

Abstract
The 440-amino acid Mtu recA intein consists of independent protein-splicing and endonuclease domains. Previously, removal of the central endonuclease domain of the intein, and selection for function, generated a 168-residue mini-intein, ΔI-SM, that had splicing activity similar to that of the full-length, wild-type protein. A D422G mutation (ΔI-CM) increased C-terminal cleavage activity. Using the I-SM mini-intein structure (presented here) as a guide, we previously generated a highly active 139-residue mini-intein, ΔΔIhh-SM, by replacing 36 amino acids in the residual endonuclease loop with a seven-residue β-turn from the autoprocessing domain of Hedgehog protein. The three-dimensional structures of ΔI-SM, ΔΔIhh-SM, and two variants, ΔΔIhh-CM and ΔΔIhh, have been determined to evaluate the effects of the minimization on intein integrity and to investigate the structural and functional consequences of the D422G mutation. These structural studies show that Asp422 is capable of interacting with both the N- and C-termini. These interactions are lacking in the CM variant, but are replaced by contacts with water molecules. Accordingly, additional mutagenesis of residue 422, combined with mutations that isolate N-terminal and C-terminal cleavage, showed that the side chain of Asp422 plays a role in both N- and C-terminal cleavage, thereby suggesting that this highly-conserved residue regulates the balance between the two reactions.
Keywords: crystal structure, intein minimization, mutational analyses, protein splicing
Introduction
Inteins are self-splicing elements that exist as in-frame protein fusions with two flanking protein fragments, called exteins 1. The majority of the ~340 inteins identified to date each consist of two structurally and functionally independent domains: the homing endonuclease domain and the splicing domain (InBase, NEB intein data base) 2. In these bi-functional inteins, the endonuclease domain is inserted between N- and C-terminal protein-splicing subdomains (100–150 and 35–50 amino acids, respectively). It is apparent that the protein-splicing function of the intein is independent of the homing endonuclease activity, because splicing-proficient inteins that lack the endonuclease domain occur naturally and can also be engineered 3,4,5,6,7,8,9,10. Therefore, only the splicing subdomains need to be taken into consideration in structure-function studies of protein splicing.
Structural studies of inteins have shown that the N-terminal and C-terminal splicing subdomains form a single globular domain in which the N- and C-termini are located about 9 Å apart at the center of a nearly flat surface of the molecule 11,12,13,14,15,16,17. Conserved intein residues include the N-terminal cysteine, the penultimate histidine and the C-terminal asparagine (Fig. 1a). Also conserved is the first residue of the C-terminal extein, which is usually a cysteine, serine, or threonine. These residues play a defined role in the reaction mechanism, which involves several steps to accomplish cleavage at the N-terminus and the C-terminus of the intein and ligation of the exteins (Fig. 1b).
Figure 1.
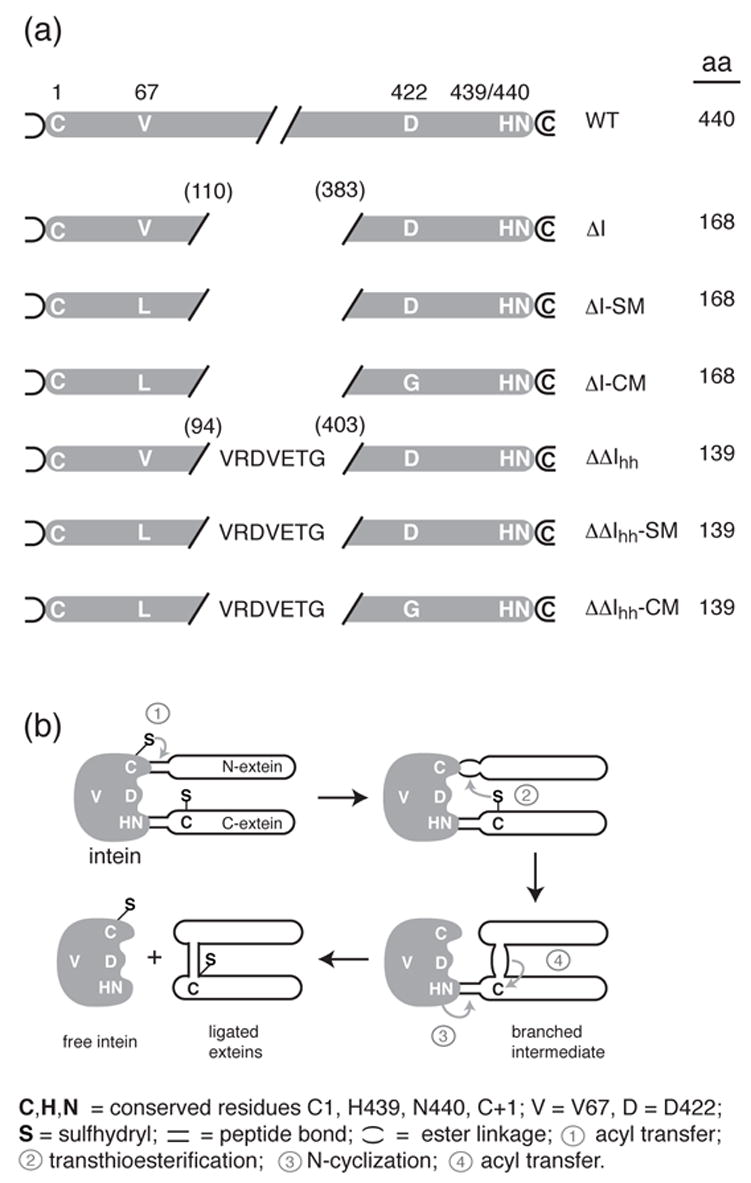
(a) Schematic diagram of the sequences of the inteins discussed in this paper with their corresponding identification codes. The residue numbers used throughout the manuscript refer to the numbering of the intact M. tuberculosis recA intein. (b) Schematic diagram of the steps involved in protein splicing. See text for details.
