

Abstract
To examine the mechanism by which growth-stimulated pancreatic β-cells dedifferentiate, somatic cell fusions were performed between MIN6, a highly differentiated mouse insulinoma, and βlox5, a cell line derived from human β-cells which progressively dedifferentiated in culture. MIN6/βlox5 somatic cells hybrids underwent silencing of insulin expression and a marked decline in PDX1, NeuroD, and MafA, indicating that βlox5 expresses a dominant trans-acting factor(s) that represses β-cell differentiation. Expression of Hes1, which inhibits endocrine differentiation was higher in hybrid cells than in parental MIN6 cells. Hes6, a repressor of Hes1, was highly expressed in primary β-cells as well as MIN6, but was repressed in hybrids. Hes6 overexpression using a retroviral vector led to a decrease in Hes1 levels, an increase in β-cell transcription factors and partial restoration of insulin expression. We conclude that the balance of Notch activators and inhibitors may play an important role in maintaining the β-cell differentiated state.
Keywords: β-cell, insulin, cell fusion, differentiation, islet, somatic cell hybrids
INTRODUCTION
Primary β-cells stimulated to enter the cell cycle quickly dedifferentiate and lose insulin expression [1], suggesting a tight inverse linkage between β-cell growth and differentiation. This study aimed to determine the mechanism underlying this. There were two possibilities for the observed loss of β-cell differentiation: loss of factor(s) that act positively upon β-cell differentiation, and/or upregulation of factors that act negatively upon β-cell differentiation.
To distinguish between those possibilities, we generated somatic cell hybrids between βlox5, an immortal human cell line generated from adult pancreatic islets [2], that expressed insulin in early passage but lost expression with continuous culture, and MIN6, a mouse insulinoma line that retains high levels of insulin expression [3]. These hybrid cell lines were analysed for expression of insulin and other β-cell genes. Three possible outcomes were postulated. 1. If MIN6 cells express a transacting activator of β-cell differentiation that is absent in βlox5, hybrid genomes should be reprogrammed to activate the human insulin gene transcription program. 2. If βlox5 cells express a transacting repressor of β-cell differentiation absent in MIN6, hybrids genomes should lose mouse insulin gene transcription. 3. If insulin expression is regulated by cisacting elements, hybrids should retain mouse insulin gene expression and lack human insulin gene expression.
Examination of insulin gene expression in the hybrids were consistent with the expression of transacting repressors in βlox5 that acted on the β-cell genes in MIN6. Shutoff of those genes correlated with upregulation of Hes1, a transcriptional repressor in the Notch pathway. Expression of Hes6, a repressor of Hes1, resulted in upregulation of β-cell genes, including PDX-1 and insulin I. However, the activation was selective for the murine alleles, consistent with a role for cis-acting control as well.
RESULTS
Hybrid cells survive and grow in double hygromycin/neomycin selection medium
To generate somatic cell hybrids, G418-resistant βlox5 were fused with hygromycin-resistant MIN6 using polyethylene glycol (Supplementary Figure 1). Cells were then cultured continuously in G418/hygromycin selection medium. A fusion between G418-resistant and hygromycin resistant MIN6, done as a control for effects of fusion on differentiation, was efficient, with hundreds of colonies per plate. Surviving cells were pooled in an uncloned population designated MMC. Fusion of MIN6 cells with βlox5 was much less efficient, (one event per 1x106 human cells). Only five stable clones were generated, each from a different well. Each clone was cultured continuously through approximately 40 doublings. Three representative hybrid clones, BMC1-3, were further analysed.
Inhibition of mouse insulin gene transcription
Initial PCR analysis examined mouse and human insulin mRNA using species-specific primers. Since chromosome loss occurs in mouse-human cell fusions [4], leading to inter- and intra-clonal heterogeneity, we also used PCR to detect genomic DNA in hybrid and parental cells, ensuring the presence of all genes of interest (Supplemental Fig. 2). Thus, we confirmed that interspecies fusion had occurred, and that each hybrid clone contained the gene of interest, so that RNA gene transcript changes reflected a transcriptional event rather than chromosome loss.
As expected, parental MIN6 homologous fusions (MMC) retained high levels of mouse insulin I and II gene transcripts (Fig. 1A), indicating that cell fusion by itself was not detrimental. In contrast, neither βlox5 nor any mixed hybrid BMC clones expressed human insulin (Fig. 1A). Mouse insulin I and insulin II expression was abolished in the BMC clones, except for a low level of insI mRNA in BMC1. Thus, a gene or genes expressed in βlox5 appear to dominantly repress insulin gene expression.
Figure 1.
