studies (29,30) reporting that TMCL exhibits a Lβ/Lα phase transition temperature (∼40°C) that is markedly higher than that of the corresponding diacyl PG (∼23°C (61)). Given the low capacity for shielding of the phosphate groups by the CL headgroup glycerol (see above), the relatively high Lβ/Lα phase transition temperature of TMCL can probably be rationalized by the combined effects of the relatively small size of its polar headgroup and the existence of a H-bonding and/or a cation interaction network, as postulated for PA bilayers (62). Our calorimetric results also indicate an enthalpy for the TMCL Lβ/Lα phase transition (12.9 kcal/mol) that is slightly higher but generally compatible with the range of values (∼11–12.5 kcal/mol) reported in the literature under comparable conditions (29,30). Also, the per chain normalized enthalpy (∼3.2 kcal/mol) is compatible with those observed at the Lβ/Lα type phase transitions of most phospho- and glycolipids that have been examined so far. Our calorimetric measurements are thus consistent with the assignment of the TMCL Lβ/Lα phase transition as a simple hydrocarbon chain-melting event like those observed in most naturally occurring glycerolipids.
These studies have also shown that TMCL readily forms a highly ordered lamellar crystal-like (Lc′) phase when cooled to temperatures below 10°C. To our knowledge, this aspect of the polymorphic phase behavior of CL membranes has not been reported previously. Our spectroscopic and x-ray diffraction data also indicate that although the temperature at which the Lc′/Lβ phase transition occurs varies with the nature of the dispersal medium, the structural properties of the Lc′ phase are essentially the same under all conditions explored here. The structural picture of the TMCL Lc′ phase that emerges from these studies is as follows. In the hydrophobic domain, the closely packed all-trans hydrocarbon chains of TMCL are slightly tilted (∼12°) to the bilayer surface and seem to be arranged in an orthorhombic subcell (x-ray diffraction data) with their zigzag planes parallel to each other (FTIR spectroscopic data). In the bilayer polar/apolar interfacial region near the backbone glycerol, there appears to be two populations of ester carbonyl groups (FTIR spectroscopic data). One of these gives rise to a sharp, higher-frequency C=O stretching band consistent with its being fairly rigid and located in a relatively nonpolar environment free of H-bonding interactions with either water or other H-bonding donor groups. The other population gives rise to a broad, lower-frequency C=O stretching band, and is probably located in a more polar and more heterogeneous environment where it is interacting via H-bonding with water or other donor groups. Given the overall low mobility of CL molecules in the Lc′ phase, the heterogeneity underlying the broad lower-frequency ester C=O band probably arises from heterogeneity in the orientation of H-bonding donor groups. This suggests that the source of the H-bonding donor groups is external to the CL molecule itself and is most probably water molecules in the dispersal medium. Our studies also indicate that the two phosphate moieties of the TMCL molecule are essentially immobilized on the 31P-NMR timescale in the Lc′ phase. However, the frequency of the phosphate O-P-O asymmetric stretching band (FTIR spectroscopic data) also suggests that these phosphate moieties are well hydrated. This latter conclusion is supported by the x-ray diffraction results, which show that the Lc′ phase lamellar repeat distance (∼54 Å) is significantly greater than the phosphate-phosphate bilayer thickness (∼43 Å), consistent with the presence of an aqueous interlamellar space of ∼11 Å. Also, given the relative ease with which 31P-NMR spectra of the Lc′ phase were acquired, it is unlikely that the phosphate moieties are involved in strong, long-lived H-bonding interactions in this phase. Thus, interactions involving these negatively charged phosphate groups at the surface of the Lc′ phase of TMCL are probably better defined by hydrogen bonding to water and by Gouy-Chapman type interactions with counterions (for references on the latter, see (55)).