Previously, the central endonuclease domain of the bipartite 440-amino acid Mycobacterium tuberculosis recA intein was removed, as part of an effort to minimize the intein and to define the functional and structural independence of the protein splicing and endonuclease activities 7,8. Guided by sequence analysis and comparison with the structures of other inteins, we generated a series of mini-inteins (ΔI). The most active ΔI was a 168-residue intein, consisting of residues 1–110 and 383–440. However, the activity of this intein was still much lower than that of the full-length protein. By directed evolution, selecting for enhanced splicing activity, it was shown that the splicing activity of the ΔI could be restored to approximately the same level as that of the full-length, wild-type intein by a single mutation, V67L (ΔI-SM, splicing mutant) 18. This hydrophobic mutation was speculated to stabilize the structure that had been perturbed by deletion of the central endonuclease domain. A second directed-evolution study, selecting for enhanced C-terminal cleavage activity, yielded three mutations, D24G, V67L and D422G. The two latter mutations were critical for enhanced, pH-sensitive, C-terminal cleavage activity (ΔI-CM, cleaving mutant) 18. Thus, the V67L mutation was in common between ΔI-SM and I-CM, and D422G was distinctive for the cleavage activity of the I-CM mini-intein. Figure 1a shows the sequence differences and nomenclature for the various inteins used in this study.
In the crystal structure of ΔI-SM, described here, 24 amino acids in the linker region between the intein subdomains, where the endonuclease domain was removed, are disordered, and only the N-terminal 1–98 and C-terminal 402–440 residues form a globular domain. Based on this observation, several minimized mini-inteins (ΔΔI), ranging in size from 135 to 152 amino acids, were prepared 19. In these constructs, 137 amino acids was the lower size limit for full protein-splicing activity. Furthermore, this study showed that the activation effect of the V67L mutation was universal for mini-inteins longer than 137 amino acids and resulted from structural stabilization of the intein. Among these ΔΔIs was a 139-residue mini-intein (ΔΔIhh-SM, named 94hh403-SM in ref. 19). The linker region of I-SM (residues 95-110 and 383-402) was replaced in ΔΔIhh-SM with a seven-residue sequence that corresponds to a β-turn found at this site in the structure of the autoprocessing domain of Drosophila Hedgehog protein, which is structurally and mechanistically related to inteins 20. ΔΔIhh-SM was shown to be at least as active as the original ΔI-SM.
Here, we report the crystal structures of the 168-residue ΔI-SM and three derived 139-residue mini-inteins: the above-mentioned ΔΔIhh-SM; a cleavage derivative thereof, ΔΔIhh-CM; and ΔΔIhh, which contains the native Val67 residue and has poor splicing activity compared to ΔΔIhh-SM. The structures, in combination with further mutational analysis of residue 422 in the context of ΔI-SM, led to the discovery that this amino acid plays a role in N-terminal as well as C-terminal cleavage, and may therefore be a pivotal residue in coordinating reactivity of the two splice sites.
Results
Crystal structure of ΔI-SM
The 168-residue ΔI-SM mini-intein, which corresponds to the splicing domain of the Mtu recA intein, was obtained by removal of residues 111–382 of the 440-residue intein7, and subsequent mutation and selection for optimal protein splicing activity18. The protein used for crystallographic studies was prepared by a DDT-induced N-terminal cleavage mechanism19 rather than splicing. Regardless, the protein corresponds precisely to the product of the intein-splicing reaction, comprising native N-terminal Cys and C-terminal Asn residues, and lacking exteins.
The crystal structure of ΔI-SM was determined by MAD-phasing methods, using a mercury derivative, and was refined to 1.7 Å resolution. The molecular conformation (Fig. 2a) consists almost exclusively of curved β-strands, forming a disc-shaped molecule that consists of two lobes joined by two covalent links at one end, and extensive hydrophobic interactions along the central interface. Overall, the structure of ΔI-SM is very similar to those of other mini-inteins or the splicing domains of inteins. Least-squares superposition shows that the Mycobacterium xenopi gyrA mini-intein 12 (PDB entry 1AM2) is the structurally most similar homolog (r.m.s.d. of 1.35 Å for 119 α-carbon atoms) (Fig. 2b). As seen in other inteins, the N- and C-terminal residues are approximately 9 Å apart and centrally located on a nearly planar surface of the molecule. In the structure of ΔI-SM, electron density is absent or poor for residues in the linker region between the two splicing subdomains and that are residual residues of the endonuclease domain insertion. The final model consists of residues 1 to 102 and 399 to 440 and residues 1 to 102 and 402 to 440 of the two molecules in the asymmetric unit (molecule A and B), respectively. Otherwise, the conformations of the two molecules are essentially identical, with an r.m.s.d. of 0.71 Å for the α-carbons of residues 1–95 and 405–439.
Figure 2.
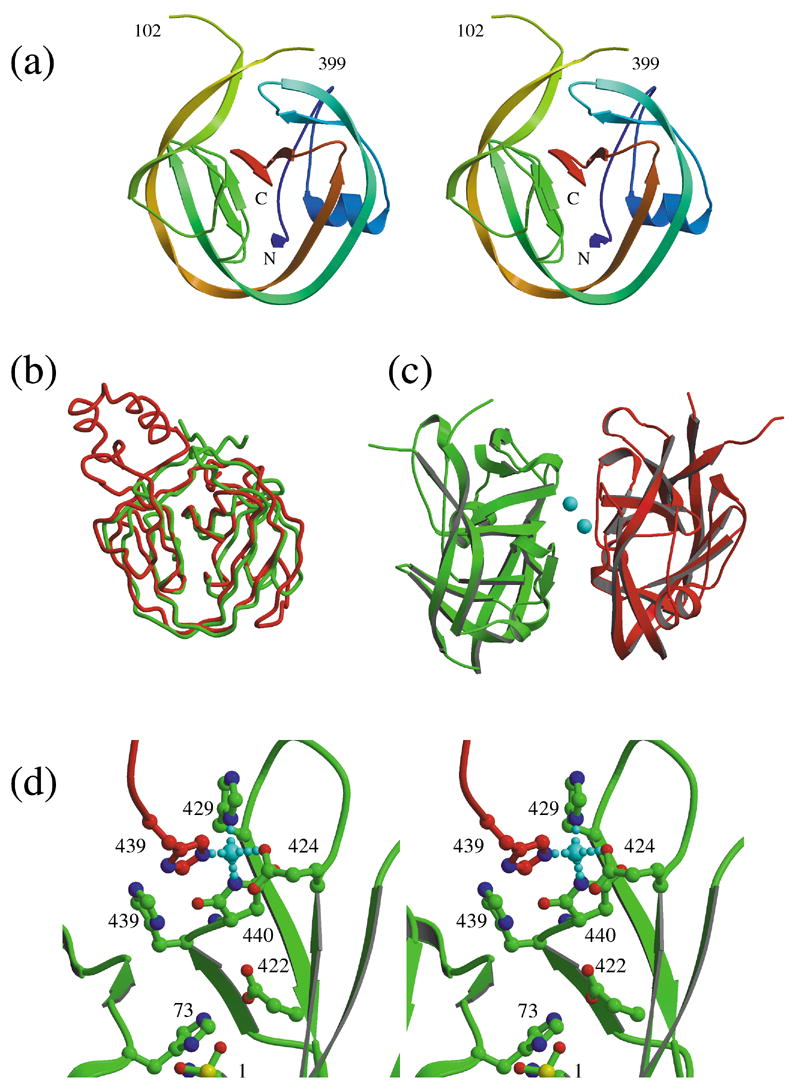
Structure of ΔI-SM. (a) Molecular structure of ΔI-SM. (b) Comparison of the conformation of ΔI-SM (green) with the GyrA intein12 (red). (c) Dimer observed in the asymmetric unit, with zinc ions shown in cyan. (d) Coordination of the zinc ion by residues Glu424, His429, and the C-terminal aminosucciminide (SU440) from one molecule (green), and His439 from the second molecule (red) in the asymmetric unit.