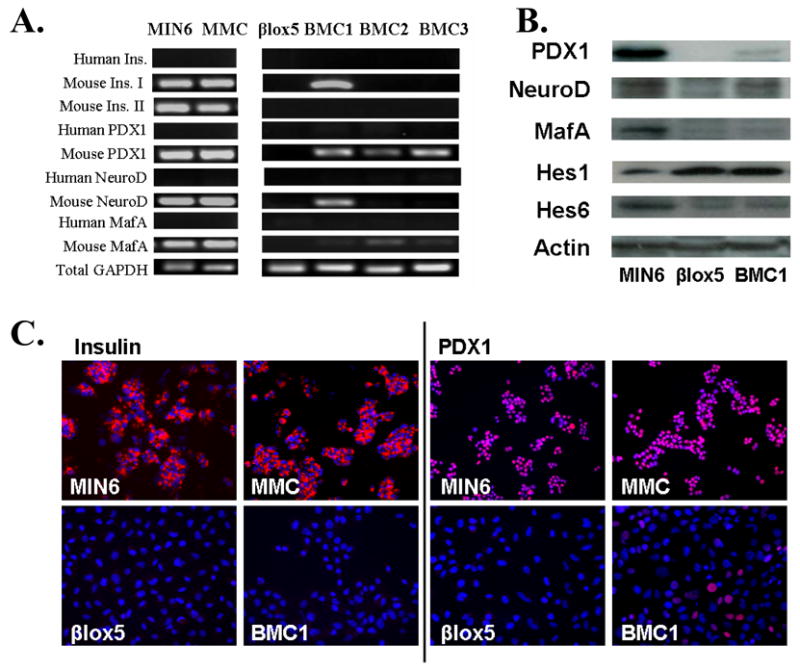
A. RT-PCR analysis of human and mouse insulin and insulin transcription factor mRNA. GAPDH was used to ensure that equal amounts of cDNA were used. B. Immunostaining for insulin (red in left panels) and PDX1 (red in right panels). Nuclei are labeled by DAPI (blue). C. Western blot analysis of PDX1, NeuroD, MafA, Hes1 and Hes6. Actin was used as a loading control.
Reprogramming of transcription factors in insulin-producing cells following cell fusion
Expression of PDX1, NeuroD and MafA was analyzed as they are important transactivators of insulin gene expression [5]. MIN6 and homologous Min6 MMC fused cells expressed high levels of mRNA for each of these factors (Fig. 1A), but neither βlox5 nor hybrids expressed mRNA for human PDX1, MafA, or NeuroD1 (Fig. 1A).
Each hybrid clone expressed detectable levels of the mouse PDX1 gene transcript, but the levels of expression were lower than in MIN6 or MMC cells (Fig. 1A). Mouse MafA was weakly detectable in each hybrid clone, while mouse NeuroD was detectable only in the hybrid BMC1 (Fig. 1A). Notably, the only hybrid clone expressing detectable mouse PDX1, NeuroD and MafA mRNA was BMC1, which was also the only clone retaining expression of mouse insulin I gene transcripts. Consistent with the gene expression data, BMC1 expressed PDX-1, NeuroD1, and MafA protein, but only at a low level, resembling the pattern in βlox5 much more closely than that in MIN6 (Fig. 1B).
Hybrid clones lose insulin and PDX1 immunostaining
To determine whether the low level of mouse INS1 and PDX-1 mRNA in BMC1 was due to homogeneous extinction by a dominant factor in βlox5, or whether there was heterogeneity in the BMC1 cells despite their having arisen from a single colony, immunostaining for those proteins was used. MIN6 and MMC cells all strongly expressed insulin, while neither βlox5 nor BMC1 displayed any insulin immunostaining, as expected (Fig. 1C).
Like insulin, PDX1 protein was strongly expressed in MIN6 and MMC cells and absent in βlox5 (Fig. 1C). However, the BMC1 hybrid had a small population of strongly PDX1 positive cells (Fig. 1C).
Notch activation is dominant in human-mouse somatic cell hybrids
Notch signalling plays an important role in pancreatic differentiation and cell fate decisions [6; 7; 8]. Overexpression of Notch1 leads to dedifferentiation of INS1 insulinoma cells as assessed by reduction of both PDX1 and insulin gene transcription [9]. Hes1 also mediates repression of insulin gene transcription in dexamethasone treated HIT insulinoma cells [10]. Thus it was a logical candidate for a regulatory role in the reprogramming of the insulin gene program in hybrid cells. Hes1 is the key downstream effector of the Notch signalling pathway in β-cells [11]. To determine if Hes1 was significant in regulating the differentiation state of pancreatic cell lines, we performed quantitative real-time RT-PCR on RNA from the parental cell lines MIN6 and βlox5, revealing that the non-insulin-producing βlox5 cells had Hes1 transcript levels 10-fold higher than insulin-producing MIN6 cells relative to GAPDH, using a set of primers that detected both human and mouse GAPDH (Fig. 2A). Because Hes1 is controlled at both transcriptional and post-transcriptional levels, Western blots for Hes1 were performed, showing that the βlox5 and hybrid cells had considerably higher levels of Hes1 protein than MIN6 cells (Fig. 1B).