The phosphate-phosphate distance (∼43 Å) and the hydrocarbon chain length (∼17. 4 Å) calculated for the Lc′ phase of TMCL also provide some insight into the possible conformations of the lipid molecules in the bilayer. There are two extreme conformations for the alignment of the backbone glycerols of TMCL, one along the bilayer normal as observed in single-crystal x-ray studies of PC and PE (63,64), and one roughly parallel to the bilayer surface as observed in single-crystal x-ray studies of PG and PA (62,65). The latter conformation can be excluded because the length of two opposed untilted hydrocarbon chains (∼35 Å, see Table 1) and the mean thickness of the headgroup and backbone moieties relative to the bilayer normal (∼2.5 Å, our estimates) are together too short to account for the observed P-P distance of ∼43 Å. However, our experimental data can accommodate a conformation in which the TMCL backbone glycerols are oriented similar to PE and PC, with a slight tilting of the hydrocarbon chains. Such a conformation is obviously favorable for an optimal intramolecular packing of the hydrocarbon chains in the Lc′ phase and is also compatible with the results of our x-ray data obtained in the Lβ phase.
Our studies have also provided a fairly clear picture of the structures and organization of the Lβ and Lα phases of TMCL and of the structural changes occurring at the calorimetrically resolved thermotropic phase transitions. When the Lβ gel phase of TMCL is cooled to low temperatures, the formation of the Lc′ phase is probably initiated by a marked but progressive reduction in the amount of water present in the interlamellar space and bilayer polar/apolar interfaces, as indicated by the decrease of the lamellar repeat distance upon cooling (data not shown). Initially, this process probably involves the formation of relatively small domains of the Lc′ phase that reconvert to the Lβ gel phase at temperatures between 10 and 20°C. This assumption is supported by WAXS data, which showed only hardly discernible Bragg peaks immediately after cooling below the Lc′/Lβ phase transition (data not shown). However, with prolonged low-temperature annealing, the growth of Lc′ phase continues and the small domains eventually coalesce into the extensive lattices that exhibit the structural properties described above. Upon heating, these structures reconvert to the Lβ gel phase at temperatures between 25 and 30°C. This process is accompanied by a modest increase in the hydration of the bilayer polar/apolar interfaces but by a considerable swelling of the interlamellar spaces. We suggest that such a large increase of the bilayer separation can only occur through a change of the membrane surface charge density, probably through the release of counterions. There is also an increase in the rates and amplitudes of reorientational fluctuations of the hydrocarbon chain segments, which are reflected by a decrease within close-contact, coupled interactions between vibrating CH2 groups (FTIR data), caused by a distortion of the orthorhombic lattices formed at low temperatures (x-ray diffraction data). The 31P-NMR spectroscopic data also suggests that although there is some increase in the mobility of the phosphate moieties when the Lc′ phase converts to the Lβ phase, the rates of such motions remain slow on the 31P-NMR timescale, presumably because of the restrictions imposed by the tethering of the phosphate moieties to the headgroup glycerol. Finally, upon conversion to the Lα phase, there is a cooperative melting of the hydrocarbon chains which is accompanied by an increase in the hydration of the bilayer polar/apolar interfaces, a small decrease in the thickness of the lipid bilayer and the interlamellar spaces, and a considerable increase in the rates and amplitudes of motion in all regions of the lipid bilayer. In this phase, the motions of phosphate moieties are also fast on the 31P-NMR timescale, but the range and amplitudes of such motions are restricted, presumably because of the tethering effect alluded to above.
