

Abstract
Receptor-mediated changes in cAMP production play an essential role in sympathetic and parasympathetic regulation of the electrical, mechanical, and metabolic activity of cardiac myocytes. However, responses to receptor activation cannot be easily ascribed to a uniform increase or decrease in cAMP activity throughout the entire cell. In this study, we used a computational approach to test the hypothesis that in cardiac ventricular myocytes the effects of β1-adrenergic receptor (β1AR) and M2 muscarinic receptor (M2R) activation involve compartmentation of cAMP. A model consisting of two submembrane (caveolar and extracaveolar) microdomains and one bulk cytosolic domain was created using published information on the location of β1ARs and M2Rs, as well as the location of stimulatory (Gs) and inhibitory (Gi) G-proteins, adenylyl cyclase isoforms inhibited (AC5/6) and stimulated (AC4/7) by Gi, and multiple phosphodiesterase isoforms (PDE2, PDE3, and PDE4). Results obtained with the model indicate that: 1), bulk basal cAMP can be high (∼1 μM) and only modestly stimulated by β1AR activation (∼2 μM), but caveolar cAMP varies in a range more appropriate for regulation of protein kinase A (∼100 nM to ∼2 μM); 2), M2R activation strongly reduces the β1AR-induced increases in caveolar cAMP, with less effect on bulk cAMP; and 3), during weak β1AR stimulation, M2R activation not only reduces caveolar cAMP, but also produces a rebound increase in caveolar cAMP following termination of M2R activity. We conclude that compartmentation of cAMP can provide a quantitative explanation for several aspects of cardiac signaling.
INTRODUCTION
It is well accepted that receptor-mediated changes in cAMP production play an essential role in autonomic regulation of cardiac function. β1-adrenergic receptors (β1ARs) increase cAMP production through stimulatory G-protein (Gs)-dependent activation of adenylyl cyclase (AC), and M2 muscarinic receptors (M2Rs) antagonize β1AR responses through inhibitory G-protein (Gi)-dependent inhibition of AC activity. M2Rs also produce a delayed stimulatory response that involves an increase in cAMP production (1).
Despite the large number of studies demonstrating an essential role for cAMP in mediating autonomic responses in the heart, several observations remain incompletely understood. For example, it has been estimated that the cellular concentration of cAMP in cardiac preparations is ∼1 μM under unstimulated conditions (2,3). Furthermore, cAMP produces most of its acute functional responses through the activation of protein kinase A (PKA). However, the regulatory subunits of PKA bind cAMP with an affinity of 100–300 nM (4,5). This suggests that PKA is almost fully activated under resting conditions. If this is true, then how can βAR stimulation of cAMP production elicit responses that are known to involve activation of PKA? One possible explanation, which has been around for over 30 years, is that the changes in cAMP responsible for generating functional responses occur in a microdomain that is somehow isolated from the bulk of the cell (6). Support for this hypothesis comes from studies demonstrating that changes in the relative amount of cAMP and PKA activity in the membrane or particulate fraction of cardiac preparations correlates more closely with the regulation of functional responses than changes in cAMP activity observed in the soluble or cytosolic fractions (7–9).
There are also unanswered questions concerning the mechanisms by which changes in cAMP are involved in muscarinic responses. For instance, it has not always been possible to demonstrate that muscarinic antagonism of β-adrenergic responses corresponds with a decrease in the total cAMP content of cardiac preparations (10). Furthermore, M2R activation can produce cAMP-dependent stimulatory responses in addition to the inhibitory effects. What's more, the stimulatory effects exhibit much slower kinetics (11–13). So how can M2R activation produce both inhibition and stimulation of cAMP-dependent effects, and why are there differences in the kinetics of the two types of responses? At least part of the answer lies in the fact that cardiac myocytes express multiple isoforms of AC. These include AC5 and AC6 as well as AC4 and AC7 (14,15). Yet, not all AC isoforms respond to Gi activation in the same manner. The activated α-subunit of Gi (Giα) directly inhibits AC5/6, but not AC4/7 (16–18). In fact, AC4/7 is actually stimulated by free βγ-subunits released upon activation of Gi (19,20).
The opposing effects of Gi activation on different types of AC activity can explain the inhibitory and stimulatory effects that M2R activation has on cAMP-dependent responses in ventricular myocytes (11). However, it doesn't explain the disparity in the kinetics of the responses. Again, compartmentation of cAMP production may be involved. Evidence in support of this idea comes from studies demonstrating how different components of the signaling pathways are organized in the membrane. For example, AC5/6 is consistently associated with caveolar domains of the plasma membrane, whereas AC4/7 activity is associated with extracaveolar domains (21). This indicates that muscarinic inhibition and stimulation of cAMP occur in different subcellular locations. This has led us to hypothesize that compartmentation of cAMP can explain the complex temporal response produced by M2R activation.
In this study, we used a computational approach to determine whether or not compartmentation of cAMP may be important in answering the above questions. However, earlier models of cAMP signaling in the heart did not consider compartmentation or muscarinic modulation of β-adrenergic responses (22,23). Therefore, we developed a new theoretical framework that incorporates existing information on the subcellular location of the various elements involved in cAMP production and degradation as well as the kinetics of critical reactions in both β1AR and M2R signaling pathways. The resulting model was then used to predict the effects that receptor activation has on cAMP concentrations in caveolar, extracaveolar, and bulk cytoplasmic compartments. The results demonstrate that basal concentrations of cAMP in the caveolar compartment can be maintained at a level significantly lower than that of the total cell so that β1AR activation may then regulate the activity of PKA through changes in the concentration of cAMP in this microdomain. The results also demonstrate that even when there are only minimal changes in total cellular cAMP levels, M2R stimulation can significantly inhibit cAMP levels in the caveolar domain. Finally, the model demonstrates that the delayed stimulatory response to M2R activation is consistent with limited diffusion of cAMP from an extracaveolar domain to the caveolar domain, where PKA activity is regulated.
METHODS
Strategy for model design
Our goal was to determine whether a compartmental model can quantitatively explain the changes in cAMP in cardiac ventricular myocytes in response to activation of adrenergic and muscarinic receptors. We designed the model to be realistic, in the sense of being consistent with available information on the molecules involved. In particular, the model includes multiple isoforms of AC and phosphodiesterase (PDE), which have been proposed to be critical for regulation of cAMP levels. For these reasons, the model includes many molecular entities, each of which requires several parameters to define its functional behavior. Some of the parameters are strongly constrained by existing experimental data, but others are not (as discussed further below). We manually varied the parameters, within the range consistent with existing data, to best describe experimental results on cAMP levels in basal and stimulated conditions. From a mathematical perspective, the model is underdetermined, in that there are more available parameters than are likely to be strictly necessary to produce any single desired output. Consequentially, the parameter set presented here is highly unlikely to be unique. Because our goal was not to provide quantitative estimates for any particular model parameters, we have not performed a formal sensitivity analysis, but we note in the Discussion some of the values that strongly affected key features of the model output. In essence, we present this model as a “proof of concept”, to demonstrate that a molecularly realistic compartmental model can account for regulation of cAMP levels under a variety of experimental conditions. We expect that this model will also provide a basis for interpreting future studies on molecular perturbations of the system (e.g., inhibition of a particular PDE isoform).
Cell size and composition
A mathematical model of β1-adrenergic and M2 muscarinic receptor regulation of cAMP production in a cardiac ventricular myocyte was developed for this study. As in previous theoretical studies (24), the myocyte is assumed to be a cylinder with the following characteristics: length = 100 μm, radius = 10 μm, surface area = 6.9 × 104 μm2, and volume = 38 × 10−6 μl.
Existing kinetic data on the signaling pathways involved in regulating cAMP production and degradation were incorporated into a model consisting of three different compartments (Fig. 1). The first is the subsarcolemmal space reflecting the caveolar domains of the cell. The key signaling elements included in that compartment have been found in cholesterol-rich membrane fractions that are associated with caveolin 3, the muscle specific form of caveolin that is involved in creating signaling complexes necessary for producing functional responses (25–27). Major signaling components placed in the caveolar domain are β1ARs, M2Rs, Gs, Gi, AC5/6, as well as phosphodiesterase types 2 (PDE2), 3 (PDE3), and 4 (PDE4). The size of this compartment is ∼1% of the cytosolic volume, and it encompasses 10% of the plasma membrane surface area (28).
FIGURE 1.

Compartmentation of cAMP signaling pathways in a cardiac ventricular myocyte. β1-adrenergic receptor (β1); M2 muscarinic receptor (M2); stimulatory (Gs) and inhibitory G-proteins (Gi); adenylyl cyclase type 5 or 6 (AC5/6) and 4 or 7 (AC4/7); phosphodiesterase type 2 (PDE2), 3 (PDE3), and 4 (PDE4); cAMP flux between caveolar and extracaveolar (JCav/Ecav), extracaveolar and bulk cytosolic (JEcav/Cyt), and caveolar and bulk cytosolic (JCav/Cyt) compartments.