Figure 2.
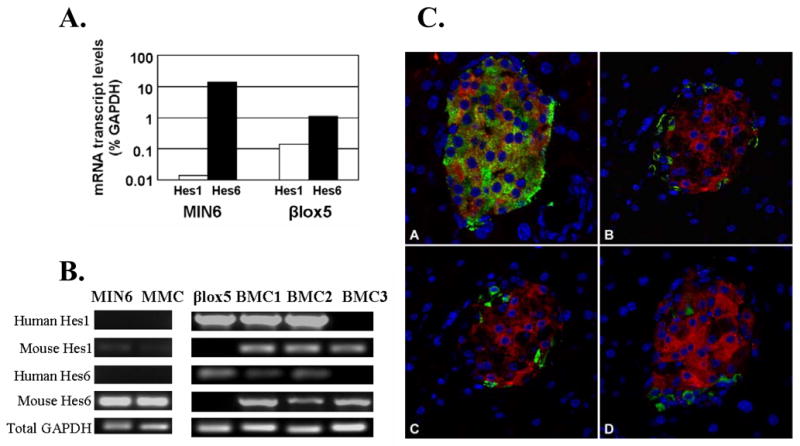
A. Quantitative real-time RT-PCR of Hes1 and Hes6 mRNA in parental cell lines.
B. RT-PCR analysis of human and mouse Hes1 and Hes6 mRNA.
C. Hes6 is highly expressed in β-cells. Immunohistochemistry for lacZ (red), and pancreatic hormones (red): A. insulin. B glucagon. C somatostatin. D. pancreatic polypeptide was done in the Hes6taulacZ knockin mouse (20).
Based on the pattern of Hes1 expression in the parental cell lines, Notch signalling became a candidate for a dominant factor that repressed β-cell differentiation in the hybrids. If so, Hes1 levels in the BMC hybrids should be similar to those in βlox5 rather than Min6. RT-PCR analysis (Fig. 2B) and Western blot analysis (Fig. 1B), demonstrated that the state of Notch activation in βlox5 is dominant over that in Min6.
The Notch pathway inhibitor Hes6 is highly expressed in islets and is differentially expressed in βlox5, Min6, and hybrids
To determine whether Hes1 was playing an active role in the nuclear reprogramming and repression of differentiation in the hybrid clones, it was necessary to repress Hes1 function. In a number of cell systems, Hes6 inhibits Hes1. The genes share substantial sequence similarity to Hes1 except in the loop region within the bHLH domain. [12; 13]. Unlike Hes1, Hes6 does not bind to N-box sequences and in fact has been found to act as a potent repressor of Hes1 by multiple mechanisms, including inhibiting Hes1 binding to coactivators and by targeting Hes1 protein for degradation [13].
The role of Hes6 in the pancreas has not been studied in depth. Immunohistochemistry revealed that Hes6 is highly expressed in islets. Strikingly, it is limited to β-cells, being absent in α-, δ-, and PP cells (Fig. 2C). Consistent with a role for Hes6 in maintaining β-cell differentiation, both Western blot (Fig. 1B) and quantitative RT-PCR (Fig. 2A) revealed that insulin-producing MIN6 cells expressed a higher level of Hes6 than did βlox5 cells.
Both mouse and human Hes6 mRNA levels dropped substantially in the hybrids compared with the parental cells (Fig. 2B) and was consistent with Western blot analysis, which found that βlox5 and BMC1 cells had markedly lower levels of Hes6 than MIN6 (Fig. 1B).