The two protein molecules in the asymmetric unit are arranged in a pseudo-dimer (Fig. 2c) that is mediated by chelation of zinc ions. Two zinc ions are sandwiched between the two protein molecules in a two-fold symmetric geometry. Each zinc ion is coordinated by His439 of one molecule and by three residues from the other molecule: Glu424, His429, and the C-terminal residue (440), which is present as an aminosucciminide rather than an Asn (see below) (Fig. 2d). The origin of the zinc ions is unclear. No zinc ions were added to any of the solutions used during purification or crystallization. Since the mercury ions used for phasing are found at the sites of the zinc ions in the native structure, the zinc ions are bound weakly enough that they can be substituted in relatively high concentration solutions (2 mM) of the alternate divalent cation. However, efforts to remove zinc by EDTA treatment of the protein solution prior to crystallization did not result in a zinc-free crystal structure. Furthermore, the protein behaves as a monomer in size-exclusion chromatography (data not shown). This suggests either that the zinc ions originate from the original culture conditions and can remain stably bound throughout the purification, or that they are recruited from the glass cover slips and/or contaminants in the crystallization buffers during crystallization. Consistent with the latter explanation, crystals only appear several weeks after set-up and grow slowly, and only relatively small amounts of protein crystallize.
However, several interesting observations relate to this crystallization artifact. First, zinc ions are known inhibitors of protein splicing 21,22,23. The observation of the zinc in this structure and in the structures of two other inteins, leads to an understanding of the basis of this inhibition mechanism, as discussed below. Second, the electron density for the C-terminal residue, Asn440, shows this residue not to be in its native state, but rather to be an aminosuccinimide. Asn440 undergoes transformation to an aminosuccinimide during the C-terminal cleavage step of the protein splicing mechanism, but this modification does not otherwise occur spontaneously. Therefore, the presence of the aminosuccinimide appears to result from, or to be stabilized by, the interaction with the zinc ion, confirming that the residue is highly reactive due to environmental factors, as discussed further below.
Crystal structure of the ΔΔIhh-SM mini-intein
The absence of observable electron density for residues 103–110 and 383–398 in the crystal structure of ΔI-SM demonstrates that these residues form a flexible loop. In an effort to define the smallest form of the intein splicing domain 19, we replaced this loop (36 residues, 95 to 402) with a seven-residue peptide (VRDVETG) that forms a β-turn at the corresponding site in the structure (PDB entry 1AT0) of the autoprocessing domain of Hedgehog protein 20. This resulted in a 139-residue intein (ΔΔIhh-SM) that has splicing activity and stability comparable to that of ΔI-SM.
The crystal structure of the ΔΔIhh-SM mini-intein was determined to 1.7 Å resolution. In contrast to what was observed in the structure of I-SM, there is only one molecule in the asymmetric unit, the C-terminal residue is an Asn, and there is no zinc ion bound to the protein. The β-turn insertion (Fig. 3a) has a conformation that is very similar to that observed in the original structure of the Hedgehog protein (r.m.s.d of 0.48 Å for the main-chain atoms of residues 95–102 and 404–405). The substitutions allow the loop, starting at residue 95, to be in closer proximity to residues of the other lobe of the molecule compared to the corresponding segment in the structure of ΔI-SM. Indeed, an additional hydrogen bond, between the nitrogen of Arg96 and the carbonyl oxygen of Gly435, is observed (Fig. 3b). In addition, the P96R mutation introduces a stacking contact of the Arg96 side chain with the side chain of Phe9, increasing the interactions between the two lobes of the molecule and thus stabilizing the structure.
Figure 3.
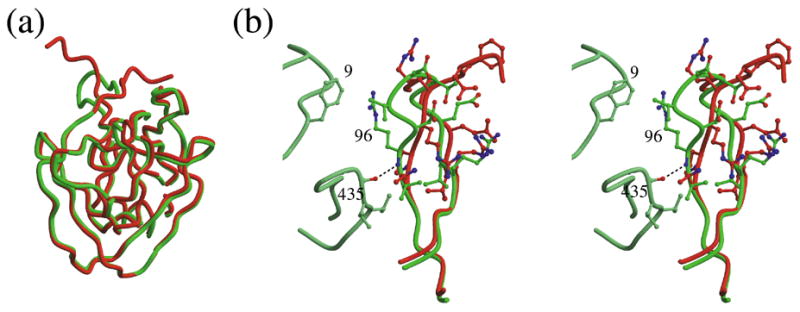
Structure of ΔΔIhh mini-intein. (a) Superposition of the structures of ΔI-SM (red) and ΔΔIhh-SM (green). In ΔΔIhh-SM, a β-turn defined on the basis of a corresponding turn in the autoprocessing domain of Hedgehog protein replaces the disordered loop of ΔI-SM (top left). (b) Detailed conformational comparison of the Hedgehog protein turn in ΔΔIhh-SM (green) with the few observable residues of the linker loop in ΔI-SM (red). Two loops from the other lobe of the molecule, residues 5–15 and 432–438, which include residues that make contacts with the β-turn, are shown in light green. These contacts are a hydrogen bond between residues 96 and 435 and a stacking interaction of the side chains of residues Phe9 and Arg96.