The second compartment reflects the subsarcolemmal space associated with cholesterol-rich lipid rafts that do not include caveolin. In the model, this extracaveolar domain contains β1ARs, M2Rs, Gi, Gs, AC4/7, as well as PDE2 and PDE4. The size of this compartment is ∼2% of the cytosolic volume, and it encompasses 20% of the plasma membrane. The final domain is the bulk cytoplasmic compartment, which makes up ∼50% of the total cell volume (28,29) and is associated with plasma membrane that contains many of the components described above, but to a lesser degree.
The signaling elements included in the model are listed in Table 1. They were placed in specific domains based on studies using biochemical methods to detect the presence of protein in different cell or membrane fractions. Due to limited quantitative information, elements that have been reported to exist in more than one domain were assumed to be distributed uniformly between relevant compartments. In some instances, functional studies were used as evidence that a particular signaling element is not uniformly distributed (see below).
TABLE 1.
Subcellular distribution of cAMP signaling elements
Most numerical values used for the parameters found in the model are either taken directly from a single experimental source or constrained by a range defined by multiple experimental sources (see Appendix I). However, no published quantitative data was available for the rate constants for Gi activation, the amount of each PDE isoform found in different subcellular compartments, or the cAMP flux rates.
To convert the amount of a specific protein described as a fraction of total membrane protein to the amount of that protein in a single ventricular myocyte, it is assumed that 1 mg of total membrane protein = 7.5 × 105 myocytes (30).
Computational components
Ligand/receptor/G-protein modules
The ternary complex model was used to describe the interaction between ligand, receptor, and G-protein (31). Briefly, the model characterizes the ability of the agonist to promote and stabilize the formation of a high affinity active ternary complex (LRG), starting from a low affinity binary ligand-receptor (LR) complex and G-protein (G). Due to the speed of these reactions (when compared to the other reactions present in the model), they are assumed to be at quasiequilibrium and are represented by algebraic equations. The same formalism was used to describe both β1-adrenergic receptor and M2 muscarinic activation in all relevant compartments.
- Module input:
- Ligand concentration (constant during a simulation).
- Free G-protein concentration (Gαβg = Gfree).
- Module output:
- Receptor bound G-protein concentration (RG + LRG).
Isoproterenol/β1 adrenergic receptor/Gs module
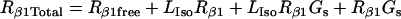 |
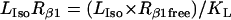 |
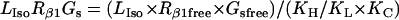 |
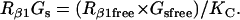 |
Acetylcholine/M2 muscarinic receptor/Gi module
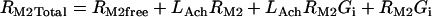 |
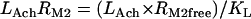 |
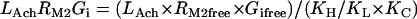 |
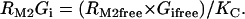 |
G-protein activation module
The ratio between the total number of Gi and Gs molecules in cardiac cells is ∼2:1 (30,32,33). Experimental evidence suggests that there is approximately equal distribution of Gs between caveolar and noncaveolar membrane domains, whereas Gi is almost entirely present in the caveolar domain (34). The activation kinetics of Gs were derived from published data (35) and modified to obtain subsecond kinetics for G-protein activation, in agreement with previous studies (36). Although the absolute rate constants for Gi activation have not been determined, the rate of Gi activation has been reported to be slower than that of Gs. Based on this information, it was assumed that the Gi activation rate is 50% of that determined for Gs.
- Module input:
- Receptor bound G-protein concentration (RG + LRG).
- Module output:
- G-protein subunits concentration (Gα-GTP, Gα-GDP, Gβγ).
- Free G-protein concentration (Gαβγ = Gfree).
The following formulations are applicable both to Gs and Gi and their corresponding receptors (β1AR and M2R).
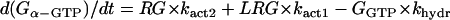 |
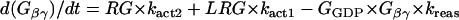 |
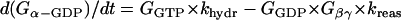 |
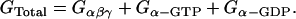 |
The output of the combined LRG and G-protein activation modules was validated by comparing the simulated concentration-response curve for isoproterenol stimulation of Gsα accumulation (not shown) with published experimental data (37).
Adenylyl cyclase modules
Adenylyl cyclase activities were divided into two functionally distinct categories: AC5/6 and AC4/7. Both categories are stimulated by Gs activation, but with very different affinities. The EC50 for Gsα stimulation of AC4/7 is ∼32 nM, whereas for AC5/6 it is ∼200 nM (38,39). This significantly affects the sensitivity of each AC category to β1AR stimulation. Each category of AC activity is also affected differently by Gi activation. AC5/6 is inhibited by Giα, whereas AC4/7 is not. Furthermore, AC4/7 is stimulated synergistically by Giβγ in the presence of Gsα activation (39). As described below, parameter values were obtained by using a Hill equation to fit the experimentally determined relationship between G-protein subunits and AC activity.
Adenylyl cyclase 5/6 module
The activity of AC5/6 and its regulation by Gs and Gi were simulated using a formulation to describe data obtained from isolated membrane preparations (37,40). This activity was then scaled up using an amplification factor (AF5/6) that was defined by the ratio of the activity produced by equal amounts (1 mg) of purified and membrane protein in the presence of a maximally stimulating concentration of Gsα.
- Module input:
- Gsα and Giα concentration (Gsα-GTP, Giα-GTP).
- Module output:
- cAMP produced by AC5/6.
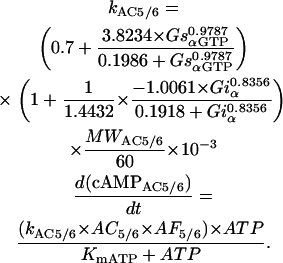 |
Adenylyl cyclase 4/7 module
AC4/7 activity and its regulation by Gs and Gi were simulated using a formulation based on published experimental data (41,39,42). Because of their structural and functional similarities (43), the kinetic properties of AC2 were used to describe the behavior of AC4/7, where necessary. The total amount of AC4/7 present in the cell was assumed to be ∼10% of AC5/6 (44,45). An amplification factor (AF4/7) was calculated as it was for AC5/6.
- Module input:
- Gsα and Giα concentration (Gsα-GTP, Giβγ).
- Module output:
- cAMP produced by AC4/7.
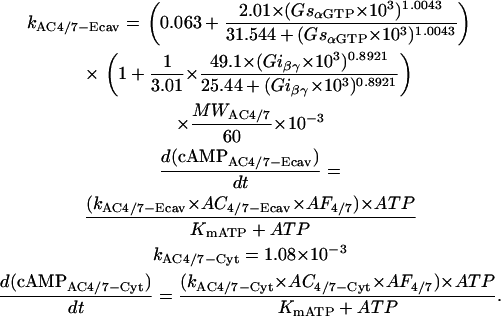 |
Phosphodiesterase modules
The total cellular protein content of the various PDE isoforms found in cardiac myocytes has been reported. However, the only information available on the subcellular location of the different PDE isoforms describes their presence in membrane (particulate) and/or cytosolic (soluble) fractions. How the membrane associated PDE activity is distributed between caveolar and extracaveolar domains is not known. The available descriptive information, along with functional studies, was used as a guide in determining the relative ratio of each PDE isoform to be included in the various compartments.
- Module input:
- cAMP concentration for the specific compartment.
- Module output:
- cAMP degraded by PDEs.
The general formulation used for each PDE isoform (PDEx) is:
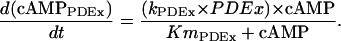 |
cAMP flux modules
Initial rates for flux of cAMP between compartments were based on previously published estimates (46).
- Module input:
- cAMP concentration.
- Module output:
- cAMP concentration.
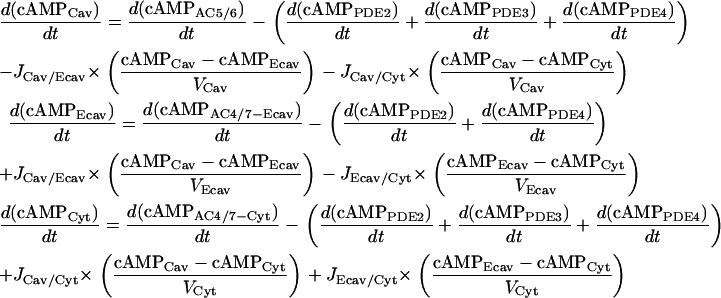 |
Implementation and validation
The model was implemented in Java (J2SE). Differential equations were solved using an iterative Euler approach. Predictions made by the model are compared to two types of previously published data. The first are cAMP measurements obtained using biochemical methods to determine the cAMP content of homogenized tissue or whole cell lysates. These data are assumed to reflect what is happening to cAMP at the level of the whole cell. The second are measurements of changes in cAMP activity in intact ventricular myocytes using a FRET-based biosensor (47). This probe consists of two separate proteins: the catalytic subunit of protein kinase A (PKA) labeled with yellow fluorescent protein (Cat-YFP) and the type II regulatory subunit labeled with cyan fluorescent protein (RII-CFP). FRET occurs when PKA is in the inactive state, and Cat-YFP and RII-CFP are bound to one another. An increase in cAMP causes molecular rearrangement of the subunits, which results in a decrease in FRET. By convention the FRET response is defined as the change in CFP/YFP fluorescence ratio relative to the baseline ratio (ΔR/Ro), so that an increase in cAMP results in an increasing “FRET response”, even though there is actually a decrease in FRET. These data are assumed to reflect what is happening to cAMP activity in the caveolar domain, where cAMP levels are expected to correlate most directly with functional responses regulated by type II PKA.
