

Abstract
A major bottleneck for validation of new clinical diagnostics is the development of highly sensitive and specific assays for quantifying proteins. We previously described a method, Stable Isotope Standards with Capture by Anti-Peptide Antibodies, wherein a specific tryptic peptide is selected as a stoichiometric representative of the protein from which it is cleaved, is enriched from biological samples using immobilized antibodies, and is quantitated using mass spectrometry against a spiked internal standard to yield a measure of protein concentration. In this report, we optimize a magnetic bead-based platform amenable to high throughput peptide capture and demonstrate that antibody capture followed by mass spectrometry can achieve ion signal enhancements on the order of 103, with precision (CVs <10%) and accuracy (relative error ~20%) sufficient for quantifying biomarkers in the physiologically relevant ng/mL range. These methods are generally applicable to any protein or biological fluid of interest and hold great potential for providing a desperately needed bridging technology between biomarker discovery and clinical application.
Keywords: Biomarkers, Anti-peptide antibody, LC/MS/MS, Targeted proteomics, Stable isotope dilution, Peptide quantitation
Introduction
Protein biomarkers have had tremendous impact on clinical management of human disease, especially cancer. For example, proteins differentially present or mutated in tumor cells have facilitated both the development and appropriate use of targeted therapeutics[1–4], as well as prognostication of disease outcomes[5, 6]. The application of genomics and proteomics technologies to protein biomarker discovery has enabled hundreds of biomarker candidates to be identified in a single discovery effort. However, as yet the promise of these discovery tools has not been fulfilled, in part due to the downstream bottleneck of clinical validation.
The current gold standard for validating putative biomarkers is the Enzyme-Linked Immunosorbent Assay (ELISA). A well functioning ELISA can be run at high-throughput and has extraordinary sensitivity and specificity for quantifying the target analyte. However, ELISA development is costly ($100,000–$2 million per biomarker candidate) and associated with a long development lead time (>1 year) and a high failure rate. The high cost and long lead time are a bottleneck to biomarker testing, and make it impractical to develop an ELISA for all putative biomarkers. It is not surprising that the number of new biomarkers validated in the last five years has been remarkably small[7].
Although the ELISA may remain the gold standard for clinical application, relieving this bottleneck to preclinical biomarker testing will require developing more affordable “bridging” methodologies with shorter development lead times. Mass spectrometry (MS) is a well established tool for quantification of metabolites in clinical samples[8, 9]. With the development of soft ionization techniques[10, 11], it is now possible to extend its application to quantifying peptide components of biomarker candidates in clinical samples. A specific tryptic peptide can be selected as a stoichiometric representative of the protein from which it is cleaved (a monitor peptide), and can be quantitated against a spiked internal standard (a synthetic stable-isotope labeled peptide) to yield a measure of protein concentration[12–14]. As already used in analytical chemistry to quantify drug metabolites and other small molecules[8], mass spectrometry offers high precision (coefficients of variation (CVs) below 5%), a good linear response range (>103), and high sensitivity of detection (less than 1 ng/mL).
Although selected reaction monitoring (SRM-MS) has been applied to the quantitation of plasma protein-derived peptides[15–21], a major limitation is that, due to ion suppression, the majority of biomarker proteins (e.g. PSA, CEA, and AFP present at ng/mL) cannot be detected in plasma in a mass spectrometry experiment without enrichment relative to large quantities of interfering proteins (e.g. albumin 50 mg/mL, globulin 35 mg/mL). To this end, we previously described a technology, “Stable Isotope Standards with Capture by Anti-Peptide Antibodies” (SISCAPA) in which anti-peptide antibodies immobilized on 100 nanoliter affinity columns were used to enrich specific peptides along with spiked stable-isotope-labeled internal standards of the same sequence[22]. Upon elution from the anti-peptide antibody supports, electrospray ionization (ESI) MS was used to quantify the peptides (natural and labeled). In a series of pilot experiments, binding and elution from these supports was shown to provide an average 120-fold enrichment of the antigen peptide relative to others, as measured by selected ion monitoring (SIM) or SRM-MS. These MS experiments generated peptide ion current measurements with cycle-to-cycle CVs near 5%.
In this current study, we extend the previous work by 1) optimizing a magnetic bead-based platform amenable to high throughput for peptide enrichment, 2) using this optimized platform to demonstrate for the first time that antibody enrichment followed by SRM-MS can achieve ion signal enhancements of >103, sufficient for quantifying biomarkers in plasma at the ng/mL range, and 3) demonstrating the capabilities of the commonly available linear ion trap mass spectrometer for quantitative testing of biomarker candidates. These highly sensitive and specific methods are generally applicable to any protein and biological fluid of interest.
Materials and methods
Materials
Polyclonal antibodies against α1-antichymotrypsin (AAC, accn. P01011) and tumor necrosis factor alpha (TNFα, accn. P01375) peptides were described previously[22]. Stable isotope standards of AAC (EIGELYLPK, 13C- and 15N-labeled amino acid in italics) and TNFα (DLSLISPLAQAVR) were obtained from Cell Signaling Technologies (Beverly, MA). The 12C version of TNFα peptide was synthesized by AnaSpec (San Jose, CA) and quantified by amino acid analysis. The labeled version of peptides resulted in a mass shift of +6 Da. Monoclonal antibody against human TNFα was purchased from R&D systems (Minneapolis, MN). DynabeadsR Protein G, Myone, M280, M270 streptavidin, tosylactivated, carboxyl, and epoxy magnetic beads were obtained from Dynal Biotechnology (Lake Success, NY).
Immobilization of antibody on magnetic beads
100 μg of antibody was added to 250 μL (7.5 mg) of Protein G beads and incubated at room temperature (RT) for one hour to allow antibodies to attach to the surface. Approximately 10 μg antibody per mg beads was bound (see Supplemental Material for more detail). The beads were then washed with 1 mL 0.2 M triethanolamine, pH 8.2. For crosslinking of antibody on beads, Protein G beads with immobilized polyclonal antibody were resuspended in 1 mL of freshly made DMP crosslinking buffer (20 nM dimethyl primelinidate·2 HCl in 0.2 M triethanolamine, pH 8.2). The mixture was incubated at RT for 30 minutes, placed on the magnet, and the supernatant was discarded. The beads were resuspended in 1 mL of 50 mM Tris, pH 7.5 and incubated for 15 minutes to stop the reaction. The amount of antibody bound to Protein G beads was estimated by comparing protein concentrations (measured by Bradford assay) of the supernatants before and after cross-linking. For experiments that determined the effects of trypsin inhibitor, mouse monoclonal antibodies for TNFα protein were biotinylated using the EZ-Link kit from Pierce (Rockford, IL). 3 μg of biotin was added to 100 μg antibodies and incubated at RT for two hours. The antibodies were extensively dialyzed using Slide-A-Lyzer mini dialysis units from Pierce (Rockford, IL) with 2 × 500 mL buffer changes to remove free biotin. 10 μg of biotinylated antibody was added to 1 mg of M270 streptavidin beads and incubated at RT for one hour.