Transduction with Hes6 retrovirus restores insulin expression in human/mouse hybrids
The Hes6 and Hes1 expression patterns were consistent with, but did not prove, that Hes1 and Hes6 were causal in modulating β-cell differentiation. If they were, we hypothesized that elevating the level of Hes6 would reverse the dedifferentiation observed in the human/mouse hybrids. To express Hes6, its cDNA was inserted into a retroviral vector. In βlox5, quantitative RT-PCR analysis demonstrated that the level of Hes6 mRNA increased substantially (Fig. 3A), and immunostaining with an anti-Hes6 antibody revealed that the level of Hes6 protein in infected cells was markedly increased compared with uninfected cells (Fig. 3B), but there was no significant effect on β-cell differentiation. In Min6, Hes6 overexpression decreased the level of Hes1 mRNA by 54% (P<0.05; Fig. 4A) and reduced the level of Hes1 protein as well (Fig. 3C). PDX1, NeuroD and MafA mRNA levels were all significantly increased (2.7-, 1.7-, and 1.6-fold respectively, P<0.05), but the levels of mouse insulin I and II mRNA were unchanged, suggesting that neither the insulin transactivators nor Hes1 are limiting insulin gene expression (Fig. 4A). However, exogenous Hes6 expression had a striking effect on BMC1 cells, where there was a substantial increase in the level of mouse insulin I mRNA (6.9-fold, P<0.05; Fig. 4A). In those cells Hes6 reduced the levels of both mouse and human Hes1 mRNA (39% and 42% decrease respectively, P<0.05; Fig. 4A). Mouse PDX1, NeuroD, and MafA mRNAs were significantly increased (1.8-, 1.5-, and 2.3-fold respectively, P<0.05; Fig. 4B), but human PDX1, NeuroD, MafA, and insulin mRNA levels were either absent or unchanged. Overexpression of Hes6 in the BMC2 and BMC3 hybrids, which had completely lost endogenous PDX-1 expression, had no effect on the pattern of either murine or human gene expression.
Figure 3.
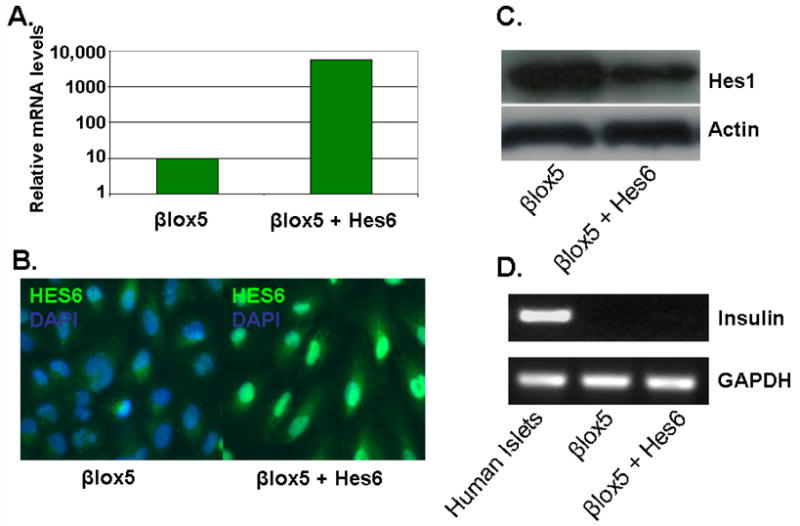
Effects of infecting parental βlox5 cells with retrovirus expressing Hes6 were determined by: A. Quantitative RT-PCR showing levels of Hes6 normalised to GAPDH (representative of three experiments), B. Immunostaining for Hes6, C. Western blotting to determine if Hes6 could induce proteolytic degradation of Hes1 and D. RT-PCR to determine if Hes6 was capable of inducing insulin gene transcription in βlox5 cells.
Figure 4.
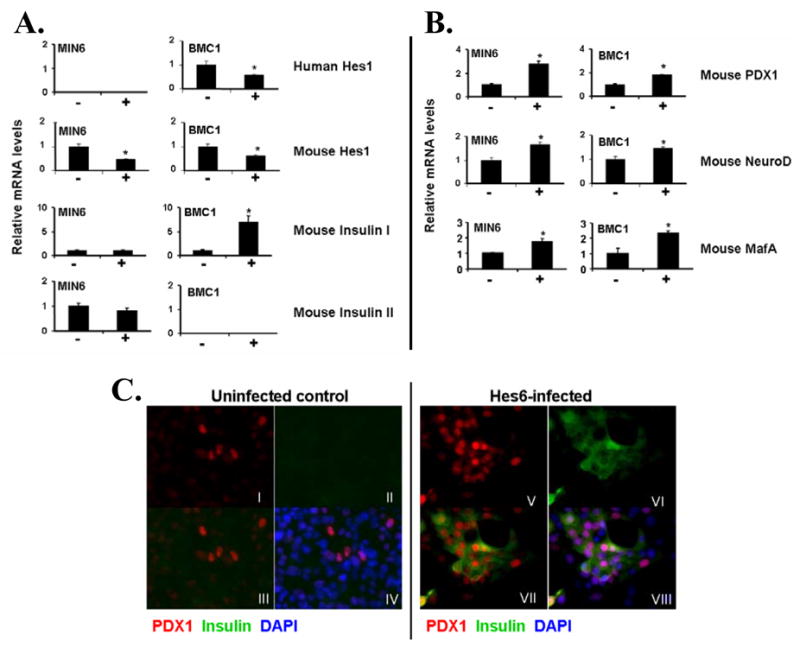
Quantitative RT-PCR showing the effects of Hes6 on insulin expression. Hes1 (A), PDX1, NeuroD and MafA (B) in uninfected (−) and infected (+) MIN6 and BMC1 cells. Data are expressed as relative mRNA levels normalised to GAPDH and represent mean ± SEM of 3–6 observations. *P<0.05.