RESULTS
Basal cAMP levels
We first validated the model's prediction of cAMP levels under basal conditions. In the absence of any agonist, previous studies have suggested that the basal concentration of cAMP in cardiac myocytes is ∼1 μM (2,3). However, this is well above the cAMP affinity of PKA (≤300 nM), which is the primary effector for this cyclic nucleotide (4,5). This apparent discrepancy may be due to cAMP measurements having been made using traditional biochemical methods involving homogenized tissue or whole cell lysates, which represent what is happening on average throughout the whole cell, but may not accurately reflect what is happening in discrete microdomains where PKA-mediated functional responses are regulated. Consistent with this idea, the model demonstrates that under basal conditions, it is possible to maintain the average concentration of cAMP across all compartments (total cAMP) at 1 μM, while in the caveolar domain, the basal concentration is ∼100 nM (see Fig. 2). Although this level of cAMP is high enough to partially activate PKA, it is still low enough to leave a concentration range over which cAMP can significantly modulate kinase activity.
FIGURE 2.

Effect of β1-adrenergic receptor activation on cAMP kinetics. Effect of submaximally (3 nM) and maximally (100 nM) stimulating concentrations of isoproterenol on the time course of changes in cAMP concentration. (A) Average concentration of cAMP in all compartments (total cAMP, simulation). (B) Concentration of cAMP in the caveolar compartment (caveolar cAMP, simulation). (C) Dependence of total and caveolar cAMP (simulations) on concentration of isoproterenol used to stimulate β1-adrenergic receptors.
Effects of β-adrenergic receptor stimulation
We next evaluated the effect that β1AR stimulation has on cAMP levels. As expected, the model predicts that β1AR stimulation causes a concentration-dependent increase in cAMP in all compartments of the cell (Fig. 2). Exposure to the β1AR agonist isoproterenol (Iso) increases total cAMP from basal (1 μM) to a maximal level ∼2.1 μM with an EC50 of ∼7 nM. This is consistent with the 50–250% increase over baseline, and EC50 of 10–80 nM observed experimentally, when using traditional biochemical methods to measure total cAMP in homogenized preparations (7,9,48–51). The model also predicts that caveolar cAMP reaches a similar concentration following maximal β1AR stimulation, but because of the lower baseline (100 nM), the overall change represents a >2000% increase.
Recently, we have used a fluorescence energy transfer (FRET)-based biosensor to measure changes in cAMP activity in intact cardiac myocytes (47). This biosensor consists of fluorescently labeled type II PKA, and in adult ventricular myocytes it is expressed in a striated pattern, suggesting that it is associated with T tubules, just like endogenous type II PKA (52,53). Caveolin-3, a marker for caveolae, is expressed in a similar striated pattern, in addition to being found in the peripheral sarcolemma (54). Biochemical studies have also demonstrated that type II PKA is highly enriched in caveolar membrane fractions (34). Therefore, it is assumed that this sensor responds to changes in cAMP activity occurring in a caveolar compartment. Consistent with this idea, we have previously demonstrated that this sensor detects β1AR stimulated changes in cAMP activity with a sensitivity that correlates directly with the β1AR sensitivity of L-type Ca2+ channels (47), which are located in caveolae associated with T tubules (55). The properties of this probe have also been well characterized in vitro (4). The EC50 for cAMP activation is 300 nM, with a Hill coefficient of 1.4. Furthermore, activation by maximally effective concentrations of cAMP produces a FRET response of ∼15% (see Fig. 3 B). However, when expressed in ventricular myocytes, this probe exhibits a smaller dynamic FRET response (6–12%) following maximal agonist stimulation (47,4). This can be explained if basal levels of cAMP are sufficient to have partially activated PKA, even before exposure to agonist. Consistent with this idea, PKA-dependent responses can be elicited from ventricular preparations by inhibiting basal phosphatase activity (56).
FIGURE 3.

Simulation of β1-adrenergic responses. (A) In vivo activation of type II protein kinase A (PKA) by submaximally (0.3 nM) and maximally (1 μM) stimulating concentrations of isoproterenol (experimental). Increase in PKA activity measured as change in FRET response (ΔR/R0) of PKA-based biosensor expressed in an adult ventricular myocyte. Data from Warrier et al. (47). (B) cAMP sensitivity of PKA-based biosensor in vitro. Data from Mongillo et al. (4). (C) Predicted response of PKA-based biosensor in a ventricular myocyte exposed to submaximally and maximally stimulating concentrations of isoproterenol (simulation). (D) Predicted isoproterenol sensitivity of PKA-based biosensor in vivo (simulation).
If the in vitro properties of the probe described above hold true in vivo, and if we assume that the average maximal FRET response that can be elicited in adult ventricular myocytes is 9%, this then indicates that ∼25% of the PKA-based probe must be in an active state under basal conditions. This corresponds to a basal level of cAMP that is close to that predicted by the model to exist in the caveolar domain. Using the relationship between Iso concentration and cAMP levels in the caveolar compartment (Fig. 2 C), we then defined the expected relationship between Iso concentration and PKA activation. According to this calculation, Iso increases PKA activity from a basal level of 25% to maximal with an EC50 of 1.3 nM (Fig. 3 D). The sensitivity of this response is in good agreement with the EC50 of 0.5 nM for Iso activation of the PKA-based probe in intact ventricular myocytes (47). With the relationship in Fig. 3 B, we can also use the model to predict the FRET response of the PKA-based probe (Fig. 3 C) and compare this to experimentally determined PKA-FRET responses (Fig. 3 A). The model's predictions are in good agreement with the experimental results. The one possible exception is the difference in the time course with which responses turn on and off. Although there may be more than one explanation, it is most likely due to the slow exchange of solutions used to add or remove drugs during the imaging experiments (47). Consistent with this conclusion, the time course of the responses predicted by the model are more in line with the time course of cAMP-dependent ion channel responses observed when rapid solution changes are possible (13).
Effects of M2R activation in the presence of maximal β-adrenergic stimulation
M2R activation is able to antagonize responses to agonists that stimulate cAMP production. Although it is now generally accepted that such inhibitory effects are due to a decrease in cAMP production, this has not always been the case (1). Many early studies measuring cAMP levels in homogenized tissue or whole cell lysates found that muscarinic inhibitory responses do not correlate with changes in cAMP. An explanation offered for this apparent paradox has been that muscarinic inhibition of cAMP activity is localized to specific subcellular compartments (10). Consistent with this idea, the model predicts that M2R stimulation has only modest effects on total cAMP levels (Fig. 4 C). However, M2R activation significantly reduces cAMP in the caveolar compartment (Fig. 4 D). Furthermore, ACh inhibition of the Iso response is reflected in the predicted FRET response (Fig. 4 B), which correlates well with the PKA FRET response observed experimentally (Fig. 4 A). Another prediction of the model is that following the initial inhibitory effect produced by ACh, there is a gradual decrease in inhibition (“escape”), as observed experimentally (13). In the model, this is due to M2Rs stimulating cAMP production in the extracaveolar compartment, which then spills over into the caveolar compartment.
FIGURE 4.

Simulation of muscarinic inhibition of β1-adrenergic response. Effect of acetylcholine ((ACh) 10 μM) on the response to a maximally stimulating concentration (200 nM) of isoproterenol (Iso). (A) In vivo PKA activity measured as change in FRET response (ΔR/R0) of PKA-based biosensor expressed in an adult ventricular myocyte (experimental). Data from Warrier et al. (47). (B) Predicted response of PKA-based biosensor in a ventricular myocyte (simulation). (C) Average concentration of cAMP in all compartments (total cAMP, simulation). (D) Concentration of cAMP in the caveolar compartment (caveolar cAMP, simulation).
Effects of M2R activation in the presence of submaximal β-adrenergic stimulation
Having demonstrated that the model can reproduce muscarinic inhibition of β1AR-mediated cAMP production, we then evaluated its ability to explain muscarinic stimulatory responses. Previous studies in ventricular myocytes have demonstrated that muscarinic stimulatory effects are only observed in the presence of submaximal concentrations of agonists such as Iso (11–13). Consistent with this, the model predicts that M2R activation would not produce a detectible rebound stimulatory response in the presence of a maximally stimulating concentration of Iso (Fig. 4 B). However, in the presence of a concentration of Iso that is near the threshold for stimulating PKA activity above basal levels, the model predicts that upon termination of M2R activation, there should be a significant rebound stimulatory increase in cAMP concentration in the caveolar compartment, which produces a rebound increase in the predicted PKA FRET response (Fig. 5 B). This type of response correlates with the PKA-FRET response observed experimentally (Fig. 5 A). Again, the slower time course of the FRET response measured experimentally may be explained by the time required to change solutions. The model also predicts an inhibitory effect that goes below baseline during exposure to ACh, but the PKA-based probe may not be able to detect changes in cAMP below basal levels (see Appendix II).
FIGURE 5.

Simulation of muscarinic stimulatory response. Rebound stimulatory effect observed following transient exposure to acetylcholine ((ACh) 10 μM) in the presence of a submaximally stimulating concentration (0.1 nM) of isoproterenol (Iso). (A) In vivo PKA activity measured as change in FRET response (ΔR/R0) of PKA-based biosensor expressed in an adult ventricular myocyte (experimental). Data from Warrier et al. (47). (B) Predicted response of PKA-based biosensor in a ventricular myocyte (simulation). (Dashed line) Predicted response if biosensor were able to detect decreases in cAMP activity below basal levels (see Appendix II).
