

Short abstract
Usual BLAST-based methods for assessing gene presence and absence lead to systematic overestimation of within-species gene gain by lateral transfer.
Abstract
The usual BLAST-based methods for assessing gene presence and absence lead to systematic overestimation of within-species gene gain by lateral transfer.
Genomes from different strains of the same bacterial species often differ substantially (up to 30%) in gene content [1-6]. There are two general ways to account for such gene content variability ('patchy distribution') among closely related genomes: strain-specific loss of genes after divergence from a common species ancestor that contained the genes, and strain-specific gain of genes after divergence from an ancestor that lacked them. Gain might be effected through lateral gene transfer (LGT), duplication (paralog creation) or, much less likely, de novo creation. Several recent publications have attempted to assess rates of within-species gain and loss using parsimony-based approaches applied to gene presence/absence data, in the context of a reference strain phylogeny [7-11]. Similar parsimony-based approaches have also been taken for inferences of gene gain/loss at larger phylogenetic distances [12-14].
In such analyses, a pattern like that shown in Figure 1a would be interpreted to indicate a single event of gain of a gene X not present in the species ancestor, after the separation of taxa 4 and 5. Explaining this distribution as the result of loss of a gene X initially present in the ancestor would, in contrast, require a minimum of four separate events, a seemingly less parsimonious scenario. However, reasoning by parsimony in such a situation requires difficult-to-test assumptions about the relative frequency of gain and loss events (that, for instance, losses are not four times more frequent than gains). Moreover, such reasoning is simply beside the point if we have some other sort of knowledge about the relevant processes that suggests we might be misled by appearances. Here we do know that gain (at least when it occurs by gene duplication or LGT) could be effectively instantaneous, but that loss will more commonly proceed gradually, through intermediates we might call pseudogenes and gene remnants (regions recognizable as gene-derived only by synteny and statistically significant sequence similarity to the parent gene). There is thus an inherent asymmetry between gain and loss both in terms of defining and of detecting them, and failure to recognize gene remnants will inevitably lead to mistaking a situation like that shown in Figure 1b (in which a gene present in the species' ancestor has deteriorated in all lineages but one) for the situation in Figure 1a (in which a gene absent from the ancestor has been gained in a single lineage). Our goal in the present analysis was to assess how often such mistakes might be made.
Figure 1.
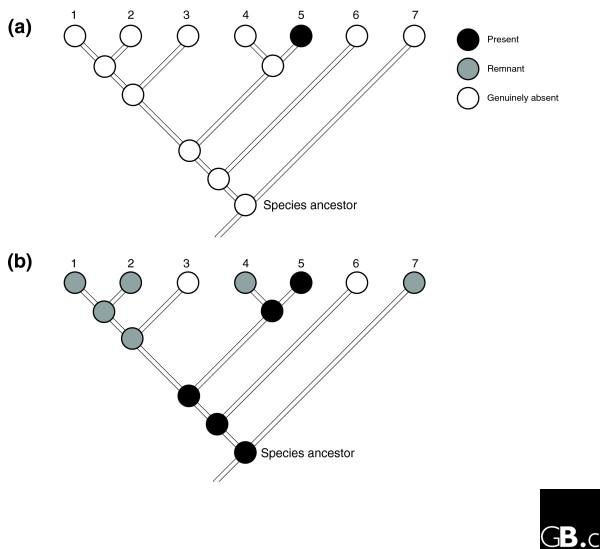
Illustration of parsimony inference from a gene/presence pattern and a reference tree topology. (a,b) Results of parsimonious inferences for the same gene family, with different criteria used to define presence/absence patterns. In (a) genes are divided into only two categories, present and absent, while in (b) the absent genes are further classified into gene remnants and genuinely absent.