Serum depletion and digestion
Human serum was purchased from Sigma-Aldrich (St. Louis, MO). The human Multiple Affinity Removal System (MARS) and 0.22 μm filters were purchased from Agilent Technologies (Palo Alto, CA) for depletion of albumin, transferrin, IgG, IgA, anti-trypsin and haptoglobin from human serum and used per the manufacturer’s recommendations. Briefly, 20 μL of human serum was diluted 5 times with MARS buffer A. Particulates in the diluted serum were moved using 0.22 μm filter before injecting to the MARS column (4.6 × 50 mm). Flow-through was collected in Buffer A at 0.25 mL/min for 4 minutes. Depleted proteins were concentrated and buffer exchanged to 50 mM ammonium bicarbonate using an Agilent Spin concentrator (5 kDa Molecular Weight Cut-off). The final depleted serum concentration was 2 mg/mL. A tryptic digest of the depleted serum was prepared as follows (See Supplemental Material for further characterization of the trypsin digestion protocol): Serum proteins were denatured by addition of methanol to a final concentration of 60% and dithiothreitol (DTT, 1M) to a final concentration of 10 mM. The mixture was incubated at 60°C for one hour. 1 M iodoacetamide was added to a final concentration of 50 mM, and the sample was incubated at room temperature for 30 minutes in the dark. The mixture was diluted with 50 mM ammonium bicarbonate to a final methanol concentration of 20% (final iodoacetamide ~16.7 mM). Trypsin (Promega, Madison, WI) was added at an initial trypsin-to-protein ratio of 1:100, and a second aliquot of trypsin was added after two hours of digestion, for a final trypsin-to-protein ratio of 1:50. The total length of trypsin digestion was 6 hours at 37°C. Lima bean inhibitor of bovine and human trypsin (Worthington, Lakewood, NJ) was added to the final digested mixture at the ratio of 1:1.3 (by weight, trypsin inhibitor:trypsin).
Antigen capture and elution
Magnetic Protein G beads with immobilized antibodies were incubated with tryptic digests of serum at 4°C overnight. The beads were washed with 10 mM Tris, 1 mM EDTA, 0.5 M NaCl. The bound peptides were eluted by incubating the beads in 8 μL of 5% acetic acid for 3 minutes at RT. The recovery of TNFα protein was analyzed by SDS-PAGE stained with SilverQuest kit from Invitrogen (Carlsbad, CA) or by Western blot.
Nano-liquid chromatography
An Agilent 1100 nano flow system (Agilent Technologies, Palo Alto, CA) equipped with a micro well-plate autosampler and an isocratic capillary pump (for sample loading) was used for liquid chromatography. Solvents used were water/0.1% formic acid (mobile phase A) and acetonitrile/0.1% formic acid (mobile phase B). An IntegraFrit trap column (2 cm × 100 μm, New Objective, Woburn, MA) was connected to a silica-based C18 monolithic column (ChromoLith CapRod, Merck, Darmstadt, Germany) via a micro-cross connector (Upchurch Scientific, Oak Harbor, WA). The trapping column was packed in house at a pressure of 500 psi using Atlantis C18 material (5 μm particle, 100 Å pore size, Waters Corporation, Milford, MA). Samples were loaded on the trapping column at 10 μL/min and desalted by washing with 2% B for 5 min. The LC gradient for the monolithic column was delivered at 800 nL/min. A linear gradient of mobile phase B was developed from 10–40% B for 10 minutes or 30 minutes depending on sample complexity.
Electrospray ionization linear ion trap mass spectrometer
The nano-LC system was connected to a linear ion trap mass spectrometer (LTQ, Thermo Electron, San Jose, CA) equipped with a nano electrospray interface operated in the positive ion mode. Typical instrument settings included a spray voltage of 1.5 kV, an ion transfer tube temperature of 200°C, and a collision gas pressure of 1.3 Torr. Voltages across the capillary and the quadrupole lenses were tuned for optimal signal intensity using the +2 ion of angiotensin I (m/z 649). For each analyte run, a MS survey scan (m/z 400–1600) was followed by two targeted MS/MS scans at the m/z of the monitor peptide and the heavy-isotope labeled version of the peptide. Quantification was performed by integrating the extracted ion chromatogram of the doubly-charged parent ion from the MS scan. Ions monitored for quantification of AAC were m/z 531.3 and 534.3 (13C- and 15N-incorporated standard); TNFα ions were m/z 692.2 and 695.2 (13C- and 15N-incorporated standard). MRM experiments were performed by integrating the areas of two transition fragment peaks from the MS/MS spectrum using XCalibur™ QualBrowser. The inclusion window for each precursor ion was ± 1 Da, the inclusion window for the fragment ion was ± 2 Da. Transitions monitored for AAC were 531.3 → 633.5, 819.5 and 534.3 → 639.5, 825.5. Transitions monitored for TNFα were 692.2 → 841.5, 954.5 and 695.2 → 847.5, 960.5.
Results
Selection of magnetic beads
Magnetic beads have been used in almost all branches of bioscience and biotechnologies for isolation, separation, and purification of various types of proteins and peptides[23]. Many varieties of magnetic beads are commercially available, with a variety of surface chemistries. We sought a bead system with high antibody coupling efficiency, good functional orientation of antibody for antigen capture, low background binding of serum peptides and proteins, and production of captured samples appropriate for mass spectrometry (i.e. without chemical contamination). To this end, a panel of seven Dynal magnetic beads were tested (see Supplemental Material for more details), including Protein G, Myone streptavidin, M280 streptavidin, M270 streptavidin, tosylactivated, epoxy, and carboxyl beads. Based on their low background binding, lack of chemical contamination, high coupling efficiency, and proper orientation of crosslinked antibodies, we chose Protein G beads for our peptide antigen capture technology.
Antigen binding, wash, and elution conditions
We sought to optimize conditions for capture and recovery of targeted peptide antigens, minimizing nonspecific binding while maximizing the recovery of the target antigens. We used a relatively small protein, tumor necrosis factor alpha (TNFα, 17 kDa), as a test antigen to monitor the capture and elution process by gel electrophoresis (SDS-PAGE). Specifically, 50 ng (2940 fmol) of TNFα was spiked into 1 mL human serum. 20 μL of anti-TNFα monoclonal antibody-coated magnetic beads were added to the serum, and the antigen was captured at 4°C overnight. The beads were washed 4 times with 1 mL 0.5 M salt. A series of elution buffers was tested on captured TNFα, and the recovery of protein was determined by Western blot. As can be seen in Figure 1a, very little antigen is lost in the flow-through or wash collections, demonstrating that the majority of antigen was successfully captured on the beads. Four of the five elution buffers tested yielded considerable recovery of antigen within 3 minutes. 5% trifluoroacetic acid (TFA) was the only exception; in this case, none of the antigen came off during the elution, although it was released by boiling the beads.
Figure 1.