A more detailed examination of the source of cAMP responsible for the rebound stimulatory response helps explain this complex behavior. This requires an appreciation of what is happening not only in the caveolar compartment, but also in adjacent compartments, especially the extracaveolar compartment. In the model, M2Rs are located in both the caveolar and extracaveolar compartments. However, the caveolar compartment contains only AC5/6 activity, while the extracaveolar compartment contains only AC4/7 activity. In the presence of ACh, Giα inhibits AC5/6, rapidly reducing cAMP in the caveolar compartment (Fig. 6, A and B). However, ACh actually increases cAMP in the extracaveolar compartment, where Giβγ stimulates AC4/7 (Fig. 6 D). The resulting concentration gradient produces a slow flux of cAMP between compartments (Fig. 6 C), but the inhibitory effect of ACh on cAMP production within the caveolar compartment dominates. Upon washout of ACh, AC5/6 inhibition quickly reverses, resulting in a rapid return of cAMP production within the caveolar compartment. This cAMP, together with flux of cAMP from the extracaveolar compartment, results in a caveolar concentration exceeding that observed in the presence of Iso alone (before application of ACh). As the concentration of cAMP in the extracaveolar compartment declines, so does the flux of cAMP into the caveolar compartment. The result is that caveolar cAMP declines to its steady-state value. Based on these results, we conclude that the diffusion of cAMP from the extracaveolar compartment into the caveolar compartment is a viable explanation for the rebound stimulatory responses observed upon washout of ACh.
FIGURE 6.

Kinetics of changes in cAMP activity associated with muscarinic stimulatory response. Changes in caveolar and extracaveolar cAMP production and concentration caused by transient exposure to acetylcholine ((ACh) 10 μM) in the presence of a submaximally stimulating concentration (0.1 nM) of isoproterenol (Iso) (simulations). (A) Concentration of cAMP in the caveolar compartment. (B) Rate of cAMP concentration change in the caveolar compartment due to the activity of AC5/6. (C) Rate of cAMP concentration change in the caveolar compartment (Cav) due to the flux from the extracaveolar compartment (Ecav). (D) Concentration of cAMP in the extracaveolar compartment. (E) Rate of cAMP concentration change in the extracaveolar compartment due to the activity of AC4/7.
DISCUSSION
Computational models of cAMP signaling have recently been used to investigate cardiac myocyte responses to β1AR stimulation (23,22). This study expands this approach by incorporating the kinetics of processes involved in β1-adrenergic as well as M2 muscarinic regulation of cAMP production and integrating this with information on the subcellular distribution of the various components of these signaling pathways. The model was tested by comparing cAMP responses predicted to occur in a caveolar compartment to previously published measurements of changes in cAMP activity detected by a FRET-based biosensor targeted to the corresponding subcellular location of intact cardiac ventricular myocytes (47). It was also tested by comparing total cAMP responses (using the weighted average of the cAMP concentrations in all three compartments) to previously published measurements of changes in cAMP obtained using traditional biochemical methods in ventricular tissue homogenates and/or whole cell lysates. This new theoretical framework can explain both simple and complex behaviors of cardiac ventricular myocytes evoked by both β1-adrenergic and M2 muscarinic signaling pathways.
The model provides a simple explanation for the observation that the total cellular cAMP level is high enough that one would expect near maximal activation of PKA, even under basal conditions. Measurements obtained using homogenized cells or tissue reflect cAMP levels averaged across the whole cell, not levels in microdomains that make up only a small fraction of the total cell volume. Although compartmentation of cAMP signaling is an obvious potential explanation, that hypothesis is difficult to evaluate experimentally. The model predicts that bulk cytoplasmic cAMP is comparable to previous estimates for the whole cell, but cAMP in the caveolar compartment operates in a range appropriate to modulate PKA activity. This obviates the need to assume that the affinity of PKA for cAMP is significantly lower in vivo than what has been measured in vitro (22,57).
The model also can explain why muscarinic inhibition of β-adrenergic functional responses has not always been found to correlate directly with changes in cAMP levels (10). Because the discrepancies have been reported in studies where cAMP levels were measured using whole cell or tissue preparations, it has been proposed that significant concentration changes actually were occurring in microdomains where PKA-dependent responses are regulated (10). In support of this idea, the model predicts that M2R activation causes only modest changes in cAMP concentration in the bulk cytoplasmic compartment (see Fig. 4 C), whereas there are significant changes in cAMP levels in the caveolar compartment (see Fig. 4 D). Furthermore, the predicted effect of M2R activation on PKA responses in the caveolar compartment (Fig. 4 B) correlates well with experimental results demonstrating that, in the presence of β1AR stimulation, M2R activation reduces cAMP activity detected by the PKA-based cAMP biosensor (Fig. 4 A).
We then used the model to determine whether compartmentation of cAMP can explain M2R-mediated delayed stimulatory responses. In ventricular myocytes, this behavior is due to Gi βγ subunits stimulating AC4/7 activity (11). However, this occurs at the same time that Giα is inhibiting AC5/6. So why does the stimulatory effect produce a rebound response upon termination of M2R activation? If AC4/7 and AC5/6 are found in different plasma membrane domains (21), we hypothesized that a time-dependent flux of cAMP from an extracaveolar compartment to a caveolar compartment could be responsible. The model demonstrates that this is a feasible explanation. It is interesting to note that the muscarinic stimulatory mechanism only affects functional responses (e.g., ion channels) in the presence of submaximally stimulating concentrations of Iso. The model actually predicts that the muscarinic stimulatory response affects cAMP levels even in the presence of maximally stimulating concentrations of Iso, but because this rebound response is above the level that maximally activates PKA, it is not expected to produce a detectable functional response (see Fig. 4).
Limitations and predictions
Although descriptive information dealing with compartmentation of cAMP signaling in cardiac myocytes is growing rapidly, the availability of quantitative data is limited. Where information does exist, it is not uncommon for there to be some variability in estimates between different studies. Because of this, there may be more than one set of parameters that produce the same behaviors we have described. In this respect, this version of the model should be viewed as just one potential configuration, albeit one that exhibits properties consistent with a number of experimental observations. Although a quantitative parameter analysis was beyond the scope of this study, we did conduct a more qualitative type of analysis of the importance of certain parameter in producing specific responses.
Except where noted, the equations used in the model's different modules were taken from previously published studies. This approach constrained many parameter values to those that resulted in module output consistent with experimental data. Less quantitative information was available for assigning values to other parameters. This is particularly true for the distribution of some signaling elements among subcellular compartments and the flux rates for cAMP between compartments. These were the parameters that turned out to have the greatest influence on the behaviors that were the focus of this study.
Experimental data also do not provide a consistent view on the location of β1ARs. Some studies have suggested that the bulk of the β1AR population exists in an extracaveolar domain (34), whereas others have concluded that the same receptor is found predominantly in a caveolar domain (58). The present version of the model assumes that β1ARs are found in both of these compartments. The validity of this assumption might be questioned, but it was necessary in order for β1AR stimulation to activate both AC5/6 and AC4/7. Stimulation of AC5/6 was necessary to produce an increase in cAMP that could then be inhibited by M2R activation. Stimulation of AC4/7 was necessary to see ACh-induced rebound stimulation of cAMP production. Because these different AC isoforms are found in different membrane domains (21,27), the simplest way to model these behaviors was to include β1ARs in those domains as well. This illustrates how the model can be used to evaluate the functional significance of biochemical data describing the location of specific elements or even differentiating between possibilities. Using this approach, our results provide support for the idea that β1ARs actually exist in caveolar as well as extracaveolar domains.
One aspect of the model that is absolutely critical for producing the ACh-induced rebound stimulatory response is the placement of AC5/6 inside and AC4/7 outside of the caveolar compartment. Although this behavior is not lost if the distribution of AC isoforms is less strict, segregation of the majority of AC activity is important. This conclusion is consistent with biochemical studies that have looked at the distribution of AC activity in different membrane fractions (27,21).
It was not necessary to include more than one type of PDE activity to obtain the basic properties of this model. What was important was the ratio of PDE to AC activity in each compartment. Furthermore, it was critical to have much higher total PDE activity in the caveolar compartment than either of the other two compartments. This was necessary to maintain a low concentration of cAMP in the caveolar domain under basal conditions, while still allowing cAMP levels in the other compartments (and thus total cAMP) to remain high. Similarly, the transient rebound response observed following washout of ACh can be reproduced by including a single type of PDE activity in the extracaveolar domain that is much lower than that in the caveolar compartment. So one might question why we included three different types of PDE activity in the model. The primary reason is that there have been numerous studies demonstrating the functional importance of PDE2, PDE3, and PDE4 activity in regulating cAMP-dependent responses in adult cardiac myocytes (59,60), and we wanted the model to reflect what is known about PDE activity in these cells. Unfortunately, there is little information available on how the different PDE isoforms are distributed. Biochemical studies have found evidence for all three isoforms in both soluble and particulate fractions of cell homogenates, and while the PDE activity in particulate fractions may be membrane associated, there is little or no information on how much of that might be caveolar and/or extracaveolar. Fortunately, the behaviors described by this model did not depend critically on exactly which PDE isoforms were present in each compartment. Another reason to include multiple PDE isoforms in the model is that each can be regulated in unique ways that may be necessary to explain other types of responses. For example, PDE4 can be activated by PKA (61). Previous work has suggested that this is involved in a feedback mechanism that can modulate the magnitude of cAMP responses detected by exogenously expressed cyclic nucleotide gated (CNG) ion channels (62). However, the subcellular location where this positive feedback activation of PDE4 may be occurring is not clear. Although it is obvious that CNG channels detect responses near the plasma membrane, they are expressed primarily in noncaveolar lipid rafts (63), and how this may relate to compartments included in this model is not known.