The crystal structures of ΔΔIhh-CM and ΔΔIhh were determined to define the structural implications of the D422G and V67L mutations. Despite very different crystallization buffers (PEG 8000 for ΔΔIhh-SM and ammonium sulfate for ΔΔIhh-CM and ΔΔIhh) all three proteins crystallized in the same crystal form. Therefore, differences in the main chain conformations of each mutant, even when relatively small, are a direct consequence of changes caused by the mutation rather than caused by crystal packing effects.
The D422G mutation in the context of the intein structure
The D422G mutation is responsible for the enhanced, pH-responsive cleavage activity of the ΔI-CM mini-intein. Asp422 is located in the area between the N- and C-termini of the intein, where it could play a role in N- or C-terminal cleavage and even in ligation of the exteins. Examination of the InBase database2 shows that this residue is highly conserved among inteins. It is an Asp in approximately 60% of the 340 entries in the database. Alternative residues at this site are almost exclusively polar residues and the most common, Cys, occurs only in about 7% of all inteins. Other residues occurring multiple times are, in decreasing order of frequency, Thr, Trp, Asn, Ser, and Arg.
Comparison of the structures of ΔΔIhh-SM (Asp422) and ΔΔIhh-CM (Gly422) shows that the mutation does not affect the overall protein conformation (r.m.s.d. 0.2 Å for all α-carbons) (Fig. 4a). However, the alpha-carbon of Gly422 is shifted by about 0.4 Å relative to that of Asp422. The location of residue 422 in the middle of a β-strand may well contribute to the limited structural effect of the introduction of a residue that affords greater flexibility to the protein conformation at this site. The absence of the relatively large Asp side chain less than 5 Å from the N- and C-terminal residues also does not greatly affect their conformations. The conformations of the N-terminal cysteine residues are identical in the two structures, as well as in the structures of ΔI-SM and ΔΔIhh. Small differences are observed in the conformations of side chains at the C-terminus (His439 and Asn440) but these are more likely attributable to the presence of a sulfate anion in the area between His439 and Asn440 in the structure of ΔΔIhh-CM, than to an effect of the mutation at residue 422.
Figure 4.
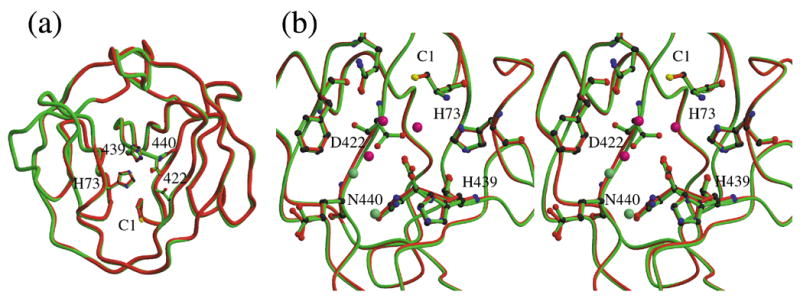
Effect of the D422G mutation. (a). Superposition of the structures of ΔΔIhh-SM (green) and ΔΔIhh-CM (red). (b) Detailed comparison of the area of residue 422. Water molecules in this area are shown as spheres, light green for ΔΔIhh-SM and rose for ΔΔIhh-CM. In ΔΔIhh-CM, two water molecules are found bound tightly to the protein in the area of the missing Asp422 side chain.
Comparison of these structures suggests two explanations for the enhanced C-terminal cleavage activity resulting from the D422G mutation, as well as for the importance of Asp422 in protein splicing. The absence of the Asp side chain at residue 422 in the ΔΔIhh-CM structure leads to a depression on the protein surface, in between the N and C-termini, that accommodates two water molecules, including one that is in hydrogen-bonding contact with the carboxyl group at the C-terminus (Fig. 4b). Since water molecules have been proposed to play significant roles in the cleavage and splicing mechanisms 16,17,24, the increased accessibility of the C-terminus in ΔΔIhh-CM may well account for the increased C-terminal cleavage activity of the CM mutant.
The second explanation is based on the analysis of the structures of ΔI-SM, ΔΔIhh-SM and ΔΔIhh. Comparison of the structures of ΔΔIhh, which has Val67 (as in the native Mtu recA intein), and ΔΔIhh-SM (Leu67) shows that this mutation does not affect protein conformation significantly (r.m.s.d 0.2 Å). In addition, it confirms that the V67L mutation increases the activity of the SM mutants, by improving packing interactions in an internal hydrophobic core. More importantly, the structure of ΔΔIhh shows Asp422 in hydrogen-bonding contact with the sulfur of the N-terminal cysteine. Thus, among the structures of ΔI-SM, ΔΔIhh and ΔΔIhh-SM, Asp422 adopts three different side chain conformations that range from an N-terminal (ΔΔIhh) to a C-terminal (ΔΔIhh-SM) contact (Fig. 6b). This flexibility could be important for activity and reinforces the idea that the Asp side chain plays a role in both the N-terminal and C-terminal cleavage reactions.
Figure 6.