In conclusion, we note that virtually all of the physical characteristics of CL bilayers as described in this and other studies can be directly or indirectly attributed to the restrictions placed on the flexibility and mobility of its polar headgroup by the tethering of the two phosphatidate moieties to a single glycerol molecule. As noted above, the low flexibility and impaired reorientational mobility of the CL headgroup diminishes its capacity for self-shielding of its negatively charged phosphate moieties and should therefore make CL-containing membranes more sensitive to interactions with the buffer. This property could be of considerable biological importance. Also, because both the intrinsic and effective sizes of the CL headgroup are small relative to the cross-sectional area of its hydrocarbon chains, CL can form inverted nonlamellar phases when its headgroup charges are either neutralized or effectively screened (22–26). This property may be biologically important because charge neutralization seems to be an important aspect of the interaction between CL and many of the CL-requiring membrane enzymes (9–10). These structural properties also seem to increase the structural integrity of CL membranes (note its relatively high Lβ/Lα phase transition temperature), presumably in part by imparting greater cohesion to the headgroup and polar/apolar interfacial regions of the membrane. In principle, these properties could explain why some organisms actually increase the CL content of their membranes under conditions of chronic resource depletion and halophilic stress (5,18–21). Moreover, these same properties may also explain why the penetration into the polar/apolar interface of CL membranes by the antibiotic peptide gramicidin S is not as efficacious as observed with membranes composed of other anionic lipids (66), and why CL-containing membranes are more resistant to the effects of some antimicrobial peptides than are membranes containing other negatively charged lipids (67–69). Thus, aside from its specific roles in supporting the function of key membrane enzymes, it also appears that CL confers upon cell membranes various physical properties that may be physiologically important.
Acknowledgments
The authors are grateful to Dr. Brian D. Sykes of the Department of Biochemistry at the University of Alberta for the generous availability of time on the NMR spectrometer.
Supported by operating and major equipment grants from the Canadian Institutes of Health Research (R.N.M.), by major equipment grants from the Alberta Heritage Foundation for Medical Research (R.N.M.), and by the Austrian Science Fund (grant No. P15657 to K.L.).
References
- 1.Daum, G. 1985. Lipids of mitochondria. Biochim. Biophys. Acta. 822:1–42. [DOI] [PubMed] [Google Scholar]
- 2.Ratledge, C., and S. G. Wilkinson. 1988. Fatty acids, related and derived lipids. In Microbial Lipids. C. Ratledge and S. G. Wilkinson, editors. Academic Press, London.
- 3.O'Leary, W. M., and S. G. Wilkinson. 1988. Gram-positive bacteria. In Microbial Lipids. C. Ratledge and S. G. Wilkinson, editors. Academic Press, London.
- 4.Wilkinson, S. G. 1988. Gram-negative bacteria. In Microbial Lipids. C. Ratledge and S. G. Wilkinson, editors. Academic Press, London.
- 5.Hoch, F. L. 1992. Cardiolipins and biomembrane function. Biochim. Biophys. Acta. 1113:71–133. [DOI] [PubMed] [Google Scholar]
- 6.Kagawa, Y., A. Kandrach, and E. Racker. 1973. Partial resolution of the enzymes catalyzing oxidative phosphorylation. XXVI. Specificity of phospholipids required for energy transfer reactions. J. Biol. Chem. 248:676–684. [PubMed] [Google Scholar]
- 7.Dale, M. P., and N. C. Robinson. 1988. Synthesis of cardiolipin derivatives with protection of the free hydroxyl: its application to the study of cardiolipin stimulation of cytochrome-c oxidase. Biochemistry. 27:8270–8275. [DOI] [PubMed] [Google Scholar]