The parameters about which we have the least information relate to the flux of cAMP within a cell. Movement of cAMP between compartments is often thought of as being limited by functional barriers associated with PDE activity (7). In this model, the role of PDE activity in regulating the flux between compartments is actually through its contribution to the concentration of cAMP in each compartment, and therefore the concentration gradient driving cAMP movement between compartments. Flux of cAMP between compartments is also a function of physical factors affecting diffusion of cAMP down its concentration gradient. If it is assumed that there are no physical barriers and cAMP can move between compartments at rates that approximate free diffusion, all of the behaviors attributed to compartmentation by this model are lost. This includes the gradient of cAMP between the caveolar and extracaveolar domain under basal conditions, as well as the ACh-induced rebound stimulatory response. The complex structure of cardiac ventricular myocytes has been shown to restrict access to submembrane regions and is likely to be an important factor influencing cAMP diffusion (64), but the actual nature of the barriers between compartments proposed in the model is not known. The flux rates predicted by this model indicate that cAMP movement is far slower than free diffusion. Validation of these values awaits development of an approach to experimentally determine cAMP flux rates between specific microdomains in intact myocytes. It is noteworthy that Rich et al. (65) also found evidence for extremely slow exchange of cAMP among compartments in HEK 293 cells, indicating that limited diffusion between microdomains is not unique to cardiac myocytes. More recently, Saucerman et al. (66) have demonstrated that limited diffusion of cAMP also occurs at the macroscopic level in neonatal ventricular myocytes.
Future directions
This version of the model only contains β1ARs, whereas most cardiac preparations express both β1 and β2ARs. However, β1ARs make up ∼80% of the total βAR population, and in normal hearts β2ARs do not contribute significantly to the functional responses of ventricular myocytes to nonselective agonists such as Iso. Furthermore, the model's predictions were validated by comparison to responses obtained from guinea pig ventricular myocytes, which unlike most cardiac preparations, do not express functional β2ARs (67). In cardiac preparations where they do exist, β2ARs may play a more important role in regulating cardiac function under conditions such as heart failure. Interestingly, selective activation of β2ARs produces compartmentalized cAMP-dependent responses. Future incorporation of β2ARs into the model may provide a useful means of evaluating the potential mechanism(s) responsible for that form of compartmentation. Another key future direction for the model is inclusion of signaling mechanisms downstream of cAMP.
CONCLUSIONS
This work supports the idea that compartmentation of cAMP signaling plays a critical role in shaping cAMP-dependent effects under basal conditions, as well as in response to β1AR and M2R activation. This includes both the inhibitory and stimulatory responses associated with M2R activation.
Acknowledgments
This work was supported by grants from the National Institutes of Health Heart, Lung and Blood Institute, and the American Heart Association. R.V.I. was the recipient of a predoctoral fellowship from the Ohio Valley Affiliate of the American Heart Association.
APPENDIX I
All parameters used in the model and their respective references are listed in Tables 2–8.
TABLE 2.
Isoproterenol/β1 adrenergic receptor/Gs module
| Parameter | Value | Units | Description | Reference |
|---|---|---|---|---|
| Rβ1TotalCav | 0.633 | μM | Concentration of β1R in Cav compartment | (30) |
| Rβ1TotalEcav | 1.267 | μM | Concentration of β1R in Ecav compartment | (30) |
| KH | 0.035 | μM | High affinity binding constant (between ligand and receptor) | (78,79) |
| KL | 0.386 | μM | Low affinity binding constant (between ligand and receptor) | (79,78) |
| KC | 8.809 | μM | Affinity binding constant (between free receptor and G-protein) | (80,22) |
TABLE 3.
Acetylcholine/M2 muscarinic receptor/Gi module
| Parameter | Value | Units | Description | Reference |
|---|---|---|---|---|
| RM2TotalCav | 0.506 | μM | Concentration of M2R in Cav compartment | (81) |
| RM2TotalEcav | 0.506 | μM | Concentration of M2R in Ecav compartment | (81) |
| KH | 0.16 | μM | High affinity binding constant (between ligand and receptor) | (82) |
| KL | 11 | μM | Low affinity binding constant (between ligand and receptor) | (82) |
| KC | 30 | μM | Affinity binding constant (between free receptor and G-protein) | (80,22) |
TABLE 4.
G-protein activation module
| Parameter | Value | Units | Description | Reference |
|---|---|---|---|---|
| GsTotalCav | 10 | μM | Concentration of Gs-protein in Cav compartment | (30,34) |
| GsTotalEcav | 10 | μM | Concentration of Gs-protein in Ecav compartment | (83,34) |
| GiTotalCav | 20 | μM | Concentration of Gi-protein in Cav compartment | (33,32,34) |
| GiTotalEcav | 1 | μM | Concentration of Gi-protein in Ecav compartment | (34,33,32) |
| kact1Gi | 0.05 | s−1 | Activation rate constant for LRGi complexes | (35,22) |
| kact2Gi | 2.5 | s−1 | Activation rate constant for RGi complexes | (35,22) |
| kact1Gs | 0.1 | s−1 | Activation rate constant for LRGs complexes | (35,22) |
| kact2Gs | 5 | s−1 | Activation rate constant for RGs complexes | (35,22) |
| khydrGi | 0.8 | s−1 | Hydrolization rate constant of Giα-GTP | (35,22) |
| kreasGi | 1.21 × 103 | s−1μM−1 | kreasGi, reassociation rate constant of Giα-GDP and Gβγ | (35,22) |
| khydrGs | 0.8 | s−1 | Hydrolization rate constant of Gsα-GTP | (35,22) |
| kreasGs | 1.21 × 103 | s−1μM−1 | kreasGi, reassociation rate constant of Gsα-GDP and Gβγ | (35,22) |
TABLE 5.
Adenylyl cyclase 5/6 module
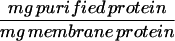
TABLE 6.
Adenylyl cyclase 4/7
| Parameter | Value | Units | Description | Reference |
|---|---|---|---|---|
| AC4/7-Ecav | 0.200 | μM | Concentration of Ecav AC4/7 | (30,45,44) |
| AC4/7-Cyt | 0.379 × 10−3 | μM | Concentration of Cyt AC4/7 | (30,45,44) |
| ATP | 5 × 103 | μM | Concentration of ATP (constant) | (28) |
| KmATP | 315 | μM | AC4/7 Km for ATP | (84) |
| AF4/7 | 130 | 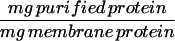 |
Amplification factor for AC4/7 | – |
| MWAC4/7 | 130 | KDa | Molecular weight of AC4/7 | – |
TABLE 7.
Phosphodiesterase modules
| Parameter | Value | Units | Description | Reference |
|---|---|---|---|---|
| KmPDE2 | 50 | μM | PDE2 Km for cAMP | (72, 85) |
| KmPDE3 | 0.08 | μM | PDE3 Km for cAMP | (76) |
| KmPDE4 | 2.2 | μM | PDE4 Km for cAMP | (86) |
| kPDE2 | 20 | s−1 | Rate constant for PDE2 | (72) |
| kPDE3 | 1.25 | s−1 | Rate constant for PDE3 | (72) |
| kPDE4 | 2.5 | s−1 | Rate constant for PDE4 | (72) |
| PDE2Cav | 4.5 | μM | PDE2 concentration in Cav compartment | (87,85) |
| PDE2Ecav | 0.02 | μM | PDE2 concentration in Ecav compartment | (87,85) |
| PDE2Cyt | 5 × 10−3 | μM | PDE2 concentration in Cyt compartment | (85) |
| PDE3Cav | 5.6 | μM | PDE3 concentration in Cav compartment | (76,23,85) |
| PDE3Cyt | 7.5 × 10−3 | μM | PDE3 concentration in Cyt compartment | (85,76,23) |
| PDE4Cav | 2.0 | μM | PDE4 concentration in Cav compartment | (85,88,23) |
| PDE4Ecav | 0.16 | μM | PDE4 concentration in Ecav compartment | (85,88,23) |
| PDE4Cyt | 5 × 10−3 | μM | PDE4 concentration in Cyt compartment | (85,88,23) |
TABLE 8.
cAMP flux modules
APPENDIX II: EFFECTS OF M2R ACTIVATION IN THE ABSENCE OF β-ADRENERGIC STIMULATION
In ventricular myocytes, M2R activation alone produces no functional response as a consequence of changes in cAMP activity (1). This does not necessarily mean that exposure to a muscarinic agonist such as acetylcholine (ACh) does not affect cAMP concentrations in caveolar domains. If basal levels of cAMP are sufficient to at least partially activate PKA, the absence of its influence on functional responses can be explained by basal phosphatase activity (56). Consistent with this idea, the model actually predicts that exposure to ACh alone does cause a decrease in caveolar cAMP levels (Fig. 7 D), as well as a decrease in the predicted FRET-response (Fig. 7 B). However, ACh has no effect on the FRET-response of the PKA-based biosensor in intact cardiac ventricular myocytes (Fig. 7 A). This apparent discrepancy is most likely due to the inability of the sensor to detect decreases in basal cAMP activity.