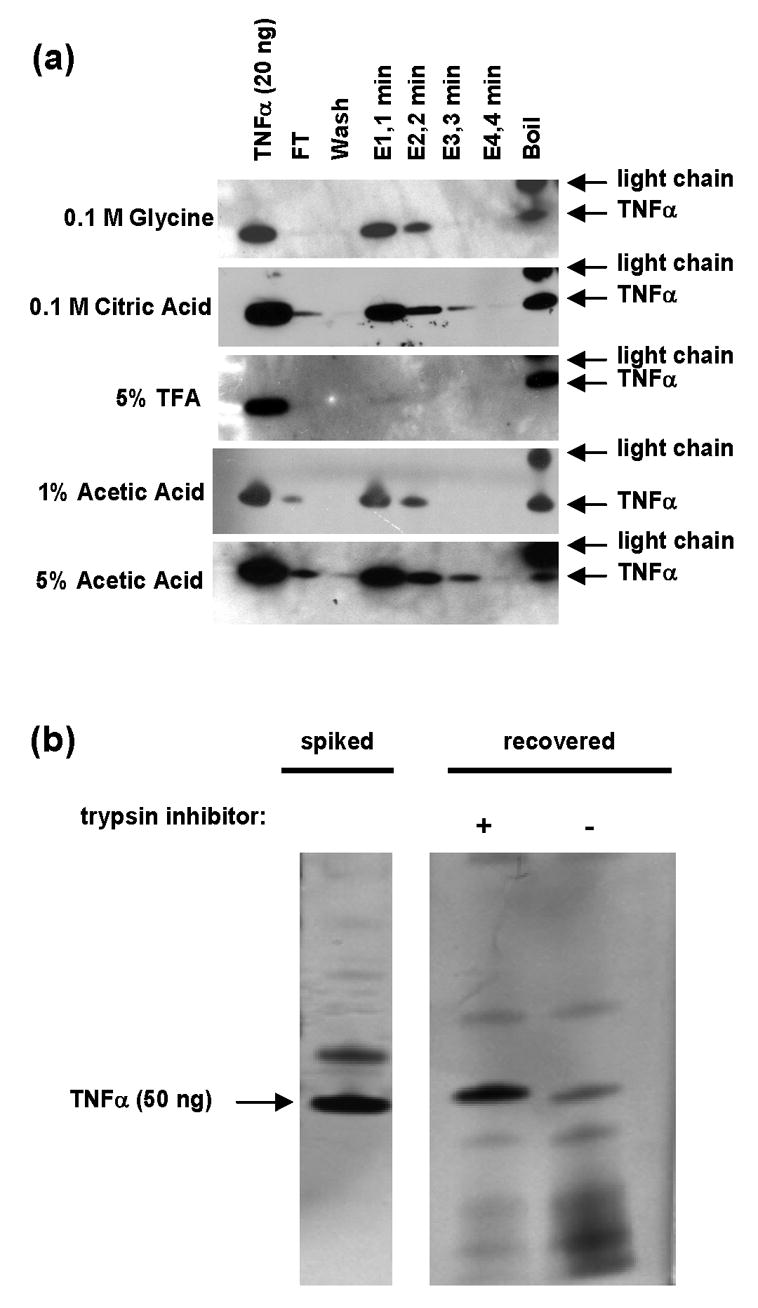
Optimization of conditions for maximum recovery of captured antigen. (a) Western blot (cropped) showing recovery of 50 ng (2940 fmol) TNFα spiked into 1 mL human serum, captured at 4°C overnight by 20 μL of anti-TNFα polyclonal antibody-coated beads. ‘FT’ refers to flow through. ‘Wash’ refers to 4 times washing of beads with 1 mL of 0.5 M NaCl. Multiple elution buffers were tested, as indicated. Four aliquots of elution buffer (labeled E1–E4, 8 μL each) were added to 20 μL of antigen-bound, antibody-coated beads at RT, in one minute intervals (total of four minutes). At the end of the elution period, the beads were boiled in SDS buffer to remove residual protein (‘Boil’). Note that the secondary antibody used is a goat anti-mouse antibody, which reveals mouse light chain fragments creleased by boiling. (b) SDS-PAGE of antibody enrichment of 50 ng of recombinant TNFα spiked into 400 μL of human serum. The gel image shows recovery of TNFα from a trypsin digest of human serum with (+) and without (−) addition of trypsin inhibitor following the digestion (prior to the antibody-capture step). Addition of trypsin inhibitor results in higher recovery of TNFα protein from trypsin-digested serum.
Addition of trypsin inhibitor to antigen captures
For peptide capture experiments, tryptic digests of serum (containing the target peptide antigen) are added to antibody-coupled beads. Theoretically, residual active trypsin in the digested serum could proteolyze and destroy the bead-bound antibodies, thereby reducing the capture efficiency of target peptide antigen. To test this possibility, we compared the recovery of recombinant TNFα spiked into proteolyzed serum in the presence or absence of trypsin inhibitor. As can be seen in Figure 1b, recovery of TNFα was significantly improved by the addition of trypsin inhibitor. Consistent with the notion that residual trypsin activity is responsible for reducing recovery, an increase in TNFα degradation products can be seen in the sample that did not receive trypsin inhibitor. Given the considerable amount of residual trypsin activity observed in this experiment, we presume antibodies on the surface of beads are also being proteolyzed. Hence, for all capture experiments using trypsin-digested serum, trypsin inhibitor is added to inhibit the digestion prior to the addition of bead-bound antibody.
Performance characteristics of SISCAPA-MS coupled measurements
For SISCAPA coupled to LC-MS to be applied to clinical samples for biomarker testing, the technology platform must be capable of precise and accurate measurements. Triple quadrupole instruments, already entrenched in clinical laboratories for measuring small molecules, have consistently provided measurements with high precision (CVs below 5%), a good linear response range (>103), and high sensitivity of detection (less than 1 ng/mL). Linear ion trap instruments, far more common in proteomics laboratories involved with biomarker discovery, have only recently been tested in this setting[24] but are nonetheless widely used for quantification[25–28]. To determine the performance characteristics of our magnetic bead-based capture system coupled to a linear ion trap, we assessed the linear range, limit of quantification, accuracy, and precision for our two test antigens, alpha-1-antichymotrypsin (AAC) and TNFα. We used the extracted ion chromatogram of the target peptide for quantification and verified the identity of the peptide by acquiring a full MS/MS spectrum.
Linear range and limit of quantification of peptides
To ensure the accuracy in quantification using stable isotope-labeled internal standards, the linear concentration ranges and limits of quantification of the instrument were determined for pure, synthetic peptides from AAC and TNFα suspended in 50 mM ammonium bicarbonate. The response for AAC and TNFα was found to be linear (R2 = 0.9984 and 0.9967, respectively) over 4 and 3 orders of magnitude, respectively (Fig. 2). The limit of quantification (LOQ, where signal-to-noise = 10) for AAC and TNFα was approximately 1 fmol (~1 pg) of peptide injected on-column.
Figure 2.
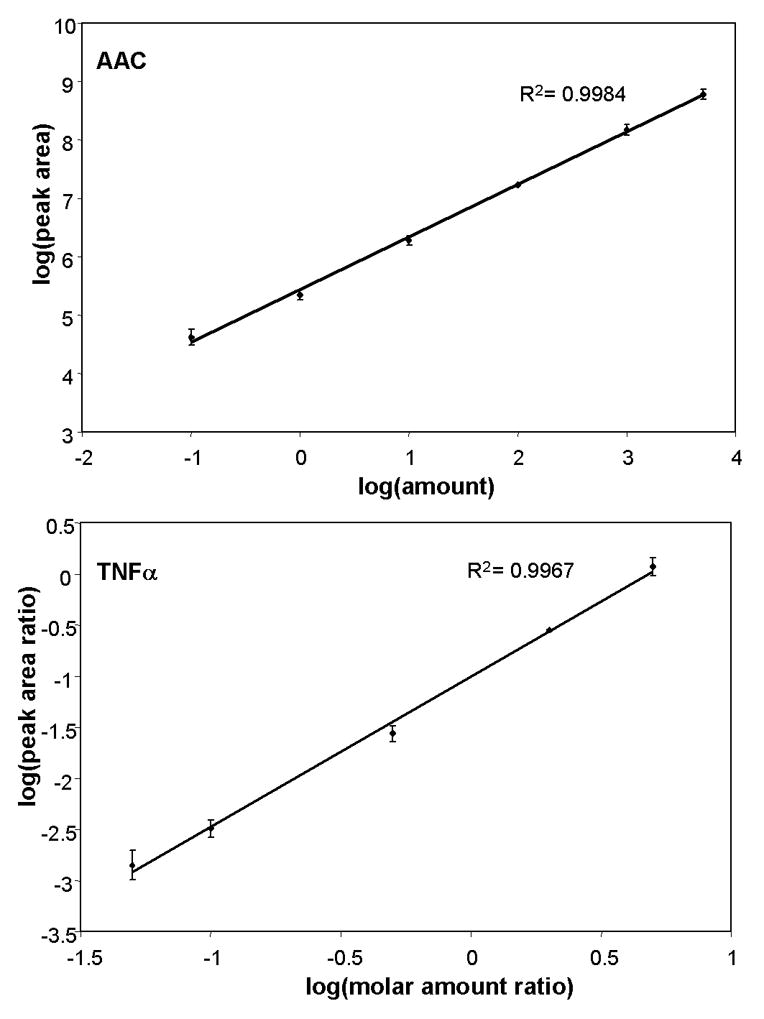
Calibration curve for quantifying heavy-labeled pure AAC and TNFα peptides. The ion signals for different amounts of pure, synthetic heavy peptides were measured using LC-MS, and used to determine the linear range of quantification on the linear ion trap instrument. Duplicate analyses were performed for each amount of peptide injected. Error bars show the range for each measurement.