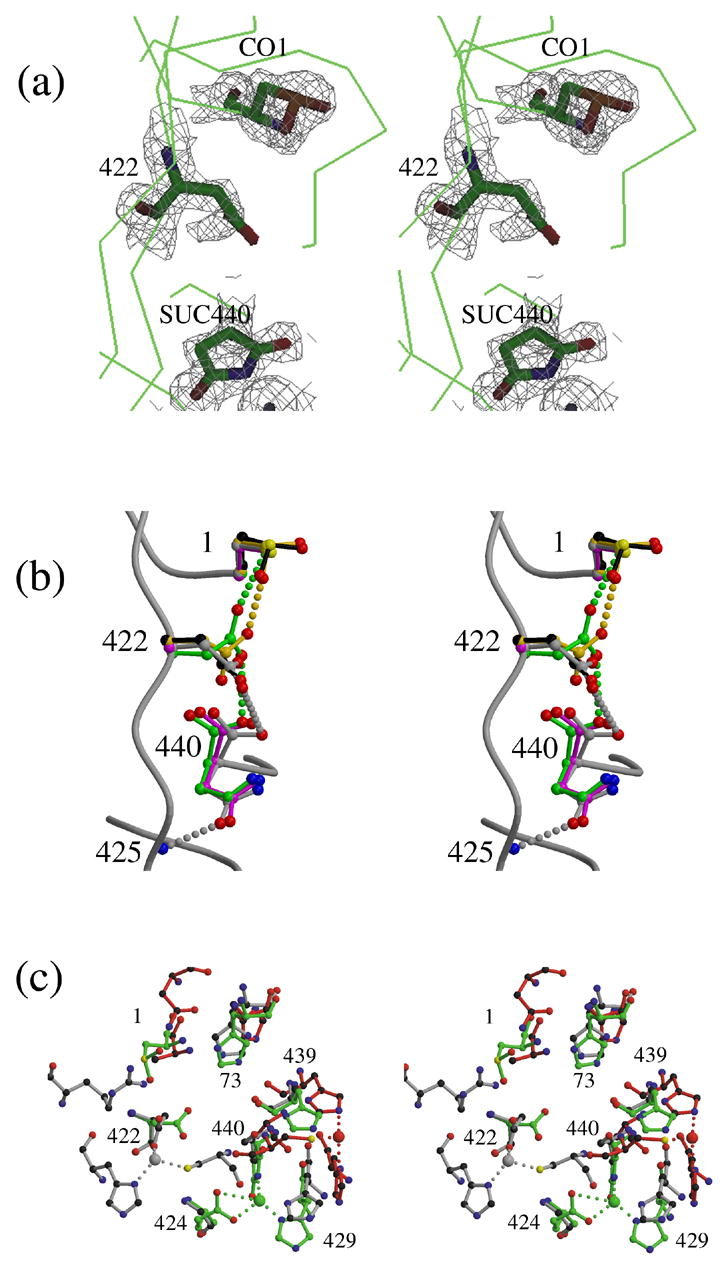
Structural comparisons of the intein N-termini and C-termini of inteins. (a) Model and electron density (contoured at 1.5 σ) of residues 1, 422 and 440 in the structure of ΔI-SM (molecule A), showing observed modifications: CO1: cysteine sulfinic acid; SUC440: aminosuccinimide. Despite inconsistent electron density, residue 422 is included in the model for I-SM as an Asp because the modification of the residue has not been characterized. (b). Conformations of Cys1, Asp422, and Asn440 in the structures of ΔΔIhh-SM (grey), ΔΔIhh-CM (magenta) and ΔΔIhh (green) and of CO1 and Asp422 in ΔI-SM molecules A (black) and B (gold). (c). Comparison of the zinc-binding sites observed in the structures of three different inteins: ΔI-SM (green), PI-SceI34 (red) and dnaE17 (grey). Residue numbers shown correspond to those of ΔI-SM.
The role of Asp422 in N-terminal and C-terminal cleavage, as defined by intein mutagenesis
In previous work, the C-terminal cleavage activities of the SM (Asp422) and CM (Gly422) variants of ΔI were compared in the presence of a C1A mutation, which inhibits splicing 18. In that context, the Gly422 mutation in the CM variant led to C-terminal cleavage activity that was increased up to 6-fold, and that was distinctly pH-responsive. To determine whether it is the loss of Asp or the gain of Gly at position 422 that leads to the CM phenotype and also to assess the role of Asp422 in intein catalysis in more detail, we now determined the effects of other residues at this position on splicing, N-, and C-terminal cleavage activities (Fig. 5).
Figure 5.
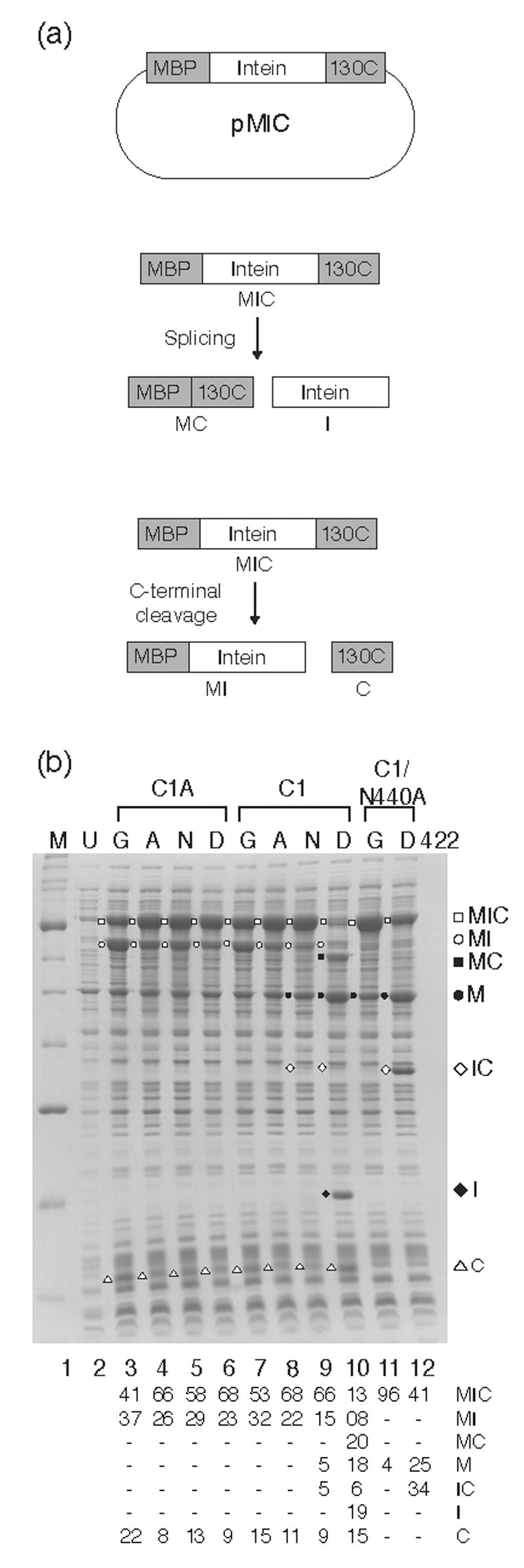
Splicing activity of mutant I-inteins for assessment of the role of Asp422. (a). pMIC vector used for expression of MIC tripartite proteins. Also shown are schematics of the splicing and C-terminal cleavage reactions. (b). In vivo splicing and cleavage reactions visualized by SDS PAGE. Induction was at 25°C. Lane M, protein markers, broad range (New England Biolabs); U, uninduced cells. Mutations at key residues are indicated above each panel with the amino acid at position 422 immediately over each lane. The splicing and cleavage products were previously identified by Western blots using antibodies to each of the three different components of the tripartite precursor7. The table below the gel represents a quantitation of the scanned images of the lanes, with all products totaling 100%. Although each experiment was repeated at least five times and the mutants behaved extremely reproducibly, the quantitation presented reflects only the data for this particular gel because of experimental variability.