FIGURE 7.

Simulation of M2 muscarinic responses in the absence of β1-adrenergic stimulation. (A) Effect of a maximally stimulating concentration of acetylcholine on the activity of the PKA-based biosensor expressed in an adult ventricular myocyte (experimental). Data from Warrier et al. (47). (B) Predicted response of PKA-based biosensor in a ventricular myocyte exposed to a maximally stimulating concentration of acetylcholine (simulation). (Dashed line) Predicted response if biosensor were able to detect decreases in cAMP activity below basal levels. (C) Change in total cAMP concentration in response to a maximally stimulating concentration of acetylcholine (simulation). (D) Change in caveolar cAMP concentration in response to a maximally stimulating concentration of acetylcholine (simulation).
In cells expressing the PKA-based biosensor, acute exposure to an agonist that stimulates cAMP production causes a decrease in FRET (increase in the FRET response) that is readily reversible (Figs. 3–5). This can be explained by the fact that cAMP binding to RII-CFP does not necessarily cause complete dissociation of Cat-YFP in the absence of substrate (47). Therefore, these labeled subunits can readily reassociate when cAMP levels decrease, resulting in an increase in FRET (decrease in FRET response). However, under basal conditions, it is more likely that the regulatory and catalytic subunits of activated PKA do completely dissociate. In this situation, there is a greater chance that fluorescently labeled subunits reassociate with unlabeled endogenous counterparts, in which case there will be no change in FRET, or the FRET response. Therefore, the absence of a decrease in the FRET response upon exposure to ACh under basal conditions does not necessarily mean that cAMP levels have not decreased.
The model also predicts that exposure to high concentrations of ACh actually causes a slight increase in total cAMP (Fig. 7 C). Although this type of response might seem counterintuitive, it is consistent with what has been observed experimentally when measuring cAMP responses with biochemical methods in ventricular tissue homogenates (68). The model can explain such effects as being due to the ability of M2R activation to stimulate cAMP production in the extracaveolar compartment.
References
- 1.Harvey, R. D., and A. E. Belevych. 2003. Muscarinic regulation of cardiac ion channels. Br. J. Pharmacol. 139:1074–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kameyama, M., F. Hofmann, and W. Trautwein. 1985. On the mechanism of β-adrenergic regulation of the Ca channel in the guinea-pig heart. Pflugers Arch. 405:285–293. [DOI] [PubMed] [Google Scholar]
- 3.Terasaki, W. L., and G. Brooker. 1977. Cardiac adenosine 3′:5′-monophosphate. Free and bound forms in the isolated rat atrium. J. Biol. Chem. 252:1041–1050. [PubMed] [Google Scholar]
- 4.Mongillo, M., T. McSorley, S. Evellin, A. Sood, V. Lissandron, A. Terrin, E. Huston, A. Hannawacker, M. J. Lohse, T. Pozzan, M. D. Houslay, and M. Zaccolo. 2004. Fluorescence resonance energy transfer-based analysis of cAMP dynamics in live neonatal rat cardiac myocytes reveals distinct functions of compartmentalized phosphodiesterases. Circ. Res. 95:67–75. [DOI] [PubMed] [Google Scholar]
- 5.Adams, S. R., A. T. Harootunian, Y. J. Buechler, S. S. Taylor, and R. Y. Tsien. 1991. Fluorescence ratio imaging of cyclic AMP in single cells. Nature. 349:694–697. [DOI] [PubMed] [Google Scholar]
- 6.Corbin, J. D., P. H. Sugden, T. M. Lincoln, and S. L. Keely. 1977. Compartmentalization of adenosine 3′:5′-monophosphate and adenosine 3′:5′-monophosphate-dependent protein kinase in heart tissue. J. Biol. Chem. 252:3854–3861. [PubMed] [Google Scholar]
- 7.Steinberg, S. F., and L. L. Brunton. 2001. Compartmentation of G protein-coupled signaling pathways in cardiac myocytes. Annu. Rev. Pharmacol. Toxicol. 41:751–773. [DOI] [PubMed] [Google Scholar]
- 8.Aass, H., T. Skomedal, and J. B. Osnes. 1988. Increase of cyclic AMP in subcellular fractions of rat heart muscle after beta-adrenergic stimulation: prenalterol and isoprenaline caused different distribution of bound cyclic AMP. J. Mol. Cell. Cardiol. 20:847–860. [DOI] [PubMed] [Google Scholar]
- 9.Hohl, C. M., and Q. Li. 1991. Compartmentation of cAMP in adult canine ventricular myocytes: relation to single-cell free Ca2+ transients. Circ. Res. 69:1369–1379. [DOI] [PubMed] [Google Scholar]
- 10.Hartzell, H. C. 1988. Regulation of cardiac ion channels by catecholamines, acetylcholine and second messenger systems. Prog. Biophys. Mol. Biol. 52:165–247. [DOI] [PubMed] [Google Scholar]
- 11.Belevych, A. E., C. Sims, and R. D. Harvey. 2001. ACh-induced rebound stimulation of L-type Ca(2+) current in guinea-pig ventricular myocytes, mediated by Gβγ-dependent activation of adenylyl cyclase. J. Physiol. (Lond.). 536:677–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belevych, A. E., and R. D. Harvey. 2000. Muscarinic inhibitory and stimulatory regulation of the L-type Ca2+ current is not altered in cardiac ventricular myocytes from mice lacking endothelial nitric oxide synthase. J. Physiol. (Lond.). 528:279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zakharov, S. I., and R. D. Harvey. 1997. Rebound stimulation of the cAMP-regulated Cl− current by acetylcholine in guinea-pig ventricular myocytes. J. Physiol. (Lond.). 499:105–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Defer, N., M. Best-Belpomme, and J. Hanoune. 2000. Tissue specificity and physiological relevance of various isoforms of adenylyl cyclase. Am. J. Physiol. 279:F400–F416. [DOI] [PubMed] [Google Scholar]
- 15.Ishikawa, Y., and C. J. Homcy. 1997. The adenylyl cyclases as integrators of transmembrane signal transduction. Circ. Res. 80:297–304. [DOI] [PubMed] [Google Scholar]
- 16.Sunahara, R. K., C. W. Dessauer, and A. G. Gilman. 1996. Complexity and diversity of mammalian adenylyl cyclases. Annu. Rev. Pharmacol. Toxicol. 36:461–480. [DOI] [PubMed] [Google Scholar]
- 17.Taussig, R., and A. G. Gilman. 1995. Mammalian membrane-bound adenylyl cyclases. J. Biol. Chem. 270:1–4. [DOI] [PubMed] [Google Scholar]
- 18.Tang, W. J., and A. G. Gilman. 1992. Adenylyl cyclases. Cell. 70:869–872. [DOI] [PubMed] [Google Scholar]
- 19.Chen, J., M. DeVivo, J. Dingus, A. Harry, J. Li, J. Sui, D. J. Carty, J. L. Blank, J. H. Exton, R. H. Stoffel, J. Inglese, R. J. Lefkowitz, et al. 1995. A region of adenylyl cyclase 2 critical for regulation by G protein beta-gamma subunits. Science. 268:1166–1169. [DOI] [PubMed] [Google Scholar]
- 20.Federman, A. D., B. R. Conklin, K. A. Schrader, R. R. Reed, and H. R. Bourne. 1992. Hormonal stimulation of adenylyl cyclase through Gi-protein βγ subunits. Nature. 356:159–161. [DOI] [PubMed] [Google Scholar]
- 21.Cooper, D. M. 2005. Compartmentalization of adenylate cyclase and cAMP signalling. Biochem. Soc. Trans. 33:1319–1322. [DOI] [PubMed] [Google Scholar]
- 22.Saucerman, J. J., L. L. Brunton, A. P. Michailova, and A. D. McCulloch. 2003. Modeling beta-adrenergic control of cardiac myocyte contractility in silico. J. Biol. Chem. 278:47997–48003. [DOI] [PubMed] [Google Scholar]
- 23.Saucerman, J. J., S. N. Healy, M. E. Belik, J. L. Puglisi, and A. D. McCulloch. 2004. Proarrhythmic consequences of a KCNQ1 AKAP-binding domain mutation: computational models of whole cells and heterogeneous tissue. Circ. Res. 95:1216–1224. [DOI] [PubMed] [Google Scholar]
- 24.Luo, C.-H., and Y. Rudy. 1994. A dynamic model of the cardiac ventricular action potential. I. Simulations of ionic currents and concentration changes. Circ. Res. 74:1071–1096. [DOI] [PubMed] [Google Scholar]
- 25.Smart, E. J., G. A. Graf, M. A. McNiven, W. C. Sessa, J. A. Engelman, P. E. Scherer, T. Okamoto, and M. P. Lisanti. 1999. Caveolins, liquid-ordered domains, and signal transduction. Mol. Cell. Biol. 19:7289–7304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Insel, P. A., B. P. Head, R. S. Ostrom, H. H. Patel, J. S. Swaney, C. M. Tang, and D. M. Roth. 2005. Caveolae and lipid rafts: G protein-coupled receptor signaling microdomains in cardiac myocytes. Ann. N. Y. Acad. Sci. 1047:166–172. [DOI] [PubMed] [Google Scholar]
- 27.Ostrom, R. S., and P. A. Insel. 2004. The evolving role of lipid rafts and caveolae in G protein-coupled receptor signaling: implications for molecular pharmacology. Br. J. Pharmacol. 143:235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bers, D. M. 2001. Excitation-Contraction Coupling and Cardiac Contractile Force. Kluwer, Dordrecht, The Netherlands.