The linear concentration range and limit of quantification for the SISCAPA assays are affected by antigen capture as well as instrument sensitivity. For example, if capture is inefficient and/or if ion suppression is significant in the eluate, the limit of quantification of a given target captured from serum will differ from the limit of quantification of the pure peptide. Hence, the linear concentration range and limit of quantification were also determined for AAC and TNFα following enrichment from serum on antibody-coated magnetic beads. Human serum was depleted of six abundant proteins and proteolyzed with trypsin. Depletion of serum has been shown to improve reproducibility of quantification[16]. Since AAC can be detected at its endogenous level, the heavy-labeled monitor peptide concentration was varied and the amount of serum was kept constant, thus allowing us to use the endogenous peptide as an internal standard. For the TNFα peptide (endogenous level below the limit of detection), synthetic (light) monitor peptide was spiked into serum at increasing concentrations keeping heavy-labeled peptide concentrations constant (for use as an internal standard). For AAC and TNFα, monitor peptides were subjected to capture by antibody coated beads and elution in 5% acetic acid. The eluates were analyzed by LC-MS and the peak area response from the extracted ion chromatogram used to construct a calibration curve (Fig. 3). Overall, the response for captured peptides was linear (R2 = 0.9969 and 0.9946 for AAC, TNFα) over about 2.5 orders of magnitude with limits of quantification around 1 fmol (~1 pg) and 10 fmol (~14 pg) of peptide on column for AAC and TNFα, respectively. As expected, the linear concentration range was greater and the limits of quantification were generally lower using pure peptides compared to captured peptides, since non-target peptides in the eluates cause some degree of ion suppression.
Figure 3.
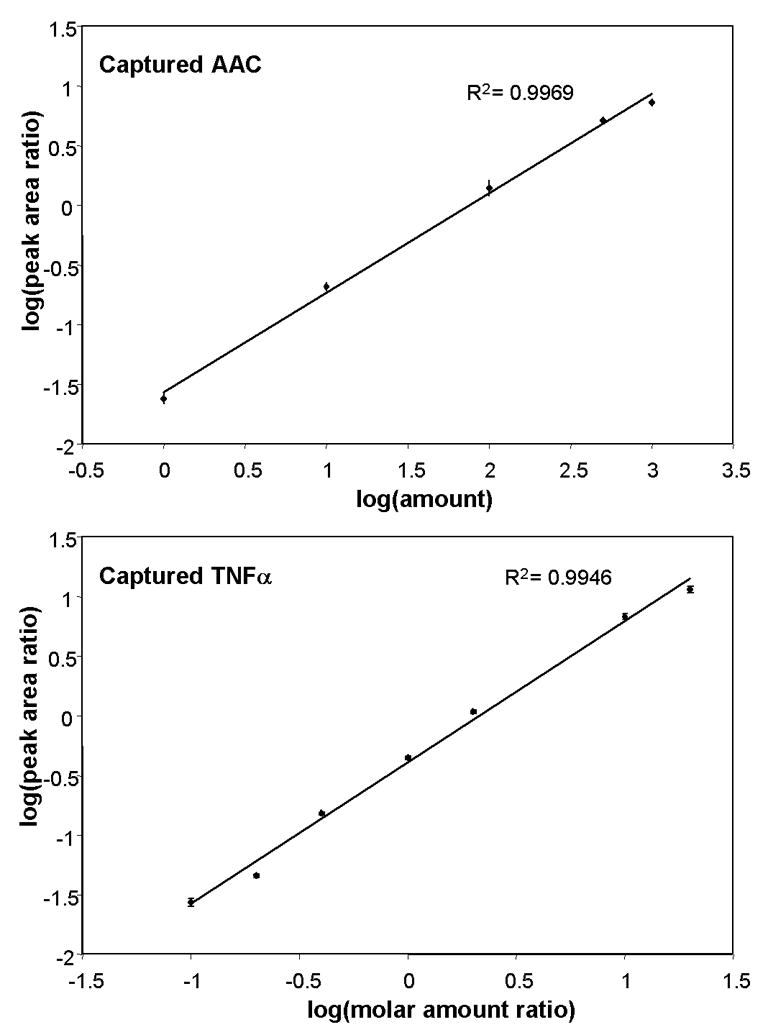
Calibration curve for quantifying enriched AAC and TNFα peptides. The ion signal ratio for different amounts of peptides spiked into serum and captured on antibody-coated beads were measured using LC-MS and used to determine the linear range of quantification. For AAC, the ratio of heavy peptide:light peptide (endogeneous) was determined from the extracted ion chromatograms and plotted versus amount of AAC spiked-in. For TNFα, the ratio of light peptide: heavy peptide was determined from the extracted ion chromatograms and plotted versus the expected molar ratio. Three replicate measurements were performed for each target. Error bars show the standard deviation of the measurements. The amounts of the peptides used for the assay characterization studies (Tables 1, 2) were selected at the same points along these curves, within the linear range of the instrument.
For the SISCAPA methodology to be of utility for testing candidate biomarkers, it must be able to quantify analytes present at ≤ ng/mL, since this is the concentration range of many known diagnostic markers. AAC is present at significantly higher concentrations (endogenous concentration 0.3–0.6 mg/mL[29]). Hence, we chose to demonstrate the capabilities of the SISCAPA-MS assay on TNFα, where the endogenous level (<10 pg/mL) was not detected by the SISCAPA assay (data not shown). 500 pg of TNFα 12C monitor peptide (equivalent to ~12 ng protein) was spiked into 1 mL of depleted and digested serum, captured by anti-TNFα magnetic beads, and the entire eluate injected for LC-MRM-MS analysis (8 μL). The resulting MRM chromatogram had a signal-to-noise > 100. Therefore, we conclude that our optimized platform can achieve limits of quantification for proteins on the order of ng/mL, sufficient to detect many biomarkers at physiologic levels (e.g. PSA, CEA, and AFP present at ng/mL).
Reproducibility and accuracy of peptide quantification
To evaluate the reproducibility of LC-MS measurements per se, three independent measurements were made on identical antigen capture samples for each of the study peptides, AAC and TNFα. A single capture was performed for each target, and each of the eluates was subjected to LC-MS analysis three times to determine reproducibility of measurements. The measured concentration, percent relative error (accuracy), and coefficients of variation (precision) are presented in Table 1. We find that the measurement is highly reproducible; CVs for the target peptides were below 10%. Additionally, the accuracy of the measurements was within 20% of the expected value.