C. Immunostaining for PDX1 and insulin in uninfected BMC1 cells (I–IV) and BMC1 cells infected with Hes6 retrovirus (V–VIII). I+V: PDX1 (red); II+VI: insulin (green); III+VII: PDX1 (red) and insulin (green); IV+VIII: PDX1 (red), insulin (green) and DAPI (blue).
As described above, PDX-1 continued to be expressed at a high levels in a subset of BMC1 hybrid cells (Figures 1C, 4C). Thus, it was of interest to determine whether the increased insulin in the hybrid cells infected with the Hes6 vector was restricted to those expressing a high level of PDX-1. BMC1 hybrids infected with the Hes6 virus exhibited an increased number of cells expressing PDX1 protein compared with uninfected cells (Fig. 4C), and most notably, a significant number of the PDX1-positive cells in the Hes6-infected hybrid cells demonstrated colocalization with insulin (Fig. 4C). Thus, a subset of cells in BMC1, i.e., those expressing PDX-1, were highly responsive to Hes6, exhibiting a large increase in insulin expression. The cells not expressing PDX-1 did not stain positively for insulin. This means that the RT-PCR data on the effects of Hes6 most likely underestimate the effects as they include both cells expressing and not expressing PDX-1. Overall, these data indicate that it is possible to partially restore the differentiation state of these cells by increasing Hes6 expression.
DISCUSSION
The finding that somatic cell fusion between Min6, a highly differentiated insulinoma line and βlox5, a β-cell line that lost insulin expression, resulted in repression of β-cell differentiation, is broadly similar to that recently reported by Cowan et al. [14], in that fusion of an undifferentiated cell type with a differentiated cell type resulted in nuclear reprogramming of a transcriptional network away from the differentiated transcriptional phenotype towards the undifferentiated. Mechanistically, Notch activation, as manifested by Hes1 expression, is a major cause of the trans-acting repression. Hes6, a downstream repressor of Notch activation which we found to be islet-restricted, could induce β-cell differentiation in the somatic cell hybrids.
Consistent with previous reports on Hes6 function [12; 13], Hes6 expression led to a decline in Hes1 protein. Interestingly, in every cell type infected with the Hes6 retrovirus we found that Hes1 mRNA levels significantly declined. This was surprising, since the Hes1 promoter binds to and is repressed by Hes1 protein in a negative feedback loop [15]. Thus, we predicted that inhibiting Hes1 protein levels and function by Hes6 would result in increased Hes1 mRNA. However, in chick embryos, Hes6 reduced Hes5 transcription [16], indicating a greater degree of complexity to the control of expression of Hes genes than has generally been recognized.
Hes6 appears to act on multiple genes that are important in β-cell differentiation, inducing upregulation of PDX1, NeuroD and MafA. In BMC1, this was accompanied by an upregulation of insulin I mRNA and induction of mature insulin protein in the subset of cells expressing detectable PDX-1 protein. The upregulation of NeuroD following inhibition of Notch signalling by Hes6 was expected [11], but our data are the first to show that expression of PDX1 and MafA can also be regulated by Hes6.
The data presented here are consistent with a model in which maintenance of a balance between Hes6 and Hes1 is important in determining and/or maintaining β-cell differentiation. Notch signalling and consequently Hes1 levels are downregulated as endocrine cells form during development [6; 7]. Adult β-cells are refractory to Notch activation, while earlier cells in the pancreatic and pancreatic endocrine lineages were highly disrupted by Notch activation [8]. However, when Hes1 was overexpressed by transient transfection, inhibition of insulin (as well as PDX-1 and NeuroD) gene expression was seen [10]. One possible explanation for these results is that the high levels of Hes6 in the islet render β-cells refractory to Notch activation, but that can be overcome with high levels of Hes1.
Overall, our data suggest that altering the balance between Hes1 and Hes6 may be a crucial component in any strategy to restore the insulin gene expression cascade in growth-stimulated pancreatic β-cells. Strategies aimed at developing insulin-producing cells from less differentiated cell types must also consider the balance of inhibitors as well as activators of Notch signaling.
MATERIALS AND METHODS
Parental cell lines and culture conditions
Cells were cultured under 10% CO2 in high-glucose DMEM containing 4.5 g/l glucose and L-glutamine, supplemented with 15% fetal bovine serum, penicillin (75 mg/l), streptomycin (50 mg/l), and β-mercaptoethanol (5 μl/l) as recommended for MIN6 cells [3].