- 29.Vinnakota, K. C., and J. B. Bassingthwaighte. 2004. Myocardial density and composition: a basis for calculating intracellular metabolite concentrations. Am. J. Physiol. Heart Circ. Physiol. 286:H1742–H1749. [DOI] [PubMed] [Google Scholar]
- 30.Post, S. R., R. Hilal-Dandan, K. Urasawa, L. L. Brunton, and P. A. Insel. 1995. Quantification of signalling components and amplification in the beta-adrenergic-receptor-adenylate cyclase pathway in isolated adult rat ventricular myocytes. Biochem. J. 311:75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Lean, A., J. M. Stadel, and R. J. Lefkowitz. 1980. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J. Biol. Chem. 255:7108–7117. [PubMed] [Google Scholar]
- 32.Bohm, M., K. Larisch, E. Erdmann, M. Camps, K. Jakobs, and P. Gierschik. 1991. Failure of [32P]ADP-ribosylation by pertussis toxin to determine Gi alpha content in membranes from various human tissues. Improved radioimmunological quantification using the 125I-labelled C-terminal decapeptide of retinal transducin. Biochem. J. 277:223–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bohm, M., P. Gierschik, and E. Erdmann. 1992. Quantification of Gi alpha-proteins in the failing and nonfailing human myocardium. Basic Res. Cardiol. 87(Suppl 1):37–50. [DOI] [PubMed] [Google Scholar]
- 34.Rybin, V. O., X. Xu, M. P. Lisanti, and S. F. Steinberg. 2000. Differential targeting of beta-adrenergic receptor subtypes and adenylyl cyclase to cardiomyocyte caveolae. A mechanism to functionally regulate the cAMP signaling pathway. J. Biol. Chem. 275:41447–41457. [DOI] [PubMed] [Google Scholar]
- 35.Brandt, D. R., and E. M. Ross. 1986. Catecholamine-stimulated GTPase cycle. Multiple sites of regulation by beta-adrenergic receptor and Mg2+ studied in reconstituted receptor-Gs vesicles. J. Biol. Chem. 261:1656–1664. [PubMed] [Google Scholar]
- 36.Frace, A. M., P.-F. Méry, R. Fischmeister, and H. C. Hartzell. 1993. Rate-limiting steps in the β-adrenergic stimulation of cardiac calcium current. J. Gen. Physiol. 101:337–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen-Goodspeed, M., A. N. Lukan, and C. W. Dessauer. 2005. Modeling of Galpha(s) and Galpha(i) regulation of human type V and VI adenylyl cyclase. J. Biol. Chem. 280:1808–1816. [DOI] [PubMed] [Google Scholar]
- 38.Sunahara, R. K., C. W. Dessauer, R. E. Whisnant, C. Kleuss, and A. G. Gilman. 1997. Interaction of Gsalpha with the cytosolic domains of mammalian adenylyl cyclase. J. Biol. Chem. 272:22265–22271. [DOI] [PubMed] [Google Scholar]
- 39.Gao, B. N., and A. G. Gilman. 1991. Cloning and expression of a widely distributed (type IV) adenylyl cyclase. Proc. Natl. Acad. Sci. USA. 88:10178–10182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Taussig, R., J. A. Iniguez-Lluhi, and A. G. Gilman. 1993. Inhibition of adenylyl cyclase by Gi alpha. Science. 261:218–221. [DOI] [PubMed] [Google Scholar]
- 41.Tang, W. J., and A. G. Gilman. 1991. Type-specific regulation of adenylyl cyclase by G protein beta gamma subunits. Science. 254:1500–1503. [DOI] [PubMed] [Google Scholar]
- 42.Taussig, R., W. J. Tang, J. R. Hepler, and A. G. Gilman. 1994. Distinct patterns of bidirectional regulation of mammalian adenylyl cyclases. J. Biol. Chem. 269:6093–6100. [PubMed] [Google Scholar]
- 43.Sunahara, R. K., and R. Taussig. 2002. Isoforms of mammalian adenylyl cyclase: multiplicities of signaling. Mol. Interv. 2:168–184. [DOI] [PubMed] [Google Scholar]
- 44.Bundey, R. A., and P. A. Insel. 2003. Quantification of adenylyl cyclase messenger RNA by real-time polymerase chain reaction. Anal. Biochem. 319:318–322. [DOI] [PubMed] [Google Scholar]
- 45.Wang, T., and M. J. Brown. 2004. Differential expression of adenylyl cyclase subtypes in human cardiovascular system. Mol. Cell. Endocrinol. 223:55–62. [DOI] [PubMed] [Google Scholar]
- 46.Rich, T. C., K. A. Fagan, H. Nakata, J. Schaack, D. M. Cooper, and J. W. Karpen. 2000. Cyclic nucleotide-gated channels colocalize with adenylyl cyclase in regions of restricted cAMP diffusion. J. Gen. Physiol. 116:147–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Warrier, S., A. E. Belevych, M. Ruse, R. L. Eckert, M. Zaccolo, T. Pozzan, and R. D. Harvey. 2005. Beta-adrenergic and muscarinic receptor induced changes in cAMP activity in adult cardiac myocytes detected using a FRET based biosensor. Am. J. Physiol. 289:C455–C461. [DOI] [PubMed] [Google Scholar]
- 48.Gao, M., P. Ping, S. Post, P. A. Insel, R. Tang, and H. K. Hammond. 1998. Increased expression of adenylylcyclase type VI proportionately increases beta-adrenergic receptor-stimulated production of cAMP in neonatal rat cardiac myocytes. Proc. Natl. Acad. Sci. USA. 95:1038–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Steinberg, S. F., H. Zhang, E. Pak, G. Pagnotta, and P. A. Boyden. 1995. Characteristics of the beta-adrenergic receptor complex in the epicardial border zone of the 5-day infarcted canine heart. Circulation. 91:2824–2833. [DOI] [PubMed] [Google Scholar]
- 50.Steinberg, S. F., S. Alcott, E. Pak, D. Hu, L. Protas, N. S. Moise, R. B. Robinson, and M. R. Rosen. 2002. beta(1)-Receptors increase cAMP and induce abnormal Ca(i) cycling in the German shepherd sudden death model. Am. J. Physiol. Heart Circ. Physiol. 282:H1181–H1188. [DOI] [PubMed] [Google Scholar]
- 51.Drazner, M. H., K. C. Peppel, S. Dyer, A. O. Grant, W. J. Koch, and R. J. Lefkowitz. 1997. Potentiation of beta-adrenergic signaling by adenoviral-mediated gene transfer in adult rabbit ventricular myocytes. J. Clin. Invest. 99:288–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ruehr, M. L., M. A. Russell, D. G. Ferguson, M. Bhat, J. Ma, D. S. Damron, J. D. Scott, and M. Bond. 2003. Targeting of protein kinase A by muscle A kinase-anchoring protein (mAKAP) regulates phosphorylation and function of the skeletal muscle ryanodine receptor. J. Biol. Chem. 278:24831–24836. [DOI] [PubMed] [Google Scholar]
- 53.Zaccolo, M., and T. Pozzan. 2002. Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science. 295:1711–1715. [DOI] [PubMed] [Google Scholar]
- 54.Head, B. P., H. H. Patel, D. M. Roth, N. C. Lai, I. R. Niesman, M. G. Farquhar, and P. A. Insel. 2005. G-protein-coupled receptor signaling components localize in both sarcolemmal and intracellular caveolin-3-associated microdomains in adult cardiac myocytes. J. Biol. Chem. 280:31036–31044. [DOI] [PubMed] [Google Scholar]
- 55.He, J. Q., R. C. Balijepalli, R. A. Haworth, and T. J. Kamp. 2005. Crosstalk of beta-adrenergic receptor subtypes through Gi blunts beta-adrenergic stimulation of L-type Ca2+ channels in canine heart failure. Circ. Res. 97:566–573. [DOI] [PubMed] [Google Scholar]
- 56.Neumann, J., P. Boknik, S. Herzig, W. Schmitz, H. Scholz, R. C. Gupta, and A. M. Watanabe. 1993. Evidence for physiological functions of protein phosphatases in the heart: evaluation with okadaic acid. Am. J. Physiol. 265:H257–H266. [DOI] [PubMed] [Google Scholar]
- 57.Beavo, J. A., P. J. Bechtel, and E. G. Krebs. 1974. Activation of protein kinase by physiological concentrations of cyclic AMP. Proc. Natl. Acad. Sci. USA. 71:3580–3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Insel, P. A., B. P. Head, H. H. Patel, D. M. Roth, R. A. Bundey, and J. S. Swaney. 2005. Compartmentation of G-protein-coupled receptors and their signalling components in lipid rafts and caveolae. Biochem. Soc. Trans. 33:1131–1134. [DOI] [PubMed] [Google Scholar]