Table 1.
Accuracy and precision for SISCAPA-MS analysis of TNFα target peptides spiked into human serum at different concentrations. The mean concentration, accuracy (percent relative error, %RE), and precision (percent coefficient of variation, %CV) are presented. Each measurement was performed in triplicate (n = 3).
| TNFα (μg/mL) | |||||||
|---|---|---|---|---|---|---|---|
| 2.32 | 4.63 | 9.26 | 23.1 | 46.3 | 231 | 463 | |
| Mean | 2.38 | 3.66 | 10.1 | 24.7 | 53.1 | 251 | 390 |
| Accuracy (%RE) | 2.6 | −20.9 | 9.2 | 7.0 | 14.7 | 8.4 | −15.9 |
| Precision (%CV) | 6.3 | 3.3 | 2.0 | 1.1 | 2.5 | 4.7 | 5.4 |
Having confirmed the reproducibility of the measurements, we next evaluated reproducibility of the antibody capture protocol. The heavy-labeled monitor peptide of AAC was captured three independent times from different serum aliquots at three spiked concentrations (keeping serum concentration the same). Eluates were analyzed by LC-MS, and the ratio of heavy peptide to light peptide was determined from the extracted ion chromatograms. The measured concentrations, percent relative error, and coefficients of variation for the replicate captures are presented in Table 2. The total mean, accuracy, and precision for captures from all three days are also shown in the table. Overall, the total CVs for the three capture experiments are excellent and range from 8% to 14%. Accuracy for the repeat capture experiments is also good and ranges from −18% to 8% relative error. Based on the known amount of spiked-in isotopically labeled standards, we empirically determined the concentrations of the endogenous AAC peptide to be 0.1 μM. The reported mean concentration of AAC in human plasma is 7 μM[29]. Although our empiric determinations are lower than that reported in the literature, pooling or processing of the reference serum used in our study (which was derived from many individuals) may have altered the concentrations, and errors in amino acid analysis could contribute to inaccuracy. Additionally, incomplete trypsin digestion could also contribute to errors in quantification. We have determined our trypsin protocol to be suitable for digestion of serum (see Supplemental Material); however, acidic residues adjacent to trypsin cleavage sites within monitor peptides may predispose to incomplete digestion. This possibility highlights a great need for internal standards to determine trypsin digestion completion. Nonetheless, based on the results presented in Tables 1 and 2 we conclude that the capture and subsequent LC-MS measurements show sufficient precision and accuracy for screening biomarker candidates[30].
Table 2.
Quantification statistics for SISCAPA-MS quantification of AAC target peptide spiked into human serum and captured with immobilized anti-peptide antibodies. The mean concentration, accuracy (percent relative error, %RE), precision (percent coefficient of variation, %CV), and number of replicates (independent captures from different aliquots of serum) for the measurements (n) are presented for quantification performed on three separate days. The total mean, accuracy, and precision were calculated using all measurements from the independent captures.
| Day | AAC (μg/mL) | |||
|---|---|---|---|---|
| 35.8 | 17.9 | 35.8 | ||
| 1 | Mean | 3.70 | 17.5 | 27.4 |
| Accuracy (%RE) | 3.3 | −2.4 | −23.4 | |
| Precision (%CV) | 4.4 | 4.1 | 2.6 | |
| n | 5 | 3 | 3 | |
| 2 | Mean | 4.40 | 20.3 | 29.4 |
| Accuracy (%RE) | 22.8 | 13.6 | −17.9 | |
| Precision (%CV) | 3.7 | 2.9 | 5.6 | |
| n | 3 | 3 | 3 | |
| 3 | Mean | 3.03 | 20.6 | 34.4 |
| Accuracy (%RE) | −15.5 | 15.0 | −3.9 | |
| Precision (%CV) | 6.8 | 0.6 | − | |
| n | 2 | 2 | 1 | |
| Total | Mean | 3.77 | 19.3 | 29.3 |
| Accuracy (%RE) | 5.4 | 7.9 | −18.2 | |
| Precision (%CV) | 14.0 | 8.4 | 9.2 | |
| n | 10 | 8 | 7 | |
Assessment of enhancement of ion signals of captured target peptides
We next assessed how well our optimized SISCAPA protocols enhance target peptide ion signals over non-target peptides in serum. We began by studying AAC, capable of being seen by mass spectrometry at its endogenous serum concentration (0.3–0.6 mg/mL[29]).
The AAC 12C monitor peptide from the endogenous serum protein AAC was captured from 20 μL processed (depleted and digested) serum on antibody-coated beads and eluted into 20 μL 5% acetic acid. Both the starting serum digest and the eluates following capture were analyzed by LC-MS. It is important to note that equivalent amounts of peptide were injected for LC-MS analysis for each sample (before and after capture). Therefore, changes in signal intensity for the target analyte are solely due to reduction in ion suppression (by high abundance serum peptides) and are not due to concentration of the peptide following capture enrichment. Figure 4a shows the total ion chromatogram for a 30 minute gradient LC-MS analysis of the total serum digest. As expected, there is considerable background ion content in the serum run. The endogenous AAC peptide was detected at trace level when analyzed by LC-MRM-MS (Fig. 4b) and its identity verified by a full MS/MS spectrum (Fig. 4c). Figure 4d shows the total ion chromatogram of the 30 minute LC-MS analysis of the anti-AAC antibody-captured sample. Notice there is a considerable decrease in background ions and the AAC peptide is the predominant peak in the chromatogram. The LC-MRM-MS chromatographic trace of the anti-AAC antibody-enriched sample is shown in Figure 4e and the AAC peptide is again verified by a full MS/MS spectrum (Fig. 4f). A large increase in the relative signal-to-noise following antibody capture and elution is apparent from examining these traces. This is due to elimination of background species which contribute to ion suppression of the AAC peptide in serum.
Figure 4.
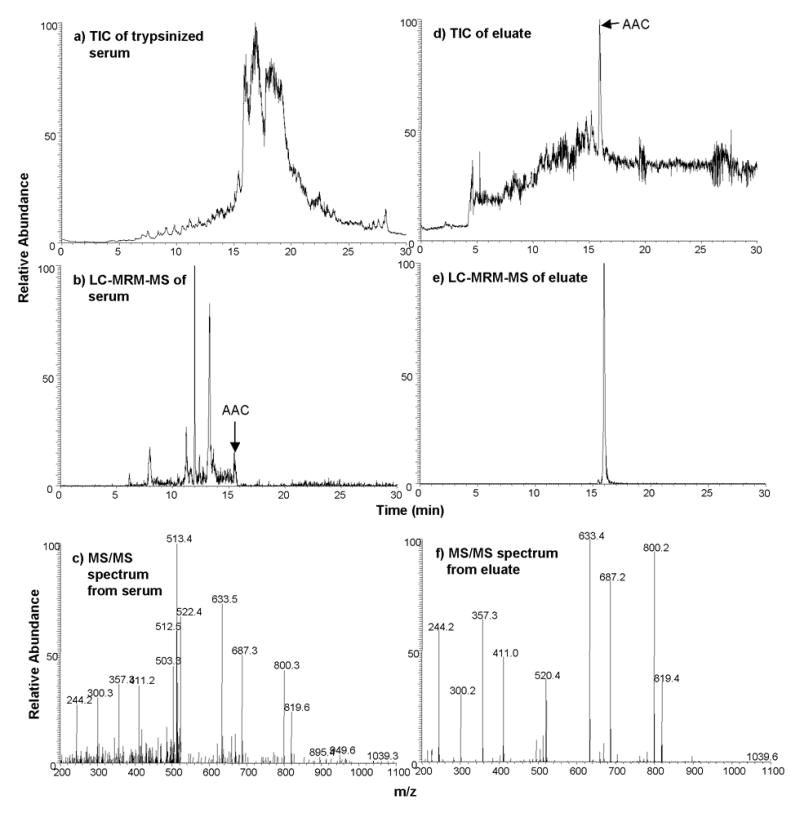
Enhancement of endogenous AAC ion signals following antibody enrichment assessed by LC-MS. (a) 30 minute LC-MS analysis of 20 μL depleted and digested human serum. (b) 30 min LC-MRM-MS analysis of AAC from same serum digest. (c) MS/MS spectrum of AAC from serum digest. (d) 30 min LC-MS analysis of antibody-enriched AAC from 20 μL depleted and digested human serum. (e) 30 min LC-MRM-MS analysis of antibody-captured AAC. (f) MS/MS spectrum of AAC from antibody-capture. Equivalent amounts of peptide were injected for LC-MS analysis for each sample (before and after capture).