Although prokaryotic genomes have traditionally been viewed as efficiently packed with functioning genes, and mutationally biased towards rapid deletion of dysfunctional regions [15], there are new indications that significant numbers of pseudogenes persist in some genomes [16-18]. In addition, detailed analyses show that in reduced genomes such as those of Rickettsia, intergenic regions often represent decaying remnants of genes [19]. Some categorization more nuanced than 'presence' versus 'absence' might thus better capture genome history. But for gain-and-loss surveys of the sort cited there may seem to be no alternative to the binary approach. A gene is considered 'present' if represented by an open reading frame (ORF) showing significant similarity in sequence (with arbitrarily chosen significance cutoff) and having similar length to a query gene; otherwise it is scored as 'absent'. We systematically screened groups of closely related genomes (see Additional data files 1-3) for gene-family presence/absence patterns using several common criteria. When potential gene remnants detectable by less stringent methods are included, the number of gene families for which events of gain or loss within a species might be inferred (because they are scored as present only in some strains) can drop by as much as 90% (or as little as 7%) - on average about 60%. The extent to which recognition of such remnants will decrease estimates of the rates of gain of genes by LGT and increase estimates of the gene content of species' ancestors will depend on how recognition affects inferred patterns of presence and absence as displayed on a phylogeny of the species' strains. Each gene family must be individually examined, and where there is frequent between-strain recombination, not only is strain phylogeny a problematic concept [20], but it will sometimes be the case that gene remnants are themselves acquired by LGT.
We have assessed the impact of more complete recognition of gene remnants in the simplest cases, those species for which only three genomes are available. We calculated the number of presence/absence patterns that change under different match-length requirements for the eight such groups in our dataset (Figure 2). Any change in any of the possible presence/absence patterns as a consequence of altered BLAST (Basic Local Alignment Search Tool) [21] criteria leads to a change in gain/loss inference on a three-taxon tree, and in all cases to a change in inferred ancestral state. Such numbers are not negligible in comparison with the total number of inferred presence/absence patterns (see the last row in Figure 2).
Figure 2.
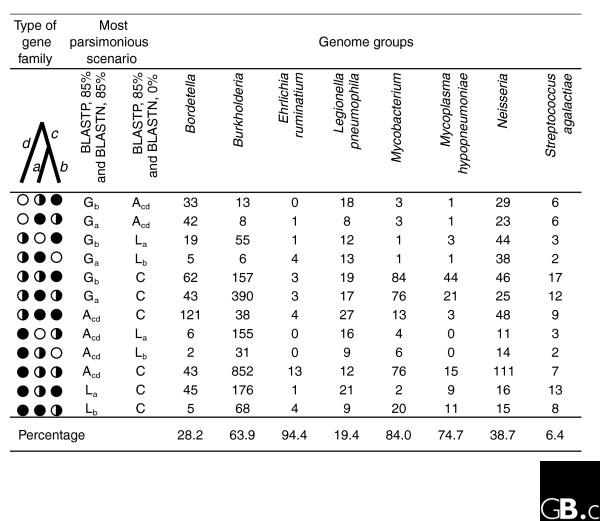
The analysis of patchily distributed gene families that change their state (present or absent) in different genomes under two different selection criteria for gene families. Eight groups of three genomes each were analyzed. In one selection scheme, a match-length requirement of 85% in BLASTN was imposed (stringent selection), while in the other there was no match-length requirement in BLASTN (relaxed selection). Corresponding gene families constructed under the two criteria were compared and classified into all possible types of gene families (total 33 = 27). Of these, only those types of gene families (12) where at least one gene is present under both criteria, and where at least one gene changes its state under the two criteria, are shown. They are coded as filled circles (present under both criteria), empty circles (absent under both criteria) and half-filled circles (absent under the stringent criterion and present under the relaxed criterion). Numbers in the figure indicate the number of patchily distributed gene families that change their state when under two different selection criteria. The last row is the total number of gene families for which differences in history might be incorrectly inferred, expressed as a percentage of total gene families detected as present in one or two, but not three, genomes in a genome group. The total number of gene families used in the calculation is listed in the second table in Additional data file 2. Branches on the three-taxon tree are denoted as a, b, c and d. G, gain; L, loss; A, ambiguous (both gain and loss are equally parsimonious); C, core (that is, present in all three genomes). The subscript refers to the branch on which the event is inferred. For the list of genomes in each group see Additional data file 3.
Therefore, without agreed-upon definitions of presence/absence and reliable methods of detection, quantitation of rates of within-species gene gain have questionable meaning. It is both a practical concern and of theoretical interest that we really do not have a definition for gene loss. It is not clear where - along the line from the appearance of the first subtly deleterious regulatory or missense mutation to the deletion of the last nucleotide - we would agree to declare a gene to be lost. Parsimony-based inferences depend on how we make that declaration, but most quantitative treatments of gene loss in evolution avoid this question altogether. Moreover, in recombinogenic species, the possibility of exchange of remnants of inactivated genes between lineages means that there will be additional difficulties in reconstructing the decay process for individual genes. Indeed, in highly recombinogenic groups such as Neisseria, where homologous recombination, not mutation, is the principal source of between-strain sequence variation [22], it should seldom be possible to reconstruct the loss of an individual gene as a linear process of decay. These problems are of practical concern, as inferences about gain and loss dominate discussion of the evolution of pathogenicity and environmental adaptation within species. They are also of theoretical interest, bearing on the use of parsimony in evolutionary reconstruction.