Generation of hybrid clones by cell fusion
To select for fusion, each partner had to be resistant to either G418 or hygromycin. βlox5 cells are G418-resistant. Two MIN6 cell strains were generated by retroviral gene transfer of the hygromycin (MIN6-HygR) or G418 resistance (MIN6-NeoR) genes. G418 was used at 50μg/ml and hygromycin at 25 μg/ml.
Cell fusion of 2x108 MIN6-HygR cells mixed with 0.5x108 NeoR cells was induced by PEG1500 (Roche) [17]. After PEG treatment, the cells were centrifuged, resuspended, and plated in 6 well plates in medium supplemented with both hygromycin and neomycin. After approximately 30 days, colonies were picked and expanded. Clones derived from MIN6-βlox5 fusion were designated ‘BMC’, and products of MIN6-MIN6 fusion referred to as ‘MMC’ (Supplemental Fig. 1).
Immunocytochemistry
Immunostaining was performed on monolayers fixed with 4% paraformaldehyde and blocked with 5% donkey serum (Jackson ImmunoResearch Laboratories) and 1% bovine serum albumin (Sigma) in PBS. Primary antibodies were mouse monoclonal anti-insulin 1:50 (Santa Cruz), rabbit polyclonal anti-PDX1 1:1500 (Chris Wright, Vanderbilt University), and goat polyclonal anti-Hes6 1:100 (Santa Cruz). Secondary antibodies were Rhodamine-red donkey anti-mouse 1:200, Rhodamine-red donkey anti-rabbit 1:375 (Jackson ImmunoResearch Laboratories), fluorophore-labeled donkey anti-mouse and donkey anti-goat, both 1:250 (Alexa Fluor 488, Molecular Probes, Inc.). Pancreases from Hes6LacZ(+/−) mice [Koyano-Nakagawa, 2000 #3292] were drop-fixed overnight in 4% PFA in 0.1M phosphate buffer, pH 7.3 followed by cryoprotection in 30% sucrose in PBS, snap-freezing in tissue freezing media and cryo-sectioning at 5–7μm. Sections were immunostained using guinea pig-anti insulin (1:350, US Biologicals), chicken anti-β-galactosidase (1:500, Cortex), goat anti-somatostatin (1:350, Santa Cruz), rabbit anti-glucagon (1:250, Chemicon) and/or rabbit anti-PP (1:200, NeoMarkers). Binding was detected with donkey anti-chicken-Rhodamine-X (1:100, Jackson Immuno), donkey anti-rabbit-Alexa488 as above and donkey anti-guinea pig-Cy5 (1:200, Jackson Immuno), followed by DAPI staining.
Western Blotting
Monolayer cultures were lysed using RIPA buffer containing a protease inhibitors (Calbiochem). Protein (20ug) was separated on 4–20% Longlife gels (Lifegels) and transferred to Immobilon-P membrane (Millipore). Primary antibodies were rabbit polyclonal anti-PDX1 1:5000; goat polyclonal anti-NeuroD 1:250 (Santa Cruz); rabbit polyclonal anti-MafA 1:2500 (Bethyl Laboratories); rabbit polyclonal anti-Hes1 1:1500 (Dr. Tatsuo Sudo, Toray Industries, Japan) and rabbit polyclonal anti-Hes6 1:250 (Santa Cruz). Anti-actin antibody (Sigma) was used to verify equivalent loading. Secondary antibodies were goat anti-rabbit HRP, goat anti-mouse HRP (both 1:1000, Pierce Biotechnology) and horse anti-goat HRP 1:400 (Vector). Chemiluminescence detection was performed using Supersignal West Dura reagents (Pierce Biotechnology).
RT-PCR
RT-PCR used cDNA corresponding to 100 ng of RNA [18]. With each primer set (Table 1), PCR conditions were 5 min at 94 °C, followed by 40 cycles of 94 °C for 45 sec, 60 °C for 45 sec, and 72 °C for 45 sec. Quantitative RT-PCR used the Opticon Real-Time System (BioRad), with SYBR Green. RT-PCR for GAPDH was used for normalization. To make comparisons between human and mouse RT-PCR analysis valid, the GAPDH primer set was the same for human and mouse, recognizing a conserved sequence. Moreover, the PCR product is highly conserved between human and mouse. The GC content of the human and mouse Hes1 primers were identical and yielded a similar product size.
Table 1.