To evaluate the increase in analyte signal relative to other components in the sample, we normalized the analyte signal to the total ion current (TIC) of the starting serum digest and compared this to the normalized analyte signal of the eluates following antibody capture. Based on the ratios of the analyte signal:TIC from the starting serum digest vs. the eluates, we estimate that antibody capture resulted in 1,453-fold enhancement of ion signal for the AAC peptide. Note that this is a conservative estimate of actual enrichment of the target peptide; the plasma sample in which these measurements were made had already been depleted of six abundant proteins using the depletion column, which is expected to contribute an additional ~5-fold enrichment[31].
We next studied a lower abundance target, TNFα. The endogenous plasma level of TNFα is <10 pg/mL, which is below the limit of detection of the instrument (when measured directly in plasma). To determine the enhancement of TNFα peptide by antibody capture and LC-MS, we spiked TNFα (light) monitor peptide (along with heavy stable isotope standard) into depleted human serum digest at a concentration of 2.3 μg/mL. The spiked-in peptides were captured by anti-TNFα antibody and analyzed by LC-MS. As illustrated in Figure 5 (a–f), a 573-fold enhancement of the TNFα peptide ion signal was achieved by antibody capture and MRM detection (monitoring two transitions).
Figure 5.
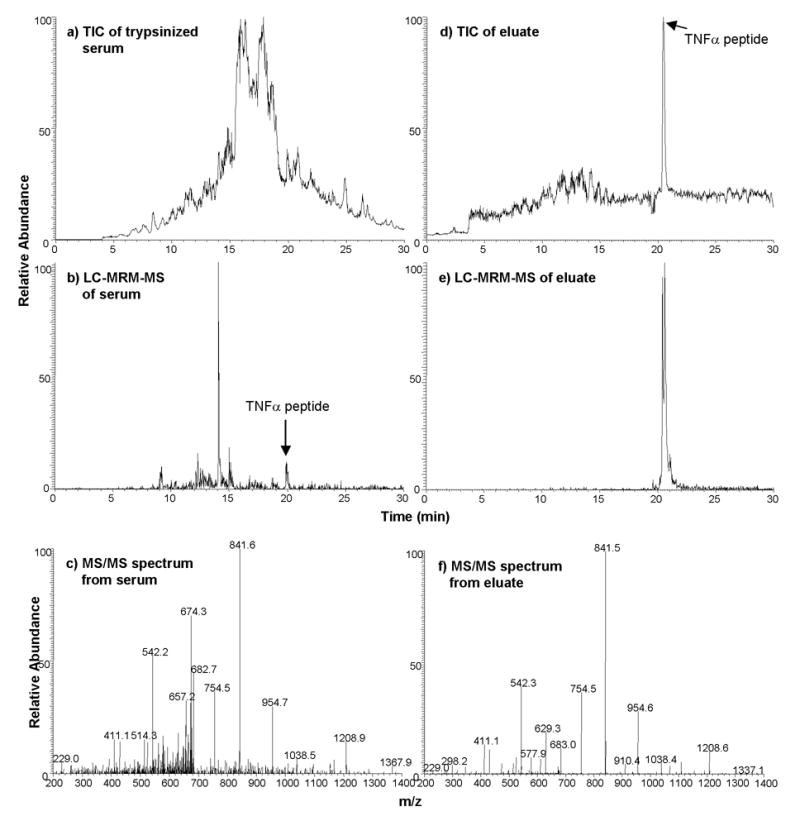
Enhancement of spiked TNFα ion signals following antibody enrichment assessed by LC-MS. (a) 30 minute LC-MS analysis of 20 μL depleted and digested human serum. (b) 30 min LC-MRM-MS analysis of TNFα from same serum digest. (c) MS/MS spectrum of TNFα from serum digest. (d) 30 min LC-MS analysis of antibody-enriched TNFα from 20 μL depleted and digested human serum. (e) 30 min LC-MRM-MS analysis of antibody-captured TNFα. (f) MS/MS spectrum of TNFα from antibody-capture. Equivalent amounts of peptide were injected for LC-MS analysis for each sample (before and after capture).
Discussion
There is tremendous opportunity to transform medicine through more and improved diagnostic biomarkers. With the application of genomics technologies, the biomarker discovery pipeline now resembles the drug discovery pipeline; there are far more candidates than can possibly be tested in reasonable time with existing resources, and there are no proven ways to prioritize them[32, 33]. Hence, although there is widespread acceptance[34] that better and more diagnostics will greatly impact patient care, the number of new diagnostics being approved for clinical use is small and rapidly vanishing[7]. We urgently need technologies that enable measurement of hundreds of biomarker candidates in thousands of clinical samples to allow rapid determination of the potential of candidates, individually or in panels, to impact clinical care.
SISCAPA has several significant advantages over the current gold standard (ELISA) in filling this gap. For instance, the need to generate pairs of antibodies compatible for sandwich assays is largely responsible for the high cost and long lead time for developing ELISAs. SISCAPA minimizes this burdensome step by requiring one antibody, not two as with the ELISA. (The mass spectrometer effectively acts as the second antibody, with exquisite specificity.) Furthermore, antibody reagents will be easier to generate for SISCAPA vs. ELISA, since there is: 1) no constraint to target non-overlapping epitopes, 2) no requirement for the antibodies to recognize the intact protein, and 3) more relaxed requirements for antibody selectivity because of the highly selective nature of the mass spectrometric analysis. For candidates showing initial promise, it may be possible to enhance sensitivity with some investment in further optimizing the assay conditions for specific antibody-antigen pairs. Likewise, for lower abundance targets where sensitivity must be optimized, carefully screening for highest affinity anti-peptide antibodies should provide benefit. (Note the difference in enrichments achieved with the TNFα vs. the AAC anti-peptide antibodies in our experiments.) SISCAPA also provides benefits over ELISA in potential throughput. The SISCAPA-MS approach can simultaneously monitor multiple targets within a single sample while the standard ELISA can generally only quantify one target per sample, making SISCAPA desirable in terms of both throughput and sample consumption. Recent data suggest that at least 200 assays could be multiplexed in a single LC-MRM-MS run[16]. Additionally, the magnetic bead-based platform described in this report is amenable to automation and high throughput, and its flexibility also allows for capture from large sample volumes allowing for further enrichment by volume reduction on the order of 102–103, extending analysis to beyond the ng/mL range, sufficient for quantifying low abundance biomarkers. Hence, we believe that SISCAPA holds great promise for facilitating the biomarker discovery pipeline.