As a matter of good practice, no claim that strains of the same species differ in gene content should be based on BLAST results alone, as differences in annotation abound and even BLASTing a single genome against itself does not recover all its annotated ORFs. No BLASTP+BLASTN-based estimate of the number of genes that a genome must have received by LGT (because they are absent from sister lineages in the same species) should be accepted without recognition that it is probably too high, possibly by several-fold. Species seem to differ in the extent to which such estimates are sensitive to BLAST parameters, and it is unlikely that optimal parameters - could these somehow be established - would be the same for all species groups. Ideally, all gene families would be examined for even highly decayed remnants.
Additional data files
The following additional data are available online with this paper. Additional data file 1 contains Materials and methods for the analyses performed. Additional data file 2 describes in detail the comparison of different BLAST-based criteria for presence/absence detection. Additional data file 3 is a table listing the composition of the analyzed genome groups.
Supplementary Material
Materials and methods.
Comparison of different BLAST criteria to detect gene presence/absence.
Thirty-two groups of genomes with ANI = 94% within each group.
Acknowledgments
Acknowledgements
This work was supported through CIHR (MOP-4467) and Genome Atlantic (Genome Canada) grants to W.F.D. O.Z. is supported through a CIHR Postdoctoral Fellowship and is an honorary Killam Postdoctoral Fellow at Dalhousie University. O.Z., C.L.N. and W.F.D. designed the study. O.Z. carried out all analyses. O.Z. and W.F.D. wrote the manuscript.
References
- Welch RA, Burland V, Plunkett G, 3rd, Redford P, Roesch P, Rasko D, Buckles EL, Liou SR, Boutin A, Hackett J, et al. Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc Natl Acad Sci USA. 2002;99:17020–17024. doi: 10.1073/pnas.252529799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasko DA, Ravel J, Okstad OA, Helgason E, Cer RZ, Jiang L, Shores KA, Fouts DE, Tourasse NJ, Angiuoli SV, et al. The genome sequence of Bacillus cereus ATCC 10987 reveals metabolic adaptations and a large plasmid related to Bacillus anthracis pXO1. Nucleic Acids Res. 2004;32:977–988. doi: 10.1093/nar/gkh258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen IT, Press CM, Ravel J, Kobayashi DY, Myers GSA, Mavrodi DV, DeBoy RT, Seshadri R, Ren Q, Madupu R, et al. Complete genome sequence of the plant commensal Pseudomonas fluorescens Pf-5. Nat Biotechnol. 2005;23:873. doi: 10.1038/nbt1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mongodin EF, Hance IR, Deboy RT, Gill SR, Daugherty S, Huber R, Fraser CM, Stetter K, Nelson KE. Gene transfer and genome plasticity in Thermotoga maritima, a model hyperthermophilic species. J Bacteriol. 2005;187:4935–4944. doi: 10.1128/JB.187.14.4935-4944.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesbø CL, Nelson KE, Doolittle WF. Suppressive subtractive hybridization detects extensive genomic diversity in Thermotoga maritima. J Bacteriol. 2002;184:4475–4488. doi: 10.1128/JB.184.16.4475-4488.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, Angiuoli SV, Crabtree J, Jones AL, Durkin AS, et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial "pan-genome". Proc Natl Acad Sci USA. 2005;102:13950–13955. doi: 10.1073/pnas.0506758102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao W, Golding GB. Patterns of bacterial gene movement. Mol Biol Evol. 2004;21:1294–1307. doi: 10.1093/molbev/msh129. [DOI] [PubMed] [Google Scholar]
- Ortutay C, Gaspari Z, Toth G, Jager E, Vida G, Orosz L, Vellai T. Speciation in Chlamydia: genomewide phylogenetic analyses identified a reliable set of acquired genes. J Mol Evol. 2003;57:672–680. doi: 10.1007/s00239-003-2517-3. [DOI] [PubMed] [Google Scholar]