We have compared the activities of the intein derivatives in the context of MIC 7, a tripartite fusion consisting of maltose-binding protein (M), intein (I), and the C-terminal DNA-binding domain of homing endonuclease I-TevI (C). We induced expression of the MIC precursor in vivo and monitored splicing products by separating and visualizing crude cell extracts on SDS-polyacrylamide gels. These experiments allow qualitative comparisons of the activities of various intein derivatives. Consistent with previous results, both ΔI-SM (Asp422) and ΔI-CM (Gly422) are incapable of splicing in the C1A context, but they do undergo C-terminal cleavage, with the Gly422 intein being more active, as evidenced by the more intense MI band (Fig. 5b, lanes 3 and 5). The results are similar whether the precursors are expressed at 37°C (data not shown) or 25 °C (Fig. 5b). In the splicing-competent Cys1 context, ΔI-SM (D422) undergoes efficient splicing, with much of the precursor converted to MC and I, the products of splicing. However, we also see products of N- and C-terminal cleavage, in particular, M (Fig. 5b, lane 10). In sharp contrast, the ΔI-CM (Gly422) derivative does not exhibit detectable splicing in this context (Fig. 5b, lane 7). Presence of an MI band indicates that the D422G mutation results in substantial C-terminal cleavage, independent of the nature of the N-terminal intein residue. This effect is explicable, as was previously suggested 18, if the presence of a Gly at position 422 in the CM protein increases C-terminal cleavage to such an extent that it proceeds before splicing can occur. Alternatively, Asp422 itself could play a role in splicing so that the loss of this residue would inhibit splicing and result in isolated cleavage reactions.
We used two approaches to evaluate these potential causes for the cleavage phenotype of D422G mutants. First, we prepared alternative mutants at position 422 to investigate if the effect was due to the unique nature of Gly, with its small side chain and allowance for greater backbone flexibility. We find that D422A and D422N derivatives display phenotypes very similar to that of D422G, despite Asn being polar and equally bulky as Asp, and both Ala and Asn being less flexible than Gly. While the extent of the C-terminal cleavage reaction differed slightly between the two mutant derivatives, both mutations inhibited splicing (Fig. 5b, lanes 8 and 9).
In the second approach, we investigated the activities of the SM (Asp422) and CM (Gly422) variants in combination with a mutation that, based on other studies of intein mechanisms25,26,27, is expected to eliminate C-terminal cleavage activity. This allowed us to isolate the splicing or N-terminal cleavage activities of residue 422 variants from the C-terminal cleavage activity. As predicted, for the wild-type Asp422 (ΔI-SM), mutation of the catalytically important C-terminal residue, Asn440, to Ala inhibited C-terminal cleavage and splicing, but maintained detectable N-terminal cleavage activity (Fig. 5b, lane 12). In contrast, ΔI-CM (Gly422) in the presence of the N440A mutation is completely inactive (Fig. 5b, lane 11). These results are consistent with the idea that Asp422 plays a role in N-terminal cleavage and possibly in splicing.
Zinc binding correlates with altered states of intein termini
In the crystal structure of I-SM, the observed electron densities for the N- and C-terminal residues were not consistent with their native states. The N-terminal cysteine appeared to be oxidized to cysteine sulfinic acid (−SO2H) (Fig. 6a, CO1), while the C-terminal asparagine had cyclized to the aminosuccinimide form (Fig. 6a, SU440). Mass spectrometry analysis of the protein used for crystallization showed no evidence of modifications (data not shown), consistent with the modifications having occurred during crystallization. The modifications confirm that both residues are highly reactive, due to their environment in the intein. The C-terminal aminosuccimide is not observed in the crystal structures of other forms of the Mtu recA intein, nor has it been reported in the crystal structures of other inteins. We propose that this modification is caused, or at least stabilized, by the presence of a zinc ion that interacts with the nitrogen of the succinimide ring. In fact, an Asn440 residue would not accommodate a zinc ion at this site. Cyclization of the C-terminal Asn to the aminosuccinimide is part of the mechanism for the C-terminal cleavage step of the intein-splicing reaction 25,26,27, but this intermediate is unstable, and it normally regenerates to an Asp through spontaneous hydrolysis 28.
Likewise, the sulfur of the N-terminal Cys plays a role in the intein splicing mechanism, as the nucleophile in the N-S acyl shift in N-terminal cleavage 25,26,27,29. While cysteine sulfinic acid is rare, it can occur as the result of over-oxidation of highly reactive cysteines in the active sites of several proteins, including peroxiredoxin 30,31 and thiocyanate hydrolase 32. The electron densities at Cys1 in the structures of the ΔΔIhh and ΔΔIhh-SM, but not ΔΔIhh-CM, also reveal possible oxidation of the Cys, although not as obviously as it seen for ΔI-SM. This leads us to conclude that the oxidation of Cys1 in ΔI-SM is due to the high reactivity of this residue that is related to environmental factors, but that it is at least partially correlated with the modification of Asn440.
Furthermore, in the structure of I-SM, the electron density for Asp422 (in both molecules in the asymmetric unit) was not fully consistent with the presence of an Asp, showing density for only a single Oδ. In contrast, electron density for this residue in the structures of ΔΔIhh-SM and ΔΔIhh indicates that it is unambiguously an Asp. This observation supports the idea that there is a correlation between the modifications at the N- and C-termini and a role in this for Asp422, which can interact with both residues.
Discussion
Conformational flexibilities of specific intein residues
The crystal structures of ΔI-SM and the three ΔΔIhh mini-inteins lead to novel insights into how the multiple steps of protein splicing could be coordinated (Fig. 1b). These molecules are all products of the splicing reaction, albeit not generated by splicing from their natural host, but rather by N-terminal cleavage of an N-extein-intein precursor (see Materials and Methods). In these structures, the N-terminal Cys maintains a single conformation, even when modified to cysteine sulfinic acid as it is in ΔI-SM. The C-terminal Asn adopts slightly different conformations that all involve a hydrogen bond between the oxygen of the carbonyl group with the main-chain nitrogen of Val425 (Fig. 6b), whereas, as described above, Asp422 adopts a range of very different conformations. Furthermore, the conformations of Asn440 observed in ΔΔIhh-SM, ΔΔIhh, and ΔΔIhh-CM differ greatly from the conformations of the C-terminal residue observed in many other intein structures 11,12,13,14,15,16,17,33. Indeed, comparison of the structures of other inteins and mini-inteins, either as products or as mutants with exteins at the N- and C-termini, shows that the conformation of the N-terminus is highly conserved, but that the C-terminus adopts a wide range of conformations 11,12,13,14,15,16,17,33. This apparent rigidity of the N-terminus and flexibility of the C-terminus and of Asp422 suggest that the capability of motion required to accomplish the steps involved in N-terminal and C-terminal cleavage and ligation is inherent in the structure and conformation of this highly conserved internal residue and of the C-terminus.