In this study we demonstrate the capabilities of the linear ion trap for quantitative assays using an extracted ion chromatogram. This mode of operation has been used for numerous quantitative proteomics studies[25–28]. Although the linear ion trap is unlikely to replace the triple quadrupole instrument (using SRM/MRM) for MS-based quantitation in the clinical setting, there are advantages of using the ion trap for early stage testing of candidate biomarkers. First, many proteomic laboratories doing MS-based biomarker discovery are using ion trap instruments and do not have ready access to triple quadrupole instruments. Second, using the same instrument for discovery and assay development facilitates choosing monitor peptides. For instance, the biomarker discovery dataset can be used to select monitor peptides that were empirically observed to ionize efficiently and produce robust fragmentation. In contrast, if the discovery data were generated on an ion trap and validation were done on a triple quadrupole, the chosen monitor peptides may not be the most optimum with respect to ionization or fragmentation, increasing the risk of assay failure. Finally, in initial validation and screening of biomarker candidates, the capability to rapidly and unambiguously verify the peptide identity (via a full MS/MS spectrum) greatly increases confidence in the data.
For this work, we adopted a scan sequence to quantify and conclusively verify the peptide target. The scan sequence consisted of a full scan MS spectrum followed by two full MS/MS spectra acquired at the mass/charge of the (light) monitor peptide and the heavy labeled monitor peptide (internal standard). The MS spectrum permits quantification of the analytes by using an extracted ion chromatogram and provides information on the background levels of other components in the sample. The full MS/MS spectra allow for unambiguous identification of the peptide. Taking into account the identification of the targeted peptide (via the MS/MS spectrum) in addition to the co-elution of the heavy-labeled internal standard peptide allows for conclusive verification of peptide identity. Additionally, integrating the peak areas of one or more fragment ions from the MS/MS spectra can be used for SRM/MRM studies. A recent study demonstrated the effectiveness of the linear ion trap operated in such a mode[24].
The capability for multiplexing many peptides into a single run is an important advantage in using SISCAPA-MS. In this paper, the entire scan sequence requires less than one second (about 200 ms each for MS scan and MS/MS scans), allowing for sufficient points across an elution peak for accurate quantification, and uniquely performs the desired analysis by providing full MS/MS spectra of the analytes. Dividing a run into multiple time segments will allow for monitoring multiple peptides. The number of possible time segments depends on chromatography and elution times of the peptides. Based on the scan sequence we have presented, one or two peptides can be monitored per time segment, allowing for a maximum of about 5 peptides to be monitored in a short run. A recent study using a single quadrupole ion trap for MRM studies (without acquiring a full MS/MS scan for peptide verification) reports scan times of about 150 ms, suggesting up to 10 peptides can be monitored in a 10-minute run[24]. In contrast, a triple quadrupole operated in MRM mode at only 18 ms per transition may be capable of monitoring significantly more peptides (e.g. up to 50–100) in a 10-minute run depending on the number of time segments[16]. Ultimately, a triple quadrupole mass spectrometer is the instrument of choice for such experiments; however, we have demonstrated the utility of an ion trap for quantitative studies, as mentioned above.
Another important aspect of the assay is complete proteolytic digestion. Because the assay depends on the enrichment and measurement of peptides, incomplete proteolysis can significantly affect detection limits and accuracy. Thus, enzymatic digestion protocols must be strictly optimized, controlled, and monitored. To this end, there is a great need for development of a proteolysis quality control (QC) standard. Such a standard could include spiking a heavy isotope-labeled synthetic polypeptide containing internal trypsin cleavage sites into serum to measure the efficiency of digestion of an endogenous or spiked polypeptide.
One can now envision an efficient, staged approach to building quantitative assays for testing biomarker candidates. For example, initially quantitative LC-SRM-MS, without antibody enrichment, could be attempted to detect peptides derived from the candidate protein in trypsin-digested plasma samples that have been depleted of abundant proteins; many moderate and high abundance candidates will likely be detectable, down to the ~μg/mL range, without the need to generate antibodies[16]. For those candidates not detectable without enrichment, the SISCAPA technology can be used. Our data indicate that candidates in the ng/mL range can be quantified on the platform described herein. For very low abundance candidates (≪ ng/mL) or for candidates refractory to the above approaches, it may still be necessary to generate an ELISA, which may achieve sensitivity in the pg/mL range with good “sandwich antibodies.” Emerging immuno-PCR adaptation of ELISAs promises even higher sensitivities[35, 36].
In conclusion, we have developed a generalizable, automatable peptide enrichment and quantification platform for high-throughput testing of biomarker candidates. This platform should provide a desperately needed bridging technology between development-intensive biomarker discovery and clinical application. Furthermore, the techniques presented here are amenable to protein quantification in any biological investigation.
Supplementary Material
Acknowledgments
The authors would like to thank John Keane, Richard Ivey, and Regine Schoenherr for advice and comments on this manuscript. This work was funded by NCI subcontract 23XS144A as well as by generous gifts from the Canary Foundation, the Keck Foundation, and the Paul G. Allen Family Foundation.
Footnotes
Category assignment: Peptides, amino acids, and amino acid derivatives
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Clemons M, Danson S, Howell A. Tamoxifen (“Nolvadex”): a review. Cancer Treat Rev. 2002;28:165–80. doi: 10.1016/s0305-7372(02)00036-1. [DOI] [PubMed] [Google Scholar]
- 2.Haber DA, Bell DW, Sordella R, Kwak EL, Godin-Heymann N, Sharma SV, Lynch TJ, Settleman J. Molecular Targeted Therapy of Lung Cancer: EGFR Mutations and Response to EGFR Inhibitors. Cold Spring Harb Symp Quant Biol. 2005;70:419–26. doi: 10.1101/sqb.2005.70.043. [DOI] [PubMed] [Google Scholar]
- 3.Ligibel JA, Winer EP. Trastuzumab/chemotherapy combinations in metastatic breast cancer. Semin Oncol. 2002;29:38–43. doi: 10.1053/sonc.2002.34054. [DOI] [PubMed] [Google Scholar]
- 4.Shawver LK, Slamon D, Ullrich A. Smart drugs: tyrosine kinase inhibitors in cancer therapy. Cancer Cell. 2002;1:117–23. doi: 10.1016/s1535-6108(02)00039-9. [DOI] [PubMed] [Google Scholar]