- 59.Stoclet, J.-C., T. Keravis, N. Komas, and C. Lugnier. 1995. Cyclic nucleotide phosphodiesterases as therapeutic targets in cardiovascular disease. Exp. Opin. Invest. Drugs. 4:1081–1100. [Google Scholar]
- 60.Maurice, D. H., D. Palmer, D. G. Tilley, H. A. Dunkerley, S. J. Netherton, D. R. Raymond, H. S. Elbatarny, and S. L. Jimmo. 2003. Cyclic nucleotide phosphodiesterase activity, expression, and targeting in cells of the cardiovascular system. Mol. Pharmacol. 64:533–546. [DOI] [PubMed] [Google Scholar]
- 61.Conti, M., G. Nemoz, C. Sette, and E. Vicini. 1995. Recent progress in understanding the hormonal regulation of phosphodiesterases. Endocr. Rev. 16:370–389. [DOI] [PubMed] [Google Scholar]
- 62.Rochais, F., G. Vandecasteele, F. Lefebvre, C. Lugnier, H. Lum, J. L. Mazet, D. M. Cooper, and R. Fischmeister. 2004. Negative feedback exerted by cAMP-dependent protein kinase and cAMP phosphodiesterase on subsarcolemmal cAMP signals in intact cardiac myocytes: an in vivo study using adenovirus-mediated expression of CNG channels. J. Biol. Chem. 279:52095–52105. [DOI] [PubMed] [Google Scholar]
- 63.Brady, J. D., T. C. Rich, X. Le, K. Stafford, C. J. Fowler, L. Lynch, J. W. Karpen, R. L. Brown, and J. R. Martens. 2004. Functional role of lipid raft microdomains in cyclic nucleotide-gated channel activation. Mol. Pharmacol. 65:503–511. [DOI] [PubMed] [Google Scholar]
- 64.Parfenov, A. S., V. Salnikov, W. J. Lederer, and V. Lukyanenko. 2006. Aqueous diffusion pathways as a part of the ventricular cell ultrastructure. Biophys. J. 90:1107–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rich, T. C., T. E. Tse, J. G. Rohan, J. Schaack, and J. W. Karpen. 2001. In vivo assessment of local phosphodiesterase activity using tailored cyclic nucleotide-gated channels as cAMP sensors. J. Gen. Physiol. 118:63–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Saucerman, J. J., J. Zhang, J. C. Martin, L. X. Peng, A. E. Stenbit, R. Y. Tsien, and A. D. McCulloch. 2006. Systems analysis of PKA-mediated phosphorylation gradients in live cardiac myocytes. Proc. Natl. Acad. Sci. USA. 103:12923–12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hool, L. C., and R. D. Harvey. 1997. Role of β1- and β2-adrenergic receptors in regulation of Cl− and Ca2+ channels in guinea pig ventricular myocytes. Am. J. Physiol. 273:H1669–H1676. [DOI] [PubMed] [Google Scholar]
- 68.Watanabe, A. M., and H. R. Besch. 1975. Interaction between cyclic adenosine monophosphate and cyclic guanosine monophosphate in guinea pig ventricular myocardium. Circ. Res. 37:309–317. [DOI] [PubMed] [Google Scholar]
- 69.Steinberg, S. F. 2004. β(2)-Adrenergic receptor signaling complexes in cardiomyocyte caveolae/lipid rafts. J. Mol. Cell. Cardiol. 37:407–415. [DOI] [PubMed] [Google Scholar]
- 70.Huang, C., J. R. Hepler, L. T. Chen, A. G. Gilman, R. G. Anderson, and S. M. Mumby. 1997. Organization of G proteins and adenylyl cyclase at the plasma membrane. Mol. Biol. Cell. 8:2365–2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cooper, D. M. 2003. Regulation and organization of adenylyl cyclases and cAMP. Biochem. J. 375:517–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bethke, T., W. Meyer, W. Schmitz, H. Scholz, B. Stein, K. Thomas, and H. Wenzlaff. 1992. Phosphodiesterase inhibition in ventricular cardiomyocytes from guinea-pig hearts. Br. J. Pharmacol. 107:127–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Verde, I., G. Vandecasteele, F. Lezoualc'h, and R. Fischmeister. 1999. Characterization of the cyclic nucleotide phosphodiesterase subtypes involved in the regulation of the L-type Ca2+ current in rat ventricular myocytes. Br. J. Pharmacol. 127:65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rich, T. C., C. W. Dessauer. 2004. Assessment of PDE activity in rat neonatal cardiac myocytes. Biophys. J. 86:229A (Abstr.).
- 75.Movsesian, M. A. 2002. PDE3 cyclic nucleotide phosphodiesterases and the compartmentation of cyclic nucleotide-mediated signalling in cardiac myocytes. Basic Res. Cardiol. 97(Suppl 1):I83–I90. [DOI] [PubMed] [Google Scholar]
- 76.Hambleton, R., J. Krall, E. Tikishvili, M. Honeggar, F. Ahmad, V. C. Manganiello, and M. A. Movsesian. 2005. Isoforms of cyclic nucleotide phosphodiesterase PDE3 and their contribution to camp hydrolytic activity in subcellular fractions of human myocardium. J. Biol. Chem. 280:39168–39174. [DOI] [PubMed] [Google Scholar]
- 77.Brunton, L. L. 2003. PDE4: arrested at the border. Sci. STKE. 2003:E44. [DOI] [PubMed]
- 78.Green, S. A., B. D. Holt, and S. B. Liggett. 1992. β1- and β2-Adrenergic receptors display subtype-selective coupling to Gs. Mol. Pharmacol. 41:889–893. [PubMed] [Google Scholar]
- 79.Garnier, V., R. Zini, R. Sapena, and J. P. Tillement. 1997. A match between binding to beta-adrenoceptors and stimulation of adenylyl cyclase parameters of (-)isoproterenol and salbutamol on rat brain. Pharmacol. Res. 35:303–312. [DOI] [PubMed] [Google Scholar]
- 80.Neubig, R. R. 1994. Membrane organization in G-protein mechanisms. FASEB J. 8:939–946. [DOI] [PubMed] [Google Scholar]
- 81.Wei, J.-W., and P. Sulakhe. 1978. Regional and subcellular distribution of myocardical muscarinic cholinergic receptors. Eur. J. Pharmacol. 52:235–238. [DOI] [PubMed] [Google Scholar]
- 82.Vogel, W. K., D. M. Sheehan, and M. I. Schimerlik. 1997. Site-directed mutagenesis on the m2 muscarinic acetylcholine receptor: the significance of Tyr403 in the binding of agonists and functional coupling. Mol. Pharmacol. 52:1087–1094. [DOI] [PubMed] [Google Scholar]
- 83.Post, S. R., H. K. Hammond, and P. A. Insel. 1999. Beta-adrenergic receptors and receptor signaling in heart failure. Annu. Rev. Pharmacol. Toxicol. 39:343–360. [DOI] [PubMed] [Google Scholar]
- 84.Dessauer, C. W., and A. G. Gilman. 1997. The catalytic mechanism of mammalian adenylyl cyclase. Equilibrium binding and kinetic analysis of P-site inhibition. J. Biol. Chem. 272:27787–27795. [DOI] [PubMed] [Google Scholar]
- 85.Beavo, J., and M. D. Houslay. 1990. Cyclic Nucleotide Phosphodiesterases: Structure, Regulation and Drug Action. Wiley, New York.
- 86.Reeves, M. L., B. K. Leigh, and P. J. England. 1987. The identification of a new cyclic nucleotide phosphodiesterase activity in human and guinea-pig cardiac ventricle. Implications for the mechanism of action of selective phosphodiesterase inhibitors. Biochem. J. 241:535–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mongillo, M., C. G. Tocchetti, A. Terrin, V. Lissandron, Y. F. Cheung, W. R. Dostmann, T. Pozzan, D. A. Kass, N. Paolocci, M. D. Houslay, and M. Zaccolo. 2006. Compartmentalized phosphodiesterase-2 activity blunts beta-adrenergic cardiac inotropy via an NO/cGMP-dependent pathway. Circ. Res. 98:226–234. [DOI] [PubMed] [Google Scholar]
- 88.Jurevicius, J., V. A. Skeberdis, and R. Fischmeister. 2003. Role of cyclic nucleotide phosphodiesterase isoforms in cAMP compartmentation following beta2-adrenergic stimulation of ICa,L in frog ventricular myocytes. J. Physiol. 551:239–252. [DOI] [PMC free article] [PubMed] [Google Scholar]