- 8.Arnold, S., and B. Kadenbach. 1997. Cell respiration is controlled by ATP, an allosteric inhibitor of cytochrome c oxidase. Eur. J. Biochem. 249:350–354. [DOI] [PubMed] [Google Scholar]
- 9.Lange, C., J. H. Nett, B. L. Trumpower, and C. Hunte. 2001. Specific roles of protein-phospholipid interactions in the yeast cytochrome bc1 complex structure. EMBO J. 20:6591–6600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McAuley, K. E., P. K. Fyfe, J. P. Ridge, N. W. Isaacs, R. J. Cogdell, and M. R. Jones. 1999. Structural details of an interaction between cardiolipin and an integral membrane protein. Proc. Natl. Acad. Sci. USA. 96:14706–14711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Serrano, R., B. J. Banner, and E. Racker. 1976. Purification and properties of the proton-translocating adenosine triphosphatase complex of bovine heart mitochondria. J. Biol. Chem. 251:2453–2461. [PubMed] [Google Scholar]
- 12.Hoch, F. L. 1998. Cardiolipins and mitochondrial proton-selective leakage. J. Bioenerg. Biomembr. 30:511–532. [DOI] [PubMed] [Google Scholar]
- 13.McAuley, K. E., P. K. Fyfe, J. P. Ridge, N. W. Isaacs, R. J. Cogdell, and M. R. Jones. 1999. Structural details of an interaction between cardiolipin and an integral membrane protein. Proc. Natl. Acad. Sci. USA. 96:14706–14711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanaka, T., and J. F. Strauss. 1982. Stimulation of luteal mitochondrial cholesterol side-chain cleavage by cardiolipin. Endocrinology. 110:1592–1598. [DOI] [PubMed] [Google Scholar]
- 15.Tuckey, R. C., and K. J. Cameron. 1993. Catalytic properties of cytochrome P-450(SCC) purified from the human placenta—comparison to bovine cytochrome P-450(SCC). Biochim. Biophys. Acta. 1163:185–194. [DOI] [PubMed] [Google Scholar]
- 16.Kisselev, P., R. C. Tuckey, S. T. Woods, T. Triantopoulos, and D. Schwartz. 1999. Enzymatic properties of vesicle-reconstituted human cytochrome P450SCC (CYP11A1)—differences in functioning of the mitochondrial electron-transfer chain using human and bovine adrenodoxin and activation by cardiolipin. Eur. J. Biochem. 260:768–773. [DOI] [PubMed] [Google Scholar]
- 17.Wakeham, M. C., R. B. Sessions, M. R. Jones, and P. K. Fyfe. 2001. Is there a conserved interaction between cardiolipin and the Type II bacterial reaction center? Biophys. J. 80:1395–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koch, H. U., R. Haas, and W. Fischer. 1984. The role of lipoteichoic acid biosynthesis in membrane lipid metabolism of growing Staphylococcus aureus. Eur. J. Biochem. 138:357–363. [DOI] [PubMed] [Google Scholar]
- 19.Short, S. A., and D. C. White. 1971. Metabolism of phosphatidylglycerol, lysylphosphatidyl-glycerol and cardiolipin of Staphylococcus aureus. J. Bacteriol. 108:219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanemasa, Y., T. Yioshioka, and H. Hayashi. 1972. Alteration of the phospholipid composition of Staphylococcus aureus cultures in medium containing NaCl. Biochim. Biophys. Acta. 280:444–450. [PubMed] [Google Scholar]
- 21.Okabe, A., Y. Hirai, H. Hayashi, and Y. Kanemasa. 1980. Alteration in phospholipid composition of Staphylococcus aureus during formation of autoplast. Biochim. Biophys. Acta. 617:28–35. [DOI] [PubMed] [Google Scholar]
- 22.Lewis, R. N. A. H., and R. N. McElhaney. 2000. Surface charge density markedly attenuates the nonlamellar phase-forming properties of lipid bilayer membranes. Biophys. J. 79:1455–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Powell, G. L., and D. Marsh. 1985. Polymorphic phase behavior of cardiolipin derivatives studied by 31P NMR and x-ray diffraction. Biochemistry. 24:2902–2908. [DOI] [PubMed] [Google Scholar]
- 24.Sankaram, M. B., G. L. Powell, and D. Marsh. 1989. Effect of acyl chain composition on salt-induced lamellar to inverted hexagonal phase transitions in cardiolipin. Biochim. Biophys. Acta. 980:389–392. [DOI] [PubMed] [Google Scholar]