- Daubin V, Lerat E, Perriere G. The source of laterally transferred genes in bacterial genomes. Genome Biol. 2003;4:R57. doi: 10.1186/gb-2003-4-9-r57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao W, Golding GB. The fate of laterally transferred genes: life in the fast lane to adaptation or death. Genome Res. 2006;16:636–643. doi: 10.1101/gr.4746406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marri PR, Bannantine JP, Paustian ML, Golding GB. Lateral gene transfer in Mycobacterium avium subspecies paratuberculosis. Can J Microbiol. 2006;52:560–569. doi: 10.1139/W06-001. [DOI] [PubMed] [Google Scholar]
- Kunin V, Ouzounis CA. The balance of driving forces during genome evolution in prokaryotes. Genome Res. 2003;13:1589–1594. doi: 10.1101/gr.1092603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirkin BG, Fenner TI, Galperin MY, Koonin EV. Algorithms for computing parsimonious evolutionary scenarios for genome evolution, the last universal common ancestor and dominance of horizontal gene transfer in the evolution of prokaryotes. BMC Evol Biol. 2003;3:2. doi: 10.1186/1471-2148-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLysaght A, Baldi PF, Gaut BS. Extensive gene gain associated with adaptive evolution of poxviruses. Proc Natl Acad Sci USA. 2003;100:15655–15660. doi: 10.1073/pnas.2136653100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mira A, Ochman H, Moran NA. Deletional bias and the evolution of bacterial genomes. Trends Genet. 2001;17:589–596. doi: 10.1016/S0168-9525(01)02447-7. [DOI] [PubMed] [Google Scholar]
- Ochman H, Davalos LM. The nature and dynamics of bacterial genomes. Science. 2006;311:1730–1733. doi: 10.1126/science.1119966. [DOI] [PubMed] [Google Scholar]
- Lerat E, Ochman H. Recognizing the pseudogenes in bacterial genomes. Nucleic Acids Res. 2005;33:3125–3132. doi: 10.1093/nar/gki631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Harrison PM, Kunin V, Gerstein M. Comprehensive analysis of pseudogenes in prokaryotes: widespread gene decay and failure of putative horizontally transferred genes. Genome Biol. 2004;5:R64. doi: 10.1186/gb-2004-5-9-r64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson JO, Andersson SG. Pseudogenes, junk DNA, and the dynamics of Rickettsia genomes. Mol Biol Evol. 2001;18:829–839. doi: 10.1093/oxfordjournals.molbev.a003864. [DOI] [PubMed] [Google Scholar]
- Feil EJ. Small change: keeping pace with microevolution. Nat Rev Microbiol. 2004;2:483–495. doi: 10.1038/nrmicro904. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanage W, Fraser C, Spratt B. Fuzzy species among recombinogenic bacteria. BMC Biol. 2005;3:6. doi: 10.1186/1741-7007-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford D. PAUP* 40 Beta Version, Phylogenetic Analysis Using Parsimony (and Other Methods) Sunderland, MA; Sinauer Associates; 1998. [Google Scholar]
- van Dongen S. Technical Report INS-R0010. Amsterdam: National Research Institute for Mathematics and Computer Science in the Netherlands; 2000. A cluster algorithm for graphs. [Google Scholar]
- Konstantinidis KT, Tiedje JM. Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci USA. 2005;102:2567–2572. doi: 10.1073/pnas.0409727102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson WR. Effective protein sequence comparison. Methods Enzymol. 1996;266:227–258. doi: 10.1016/s0076-6879(96)66017-0. [DOI] [PubMed] [Google Scholar]
- Fraser-Liggett CM. Insights on biology and evolution from microbial genome sequencing. Genome Res. 2005;15:1603–1610. doi: 10.1101/gr.3724205. [DOI] [PubMed] [Google Scholar]
- Nierman WC, DeShazer D, Kim HS, Tettelin H, Nelson KE, Feldblyum T, Ulrich RL, Ronning CM, Brinkac LM, Daugherty SC, et al. Structural flexibility in the Burkholderia mallei genome. Proc Natl Acad Sci USA. 2004;101:14246–14251. doi: 10.1073/pnas.0403306101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Materials and methods.
Comparison of different BLAST criteria to detect gene presence/absence.
Thirty-two groups of genomes with ANI = 94% within each group.