PCR primers.
| GENE PRODUCT | PRIMER SEQUENCE | PRODUCT SIZE (bp) |
|---|---|---|
| Human Insulin (genomic DNA & RNA) | CTACCTAGTGTGCGGGGAAC
GCTGGTAGAGGGAGCAGATG |
984 genomic DNA
197 RNA |
| Mouse Insulin I (genomic DNA & RNA) | CCTGTTGGTGCACTTCCTAC
TGCAGTAGTTCTCCAGCTGG |
318 genomic DNA
318 RNA |
| Mouse Insulin II (genomic DNA & RNA) | GCAGCACCTTTGTGGTTCCC
TGCAGTAGTTCTCCAGCTGG |
736 genomic DNA
248 RNA |
| Human PDX1 (genomic DNA) | CCGCAGGAACCACGATGAGA
GCCACAAACAACGCCAATCC |
298 |
| Human PDX1 (RNA) | GTCCTGGAGGAGCCCAAC
GCAGTCCTGCTCAGGCTC |
336 |
| Mouse PDX1 (genomic DNA) | GAGGACCCGTACTGCCTACA
AACATCACTGCCAGCTCCACC |
108 |
| Mouse PDX1 (RNA) | CCACCCCAGTTTACAAGCTC
ACGGGTCCTCTTGTTTTCCT |
315 |
| Human NeuroD (genomic DNA) | GAACGCAGAGGAGGACTCAC
AAGCGTCTGAACGAAGGAGA |
379 |
| Human NeuroD (RNA) | GCCCCAGGGTTATGAGACTA
GTGAGTCCTCCTCTGCGTTC |
217 |
| Mouse NeuroD (genomic DNA) | CTCGGACTTTCTTGCCTGAG
TTTCAAAGAAGGGCTCCAGA |
201 |
| Mouse NeuroD (RNA) | GCTCCAGGGTTATGAGATCG
CTCTGCATTCATGGCTTCAA |
205 |
| Human MAFA (genomic DNA & RNA) | TCAACGACTTCGACCTGATG
GGGTTGAGGTGATGCTGGTA |
322 genomic DNA
322 RNA |
| Mouse MAFA (genomic DNA & RNA) | GTACCAGCACCACCTGAACC
GAGTGATGATGGTGGGCAGT |
228 genomic DNA
228 RNA |
| Human Hes1 (genomic DNA & RNA) | TCTTTTTCGTGAAGAACTCCAA
TCAGCTGGCTCAGACTTTCA |
366 genomic DNA
235 RNA |
| Mouse Hes1 (genomic DNA & RNA) | GCGTGAAGGATTCCAAAAAT
CGCCTCTTCTCCATGATAGG |
334 genomic DNA
201 RNA |
| Human Hes6 (genomic DNA & RNA) | CAGGCCAAGCTGGAGAAC
ATGGACTCGAGCAGATGGTT |
462 genomic DNA
227 RNA |
| Mouse Hes6 (genomic DNA & RNA) | AAGCTAGAGAACGCCGAGGT
TCAGCTGAGACAGTGGCATC |
489 genomic DNA
194 RNA |
| Human/Mouse GAPDH (RNA) | ACAGTCAGCCGCATCTTCTT
AATGAAGGGGTCATTGATGG |
169 human
153 mouse |
Hes6 retrovirus
The retroviral vector encoding Hes6 was created by cloning the human Hes6 mRNA (OriGene Technologies, Rockville, MD), into the MSCV/hph retroviral vector [19].