- 25.Seddon, J. M., R. D. Kaye, and D. Marsh. 1983. Induction of lamellar-inverted hexagonal phase-transition in cardiolipin by protons and monovalent cations. Biochim. Biophys. Acta. 734:347–352. [Google Scholar]
- 26.Cullis, P. R., A. J. Verkleij, and P. H. J. T. Ververgaert. 1978. Polymorphic phase behavior of cardiolipin as detected by 31P NMR and freeze-fracture techniques—effects of calcium, dibucane and chlorpromazine. Biochim. Biophys. Acta. 513:11–20. [DOI] [PubMed] [Google Scholar]
- 27.Lindblom, G., and L. Rilfors. 1992. Nonlamellar phases formed by membrane-lipids. Adv. Colloid Interface Sci. 41:101–125. [DOI] [PubMed] [Google Scholar]
- 28.Davevcic, T., L. Rilfors, J. Strancar, G. Lindblom, and D. Stopar. 2005. Effects of lipid composition on the membrane activity and lipid phase behavior of Vibrio sp DSM14379 cells grown at various NaCl concentrations. Biochim. Biophys. Acta. 1712:1–8. [DOI] [PubMed] [Google Scholar]
- 29.Rainer, S., M. K. Jain, F. Ramirez, P. V. Ioannou, J. F. Marecek, and R. Wagner. 1979. Phase transition characteristics of diphosphatidylglycerol (cardiolipin) and stereoisomeric phosphatidyldiacylglycerol bilayers. mono-valent and divalent metal-ion effects. Biochim. Biophys. Acta. 558:187–198. [DOI] [PubMed] [Google Scholar]
- 30.Nagamachi, E., R. Kariyama, and Y. Kanemasa. 1985. The effect of headgroup structure on phase transition of phospholipids membranes as determined by differential scanning calorimetry. Physiol. Chem. Phys. Med. NMR. 17:255–260. [PubMed] [Google Scholar]
- 31.Hubner, W., H. H. Mantsch, and M. Kates. 1991. Intramolecular hydrogen-bonding in cardiolipin. Biochim. Biophys. Acta. 1066:166–174. [DOI] [PubMed] [Google Scholar]
- 32.Mantsch, H. H., C. Madec, R. N. A. H. Lewis, and R. N. McElhaney. 1985. Thermotropic phase behavior of model membranes composed of phosphatidylcholines containing isobranched fatty acids. 2. Infrared and 31P-NMR spectroscopic studies. Biochemistry. 24:2440–2446. [DOI] [PubMed] [Google Scholar]
- 33.Lewis, R. N. A. H., B. D. Sykes, and R. N. McElhaney. 1988. Thermotropic phase behavior of model membranes composed of phosphatidylcholines containing cis-monounsaturated acyl chain homologues of oleic acid. Differential scanning calorimetric and 31P-NMR spectroscopic studies. Biochemistry. 27:880–887. [DOI] [PubMed] [Google Scholar]
- 34.Pabst, G., M. Rappolt, H. Amenitsch, and P. Laggner. 2000. Structural information from multilamellar liposomes at full hydration: full q-range fitting with high quality x-ray data. Phys. Rev. E Stat. Phys. Plasmas Fluids Relat. Interdiscip. Topics. 62:4000–4009. [DOI] [PubMed] [Google Scholar]
- 35.Pabst, G., R. Koschuch, B. Pozo-Navas, M. Rappolt, K. Lohner, and P. Laggner. 2003. Structural analysis of weakly ordered membrane stacks. J. Appl. Crystallogr. 36:1378–1388. [Google Scholar]
- 36.Pabst, G. 2006. Global properties of biomimetic membranes: perspectives on molecular features. Biophys. Rev. Lett. 1:57–84. [Google Scholar]
- 37.Pabst, G., J. Katsaras, V. A. Raghunathan, and M. Rappolt. 2003. Structure and interactions in the anomalous swelling regime of phospholipid bilayers. Langmuir. 19:1716–1722. [Google Scholar]
- 38.Nagle, J. F., and S. Tristram-Nagle. 2000. Structure of lipid bilayers. Biochim. Biophys. Acta. 1469:159–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun, W.-J., S. Tristram-Nagle, and J. F. Nagle. 1996. Structure of gel phase saturated lecithin bilayers; temperature and chain length dependence. Biophys. J. 71:885–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang, R. T., R. M. Suter, and J. F. Nagle. 1994. Theory of the structure factor of lipid bilayers. Phys. Rev. E. 50:5047–5060. [DOI] [PubMed] [Google Scholar]
- 41.Guinier, A. 1963. X-ray diffraction in crystals, imperfect crystals, and amorphous bodies. W. H. Freeman, San Francisco.