Zinc as a non-specific inhibitor of protein splicing
The crystal structure of ΔI-SM contains a zinc-binding site where the cation is coordinated by the C-terminal succinimide, a Glu and two His residues. Zinc ions have now been observed in three different structures of inteins 17,34. Since zinc is known to be a potent inhibitor of intein splicing, examination of these binding sites can lead to an understanding of the zinc inhibition mechanism. Interestingly, the three binding sites are distinct (Fig. 6c). The sites in two of these inteins, ΔI-SM and the VMA precursor of PI-SceI34 (PDB entry 1EF0) are near the C-terminus of the intein, while the site in the third, dnaE17(1ZDE), is near the Cys+1 of the C-extein and the residue equivalent to Asp422. In both the VMA and dnaE structures, the zinc ion interacts with the Cys+1 of the C-terminal extein, but the protein conformations at this site differ greatly, resulting in highly divergent zinc ion locations and coordinations. These observations lead us to believe that it is the presence of multiple His and Cys residues in the area of the intein-extein junctions that is the basis for inhibition, through the fortuitous location of suitable zinc-binding sites rather than the presence of a fixed metal chelation site, or through a specific mechanism of inhibition.
A pivotal role for D422 in intein activity
The combination of structural and mutational analyses allows evaluation of the role of the Asp in the mechanism of protein splicing by inteins, which can be conceptualized as cleavage at the N-extein-intein junction coupled to extein ligation, followed by cleavage at the intein-C-extein junction (Fig. 1b). Previous studies have shown that mutations at one terminus does not prevent cleavage from occurring at the other terminus, suggesting that the activities at the two termini can be independent of each other 27. Therefore, for protein splicing to occur efficiently, the reactions must be spatially and temporally coordinated. A residue that plays a role in both N-terminal and C-terminal cleavage would be implicated in regulating the independent reactions. Our results suggest that Asp422 is such a residue.
We showed previously 18, and have confirmed here, that when C-terminal cleavage is isolated (by the C1A mutation), the D422G mutation results in greatly elevated activity (Fig. 5), indicating that Asp422 affects C-terminal cleavage. For D422G, the cleavage products MI and C account for 59% of the total intein species (Fig. 5b, lane 3), whereas for the Asp422 wild-type, these products account for only 32% (lane 6). We then blocked C-terminal cleavage and isolated N-terminal cleavage by mutating the C-terminal Asn to an Ala (N440A). In this context, Asp422 showed significant N-terminal cleavage, with M and IC accounting for 59% of the total intein species (Fig. 5b, lane 12), while D422G showed no or very little activity (lane 11). This observation clearly indicates that Asp422 plays a role in N-terminal cleavage as well. However, we note that, in vivo, N-terminal cleavage could result from cleavage either at the N-extein-intein junction in the linear thioester or at the N-extein/C-extein junction in the branched intermediate. Furthermore, we showed that Asp422 is needed for splicing, and mutating it to Gly, Ala, or Asn resulted in C-terminal cleavage only (Fig. 5, lanes 7–9). These results suggest a role for Asp422 in protein splicing, affecting both N-terminal cleavage and C-terminal cleavage.
As mentioned previously, the crystal structures reveal that Asp422 lies proximal to both termini and that it adopts different conformations. This suggests that its role in N-terminal cleavage may be occurring through an effect at the N-terminus, and not through regulation of the C-terminal cleavage step. Therefore, there are two basic models that can account for the properties of Asp422. Asp422 could have an indirect effect on the termini, or it could act directly on the terminal residues. In the first case, the acidic nature of the side chain of Asp422 is partially responsible for the local environment, which in turn affects the cleavage reactions (Fig. 7 left). This local environment effect could be purely electrostatic, involve access to water molecules, and/or could affect intermediary amino acids. Changes in the environment could affect the two cleavage reactions differently. Consistently, the D422E mutant has similar properties to the wild-type intein, although it splices less efficiently (data not shown). Alternately, Asp422 could play a more direct role in the two cleavage reactions (Fig. 7, right). The residue could be involved in important hydrogen bonding contacts that stabilize an intermediate, or it might catalyze a step in the reaction, either at each terminus separately or in a concerted manner (Fig. 7, right, top and bottom, respectively). Regardless of which model best accounts for the role of Asp422, its proximity to the intein termini and its conformational flexibility underscore a pivotal role for this residue in protein splicing that includes coordinating events at the two termini.
Figure 7.
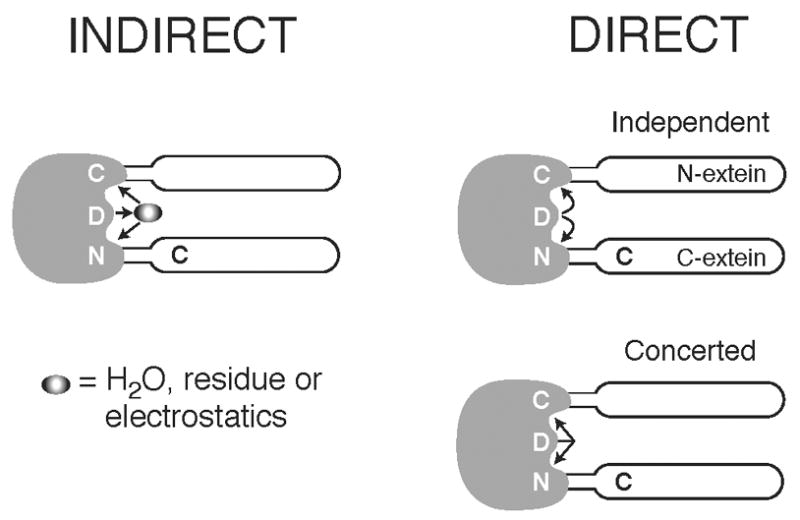
Schematic diagram showing possible interactions of Asp422 with the intein termini. Left panel: Asp422 functions indirectly at the two termini, via a water molecule, via another residue, or through an electrostatic mechanism. Right panel: top, Asp422 interacts directly at the N-terminus and C-terminus independently; bottom, Asp422 interacts directly at both termini in a concerted manner.