- 5.Fitzgibbons PL, Page DL, Weaver D, Thor AD, Allred DC, Clark GM, Ruby SG, O’Malley F, Simpson JF, Connolly JL, Hayes DF, Edge SB, Lichter A, Schnitt SJ. Prognostic factors in breast cancer. College of American Pathologists Consensus Statement 1999. Arch Pathol Lab Med. 2000;124:966–78. doi: 10.5858/2000-124-0966-PFIBC. [DOI] [PubMed] [Google Scholar]
- 6.Ross JS, Fletcher JA. The HER-2/neu oncogene in breast cancer: prognostic factor, predictive factor, and target for therapy. Stem Cells. 1998;16:413–28. doi: 10.1002/stem.160413. [DOI] [PubMed] [Google Scholar]
- 7.Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Mol Cell Proteomics. 2002;1:845–67. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- 8.Want EJ, Cravatt BF, Siuzdak G. The expanding role of mass spectrometry in metabolite profiling and characterization. Chembiochem. 2005;6:1941–51. doi: 10.1002/cbic.200500151. [DOI] [PubMed] [Google Scholar]
- 9.Chace DH, Kalas TA. A biochemical perspective on the use of tandem mass spectrometry for newborn screening and clinical testing. Clin Biochem. 2005;38:296–309. doi: 10.1016/j.clinbiochem.2005.01.017. [DOI] [PubMed] [Google Scholar]
- 10.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray ionization for mass spectrometry of large biomolecules. Science. 1989;246:64–71. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- 11.Karas M, Hillenkamp F. Laser desorption ionization of proteins with molecular masses exceeding 10,000 daltons. Anal Chem. 1988;60:2299–301. doi: 10.1021/ac00171a028. [DOI] [PubMed] [Google Scholar]
- 12.Barr JR, Maggio VL, Patterson DG, Jr, Cooper GR, Henderson LO, Turner WE, Smith SJ, Hannon WH, Needham LL, Sampson EJ. Isotope dilution--mass spectrometric quantification of specific proteins: model application with apolipoprotein A-I. Clin Chem. 1996;42:1676–82. [PubMed] [Google Scholar]
- 13.Gerber SA, Rush J, Stemman O, Kirschner MW, Gygi SP. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc Natl Acad Sci U S A. 2003;100:6940–5. doi: 10.1073/pnas.0832254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuster B, Schirle M, Mallick P, Aebersold R. Scoring proteomes with proteotypic peptide probes. Nat Rev Mol Cell Biol. 2005;6:577–83. doi: 10.1038/nrm1683. [DOI] [PubMed] [Google Scholar]
- 15.Aguiar M, Masse R, Gibbs BF. Mass spectrometric quantitation of C-reactive protein using labeled tryptic peptides. Anal Biochem. 2006;354:175–81. doi: 10.1016/j.ab.2006.03.037. [DOI] [PubMed] [Google Scholar]
- 16.Anderson L, Hunter CL. Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol Cell Proteomics. 2006;5:573–88. doi: 10.1074/mcp.M500331-MCP200. [DOI] [PubMed] [Google Scholar]
- 17.Barnidge DR, Goodmanson MK, Klee GG, Muddiman DC. Absolute quantification of the model biomarker prostate-specific antigen in serum by LC-Ms/MS using protein cleavage and isotope dilution mass spectrometry. J Proteome Res. 2004;3:644–52. doi: 10.1021/pr049963d. [DOI] [PubMed] [Google Scholar]
- 18.Berna M, Schmalz C, Duffin K, Mitchell P, Chambers M, Ackermann B. Online immunoaffinity liquid chromatography/tandem mass spectrometry determination of a type II collagen peptide biomarker in rat urine: Investigation of the impact of collision-induced dissociation fluctuation on peptide quantitation. Anal Biochem. 2006 doi: 10.1016/j.ab.2006.05.018. [DOI] [PubMed] [Google Scholar]
- 19.Kuhn E, Wu J, Karl J, Liao H, Zolg W, Guild B. Quantification of C-reactive protein in the serum of patients with rheumatoid arthritis using multiple reaction monitoring mass spectrometry and 13C-labeled peptide standards. Proteomics. 2004;4:1175–86. doi: 10.1002/pmic.200300670. [DOI] [PubMed] [Google Scholar]
- 20.Pan S, Zhang H, Rush J, Eng J, Zhang N, Patterson D, Comb MJ, Aebersold R. High throughput proteome screening for biomarker detection. Mol Cell Proteomics. 2005;4:182–90. doi: 10.1074/mcp.M400161-MCP200. [DOI] [PubMed] [Google Scholar]
- 21.Wu SL, Amato H, Biringer R, Choudhary G, Shieh P, Hancock WS. Targeted proteomics of low-level proteins in human plasma by LC/MSn: using human growth hormone as a model system. J Proteome Res. 2002;1:459–65. doi: 10.1021/pr025537l. [DOI] [PubMed] [Google Scholar]
- 22.Anderson NL, Anderson NG, Haines LR, Hardie DB, Olafson RW, Pearson TW. Mass spectrometric quantitation of peptides and proteins using Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA) J Proteome Res. 2004;3:235–44. doi: 10.1021/pr034086h. [DOI] [PubMed] [Google Scholar]
- 23.Safarik I, Safarikova M. Magnetic techniques for the isolation and purification of proteins and peptides. Biomagn Res Technol. 2004;2:7. doi: 10.1186/1477-044X-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin S, Shaler TA, Becker CH. Quantification of intermediate-abundance proteins in serum by multiple reaction monitoring mass spectrometry in a single-quadrupole ion trap. Anal Chem. 2006;78:5762–7. doi: 10.1021/ac060613f. [DOI] [PubMed] [Google Scholar]
- 25.Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17:994–9. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 26.Gygi SP, Rist B, Griffin TJ, Eng J, Aebersold R. Proteome analysis of low-abundance proteins using multidimensional chromatography and isotope-coded affinity tags. J Proteome Res. 2002;1:47–54. doi: 10.1021/pr015509n. [DOI] [PubMed] [Google Scholar]
- 27.Molloy MP, Donohoe S, Brzezinski EE, Kilby GW, Stevenson TI, Baker JD, Goodlett DR, Gage DA. Large-scale evaluation of quantitative reproducibility and proteome coverage using acid cleavable isotope coded affinity tag mass spectrometry for proteomic profiling. Proteomics. 2005;5:1204–8. doi: 10.1002/pmic.200400994. [DOI] [PubMed] [Google Scholar]
- 28.Sakai J, Kojima S, Yanagi K, Kanaoka M. 18O-labeling quantitative proteomics using an ion trap mass spectrometer. Proteomics. 2005;5:16–23. doi: 10.1002/pmic.200300885. [DOI] [PubMed] [Google Scholar]
- 29.Skeel A, Leonard EJ. alpha 1-Antichymotrypsin is the human plasma inhibitor of macrophage ectoenzymes that cleave pro-macrophage stimulating protein. J Biol Chem. 2001;276:21932–7. doi: 10.1074/jbc.M100652200. [DOI] [PubMed] [Google Scholar]
- 30.U.S. Department of Health and Human Services, Guidance for Industry. Bioanalytical Method Validation. 2001. [Google Scholar]
- 31.Brand J, Haslberger T, Zolg W, Pestlin G, Palme S. Depletion efficiency and recovery of trace markers from a multiparameter immunodepletion column. Proteomics. 2006;6:3236–42. doi: 10.1002/pmic.200500864. [DOI] [PubMed] [Google Scholar]
- 32.Butcher SP. Target discovery and validation in the post-genomic era. Neurochem Res. 2003;28:367–71. doi: 10.1023/a:1022349805831. [DOI] [PubMed] [Google Scholar]
- 33.Lindsay MA. Target discovery. Nat Rev Drug Discov. 2003;2:831–8. doi: 10.1038/nrd1202. [DOI] [PubMed] [Google Scholar]
- 34.Hartwell L, Lander E. Report to National Cancer Advisory Board, NCAB Working Group on Biomedical Technology. 2005. [Google Scholar]
- 35.Adler M. Immuno-PCR as a clinical laboratory tool. Adv Clin Chem. 2005;39:239–92. [PubMed] [Google Scholar]
- 36.Niemeyer CM, Adler M, Wacker R. Immuno-PCR: high sensitivity detection of proteins by nucleic acid amplification. Trends Biotechnol. 2005;23:208–16. doi: 10.1016/j.tibtech.2005.02.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.