- 42.Lewis, R. N. A. H., R. N. McElhaney, F. Osterberg, and S. M. Gruner. 1994. Enigmatic thermotropic phase behavior of highly asymmetric mixed-chain phosphatidylcholines which form mixed-interdigitated gel phases. Biophys. J. 66:207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lewis, R. N. A. H., I. Winter, M. Kriechbaum, K. Lohner, and R. N. McElhaney. 2001. Studies of the structure and organization of cationic lipid bilayer membranes: calorimetric, spectroscopic and x-ray diffraction studies of linear saturated P-O-ethyl phosphatidylcholines. Biophys. J. 80:1329–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mason, J. T., R. E. Cunningham, and T. J. O'Leary. 1995. Lamellar phase polymorphism in interdigitated bilayer assemblies. Biochim. Biophys. Acta. 1236:67–72. [DOI] [PubMed] [Google Scholar]
- 45.Rappolt, M., P. Laggner, and G. Pabst. 2004. Structure and elasticity of phospholipid bilayers in the Lα phase: a comparison of phosphatidylcholine and phosphatidylethanolamine membranes. In Recent Research Developments in Biophysics, Vol. 3. S. G. Pandalai, editor. Transworld Research Network, Kerala.
- 46.Tardieu, A., V. Luzzatti, and F. C. Reman. 1973. Structure and polymorphism of the hydrocarbon chains of lipids: a study of lecithin-water phases. J. Mol. Biol. 75:711–733. [DOI] [PubMed] [Google Scholar]
- 47.Maulik, P. R., M. J. Ruocco, and G. G. Shipley. 1990. Hydrocarbon chain packing modes in lipids: effect of altered subcell dimensions and chain rotation. Chem. Phys. Lipids. 56:123–133. [DOI] [PubMed] [Google Scholar]
- 48.Needham, D., and E. Evans. 1988. Structure and mechanical properties of giant lipid (DMPC) vesicle bilayers from 20°C below to 10°C above the liquid crystal-crystalline phase transition at 24°C. Biochemistry. 27:8261–8269. [DOI] [PubMed] [Google Scholar]
- 49.Lewis, R. N. A. H., and R. N. McElhaney. 2002. Vibrational spectroscopy of lipids. In The Handbook of Vibrational Spectroscopy. J. M. Chalmers and P. R. Griffiths, editors. John Wiley & Sons, New York. 5:3447–3464.
- 50.Lewis, R. N. A. H., and R. N. McElhaney. 1996. FTIR spectroscopy in the study of hydrated lipids and lipid bilayer membranes. In Infrared Spectroscopy of Biomolecules. H. H. Mantsch and D. Chapman, editors. John Wiley & Sons, New York.
- 51.Blume, A., W. Hübner, and G. Messner. 1988. Fourier transform infrared spectroscopy of 13C=O labeled phospholipids. Hydrogen bonding to carbonyl groups. Biochemistry. 27:8239–8249. [DOI] [PubMed] [Google Scholar]
- 52.Lewis, R. N. A. H., R. N. McElhaney, W. Pohle, and H. H. Mantsch. 1994. The components of the carbonyl stretching band in the infrared spectra of hydrated 1,2-diacylglycerolipid bilayers: a reevaluation. Biophys. J. 67:2367–2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Campbell, R. F., F. E. Meirovitch, and J. H. Freed. 1979. Slow motional line NMR line shapes for very anisotropic rotational diffusion phosphorous-31 NMR of phospholipids. J. Phys. Chem. 83:525–533. [Google Scholar]
- 54.Seelig, J. 1978. 31P Nuclear magnetic resonance and the headgroup structure of phospholipid membranes. Biochim. Biophys. Acta. 515:105–140. [DOI] [PubMed] [Google Scholar]
- 55.Zhang, Y.-P., R. N. A. H. Lewis, and R. N. McElhaney. 1997. Calorimetric and spectroscopic studies of the thermotropic phase behavior of the n-saturated 1,2-diacyl-phosphatidylglycerols. Biophys. J. 72:779–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lewis, R. N. A. H., and R. N. McElhaney. 1993. Calorimetric and spectroscopic studies of the polymorphic phase behavior of a homologous series of n-saturated 1,2-diacyl phosphatidylethanolamines. Biophys. J. 64:1081–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cevc, G., A. Watts, and D. Marsh. 1981. Titration of the phase transition of phosphatidylserine bilayer membranes: effects of pH, surface electrostatics, ion binding and headgroup hydration. Biochemistry. 20:4955–4965. [DOI] [PubMed] [Google Scholar]
- 58.Cevc, G., and D. Marsh. 1987. Phospholipid Bilayers: Physical Principles and Models. Wiley, New York.