Supplementary Material
Acknowledgments
Funding was from the NIH (NIDDK) (FL) and the Larry L. Hillblom Foundation (BT).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Beattie GM, Itkin-Ansari P, Cirulli V, Leibowitz G, Lopez AD, Bossie S, Mally MI, Levine F, Hayek A. Sustained proliferation of PDX-1+ cells derived from human islets. Diabetes. 1999;48:1013–9. doi: 10.2337/diabetes.48.5.1013. [DOI] [PubMed] [Google Scholar]
- 2.de la Tour D Dufayet, Demeterco C, Yoo S-J, Halvorsen T, Itkin-Ansari P, Bossie S, Lopez A, Loy M, Beattie G, Hayek A, Levine F. Glucose-responsive insulin secretion from a human pancreatic β-cell line. Diabetes. 2000;49:A31. [Google Scholar]
- 3.Miyazaki J, Araki K, Yamato E, Ikegami H, Asano T, Shibasaki Y, Oka Y, Yamamura K. Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology. 1990;127:126–32. doi: 10.1210/endo-127-1-126. [DOI] [PubMed] [Google Scholar]
- 4.Weiss MC, Green H. Human-mouse hybrid cell lines containing partial complements of human chromosomes and functioning human genes. Proc Natl Acad Sci U S A. 1967;58:1104–11. doi: 10.1073/pnas.58.3.1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ohneda K, Ee H, German M. Regulation of insulin gene transcription. Semin Cell Dev Biol. 2000;11:227–33. doi: 10.1006/scdb.2000.0171. [DOI] [PubMed] [Google Scholar]
- 6.Apelqvist A, Li H, Sommer L, Beatus P, Anderson DJ, Honjo T, Hrabe de Angelis M, Lendahl U, Edlund H. Notch signalling controls pancreatic cell differentiation. Nature. 1999;400:877–81. doi: 10.1038/23716. [DOI] [PubMed] [Google Scholar]
- 7.Jensen J, Pedersen EE, Galante P, Hald J, Heller RS, Ishibashi M, Kageyama R, Guillemot F, Serup P, Madsen OD. Control of endodermal endocrine development by Hes-1. Nat Genet. 2000;24:36–44. doi: 10.1038/71657. [DOI] [PubMed] [Google Scholar]
- 8.Murtaugh LC, Stanger BZ, Kwan KM, Melton DA. Notch signaling controls multiple steps of pancreatic differentiation. Proc Natl Acad Sci U S A. 2003;100:14920–5. doi: 10.1073/pnas.2436557100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Darville MI, Eizirik DL. Notch signaling: a mediator of beta-cell de-differentiation in diabetes? Biochem Biophys Res Commun. 2006;339:1063–8. doi: 10.1016/j.bbrc.2005.11.111. [DOI] [PubMed] [Google Scholar]
- 10.Shinozuka Y, Okada M, Oki T, Sagane K, Mizui Y, Tanaka I, Katayama K, Murakami-Murofushi K. Altered expression of HES-1, BETA2/NeuroD, and PDX-1 is involved in impaired insulin synthesis induced by glucocorticoids in HIT-T15 cells. Biochem Biophys Res Commun. 2001;287:229–35. doi: 10.1006/bbrc.2001.5573. [DOI] [PubMed] [Google Scholar]
- 11.Jensen J. Gene regulatory factors in pancreatic development. Dev Dyn. 2004;229:176–200. doi: 10.1002/dvdy.10460. [DOI] [PubMed] [Google Scholar]
- 12.Bae S, Bessho Y, Hojo M, Kageyama R. The bHLH gene Hes6, an inhibitor of Hes1, promotes neuronal differentiation. Development. 2000;127:2933–43. doi: 10.1242/dev.127.13.2933. [DOI] [PubMed] [Google Scholar]
- 13.Gratton MO, Torban E, Jasmin SB, Theriault FM, German MS, Stifani S. Hes6 promotes cortical neurogenesis and inhibits Hes1 transcription repression activity by multiple mechanisms. Mol Cell Biol. 2003;23:6922–35. doi: 10.1128/MCB.23.19.6922-6935.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cowan CA, Atienza J, Melton DA, Eggan K. Nuclear reprogramming of somatic cells after fusion with human embryonic stem cells. Science. 2005;309:1369–73. doi: 10.1126/science.1116447. [DOI] [PubMed] [Google Scholar]
- 15.Hirata H, Yoshiura S, Ohtsuka T, Bessho Y, Harada T, Yoshikawa K, Kageyama R. Oscillatory expression of the bHLH factor Hes1 regulated by a negative feedback loop. Science. 2002;298:840–3. doi: 10.1126/science.1074560. [DOI] [PubMed] [Google Scholar]
- 16.Fior R, Henrique D. A novel hes5/hes6 circuitry of negative regulation controls Notch activity during neurogenesis. Dev Biol. 2005;281:318–33. doi: 10.1016/j.ydbio.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 17.Klebe RJ, Mancuso MG. Chemicals which promote cell hybridization. Somatic Cell Genet. 1981;7:473–88. doi: 10.1007/BF01542991. [DOI] [PubMed] [Google Scholar]
- 18.de la Tour D Dufayet, Halvorsen T, Demeterco C, Tyrberg B, Itkin-Ansari P, Loy M, Yoo S-J, Hao E, Bossie S, Levine F. β-cell differentiation from a human pancreatic cell line in vitro and in vivo. Molecular Endocrinology. 2001;15:476–483. doi: 10.1210/mend.15.3.0604. [DOI] [PubMed] [Google Scholar]
- 19.Yee JK, Miyanohara A, LaPorte P, Bouic K, Burns JC, Friedmann T. A general method for the generation of high-titer, pantropic retroviral vectors: highly efficient infection of primary hepatocytes. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:9564–8. doi: 10.1073/pnas.91.20.9564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koyano-Nakagawa N, Kim J, Anderson D, Kintner C. Hes6 acts in a positive feedback loop with the neurogenins to promote neuronal differentiation. Development. 2000;127:4203–16. doi: 10.1242/dev.127.19.4203. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.