- 59.Cevc, G. 1990. How membrane chain-melting properties are regulated by the polar surface of lipid bilayers. Biochemistry. 26:6305–6310. [DOI] [PubMed] [Google Scholar]
- 60.Powell, G. L., and D. Marsh. 1985. Polymorphic phase behavior of cardiolipin derivatives studied by 31P NMR and x-ray diffraction. Biochemistry. 24:2902–2908. [DOI] [PubMed] [Google Scholar]
- 61.Zhang, Y. P., R. N. A. H. Lewis, and R. N. McElhaney. 1997. Calorimetric and spectroscopic studies of the thermotropic phase behavior of the n-saturated 1,2-diacylphosphatidylglycerols. Biophys. J. 72:779–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harlos, K., H. Eibl, I. Pascher, and S. Sundell. 1984. Conformation and packing properties of phosphatidic acid: the crystal structure of monosodium dimyristoylphosphatidate. Chem. Phys. Lipids. 34:115–126. [Google Scholar]
- 63.Pearson, R. H., and I. Pascher. 1979. The molecular structure of lecithin dehydrate. Nature. 281:499–501. [DOI] [PubMed] [Google Scholar]
- 64.Hitchcock, P. B., R. Mason, K. M. Thomas, and G. G. Shipley. 1974. Structural chemistry of 1,2 dilauroyl-dl-phosphatidylethanolamine: molecular conformation and intermolecular packing of phospholipids. Proc. Natl. Acad. Sci. USA. 71:3036–3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pascher, I., S. Sundell, K. Harlos, and H. Eibl. 1987. Conformation and packing of membrane phospholipids: the crystal structure of sodium dimyristoylphosphatidylglycerol. Biochim. Biophys. Acta. 896:77–88. [DOI] [PubMed] [Google Scholar]
- 66.Lewis, R. N. A. H., E. J. Prenner, L. H. Kondejewski, C. R. Flach, R. Mendelsohn, R. S. Hodges, and R. N. McElhaney. 1999. Fourier transform infrared spectroscopic studies of the interaction of the antimicrobial peptide Gramicidin S with lipid micelles and with lipid monolayer and bilayer membranes. Biochemistry. 38:15193–15203. [DOI] [PubMed] [Google Scholar]
- 67.Giffard, C. J., S. Ladha, A. R. Mackie, D. C. Clark, and D. Sanders. 1996. Interaction of nisin with planar lipid bilayers monitored by fluorescence recovery after photobleaching. J. Membr. Biol. 151:293–300. [DOI] [PubMed] [Google Scholar]
- 68.Demel, R. A., T. Peelen, R. J. Siezen, B. de Kruijff, and O. P. Kuipers. 1996. Nisin Z, mutant nisin Z and lacticin 481 interactions with anionic lipid correlate with antimicrobial activity. A monolayer study. Eur. J. Biochem. 235:267–274. [DOI] [PubMed] [Google Scholar]
- 69.Matsuzaki, K., K. Sugishita, N. Ishibe, M. Ueda, S. Nakata, K. Miyajima, and R. M. Epand. 1998. Relationship of membrane curvature to the formation of pores by magainin 2. Biochemistry. 37:11856–11863. [DOI] [PubMed] [Google Scholar]