Materials and Methods
Intein expression and purification for structural studies
The inteins studied in this paper, which have been described previously 18,19, are shown schematically in Figure 1. The intein genes were cloned downstream of the chitin-binding domain (CBD) gene in the pX vector, to create the pX-I series which are used to express and purify the inteins for structural studies. X represents the CBD with N-terminal fusion of the 10 N-terminal amino acids of maltose binding protein, MBP, to improve the solubility of recombinant protein. The fusion proteins were overexpressed in E. coli JM101 and purified using affinity chromatography with chitin beads. The inteins were released from the column by incubation with DTT (50 or 200 mM), which overcomes poor equilibrium and induces N-terminal cleavage, as previously described 19. Proteins for crystallization were further purified by size-exclusion chromatography and concentrated to about 8 mg/ml in a buffer containing 20 mM Tris pH 8.0, 100 mM NaCl and 2 mM DTT.
In vivo splicing and cleavage assays by protein gel analysis
The pMIC plasmids 7 were used to express tripartite fusion proteins (MBP [M] – intein [I] – C-terminal domain of I-TevI [C]) for visualization of in vivo splicing and cleavage activity by SDS-PAGE. The pMIC plasmids were transformed into E. coli JM109, and grown to OD600 = 0.4–0.6 at 30 or 37 °C. The MIC precursors were expressed by addition of 1 mM IPTG and incubation at 25 or 37 °C for 3 h. Cells were harvested by centrifugation, resuspended in lysis buffer (50 mM Tris-HCl pH8.0, 2 mM EDTA), and lysed by sonication. Splicing and cleavage products were visualized through separation of cell lysates via SDS-PAGE followed by staining with Coomassie blue. For quantitation, the gel was scanned with an Alpha Innotech FluorChemTM 8900. Each lane was scanned independently, measuring the intensity of the bands corresponding to the intein-related products. The inteins in lanes 3–6 do not yield any M product, and so the band at this location, which corresponds to an unrelated protein, was not included in quantitation for these lanes. For lanes 7–12, the intensity of the M band was corrected by subtracting the average intensity of M in lanes 3–6. For all lanes, the intensities of all intein-related bands was normalized and individual band intensities reported as a percentage of the lane’s total.
Crystallization, structure determination and refinement
Crystals of all four inteins were grown by hanging-drop vapor-diffusion methods. Crystallization buffers ΔI-SM and for ΔΔIhh-SM contained 6–8% PEG8000, 0.1M Tris pH 8.5 and 5% PEG400. Buffers for ΔΔIhh-CM and ΔΔIhh contained 65% ammonium sulfate, 0.1 M Tris pH 8.5.
For structure determination, a heavy atom derivative of ΔI-SM was prepared by soaking crystals in a stabilizing solution containing 2 mM mercury acetate. The crystals were flash-cooled in the cold nitrogen-gas stream after transfer to a stabilizing buffer containing 20% glycerol. Three-wavelength MAD data were measured at beamline X12c of the National Synchrotron Light Source, Brookhaven National Laboratory, and were processed using HKL2000 35. The structure was determined using the program SOLVE36 and phases were improved by solvent modification using RESOLVE 37. The heavy atom search revealed the presence of two heavy atom sites in the asymmetric unit, about 4.5 Å apart. Statistics for the data collection and phasing statistics for the MAD phasing are listed in Table 1. The resulting map showed good electron density for about 90% of the model. The structure was refined using CNS 38, and the model was built and evaluated using O 39. Data collection and refinement statistics of the native structure are listed in Table 2. Only one residue, Glu155, falls outside the additionally allowed region of the Ramachandran plot as determined with Procheck 40.
Table 1.
Data collection statistics for the structure determination of ΔI-SM
| INFLECTION | PEAK | REMOTE | |
|---|---|---|---|
| Wavelength (Å) | 1.011 | 1.091 | 0.94 |
| Resolution (Å) | 50.–1.59 | 50.–1.57 | 50.–1.48 |
| Redundancy | 6.4 | 5.7 | 4.8 |
| Completeness | 96.2(73.9) | 94.7(62.6) | 93.8(63.5) |
| <I/σ(I)> | 47.6(3.9) | 41.3(2.8) | 32.1(1.7) |
| Rmer | 0.044(0.443) | 0.044(0.482) | 0.048(0.677) |
Table 2.
Data collection and refinement statistics. Values in parentheses are for the highest resolution shell.
| ΔI-SM | ΔΔIhh-SM | ΔΔIhh-CM | ΔΔIhh | |
|---|---|---|---|---|
| Data collection | ||||
| Space group | P21212 | P212121 | P212121 | P212121 |
| Cell parameters (Å) | ||||
| a | 82.43 | 36.84 | 36.88 | 36.73 |
| b | 93.63 | 47.46 | 49.27 | 49.43 |
| c | 40.14 | 64.63 | 64.47 | 64.26 |
| Resolution (Å) | 50.–1.7 (1.75–1.7) | 29.–1.6 (1.65–1.6) | 30.–1.7 (1.75–1.7) | 49.–1.8 (1.86–1.8) |
| Total unique reflections | 40459 | 15495 | 13488 | 11342 |
| Redundancy | 4.0 (3.6) | 4.1 (3.4) | 3.7 (3.4) | 4.1 (3.8) |
| Completeness | 89.4 (92.1) | 99.1 (92.5) | 98.6 (92.3) | 99.4 (98.3) |
| <I/σ(I)> | 27.1 (4.6) | 7.1 (2.1) | 8.9 (3.1) | 15.4 (5.1) |
| Rmer | 0.043 (0.207) | 0.087 (0.489) | 0.068 (0.354) | 0.050 (0.230) |
| Refinement | ||||
| Number of reflections | ||||
| Resolution | 50.–1.7 | 29.0–1.6 | 30.–1.7 | 30.–1.8 |
| Rcryst | 0.183 | 0.245 | 0.252 | 0.258 |
| Rfree | 0.208 | 0.275 | 0.292 | 0.278 |
| r.m.s.d. from ideality | ||||
| bond length (Å) | 0.004 | 0.005 | 0.005 | 0.005 |
| bond angles (deg) | 1.3 | 1.3 | 1.4 | 1.3 |
| dihedrals (deg) | 25.3 | 25.0 | 25.1 | 24.9 |
Data for ΔΔIhh-SM, ΔΔIhh-CM, and ΔΔIhh were measured using the Wadsworth Center’s in-house diffraction facility (Rigaku Micromax 007 generator and R-axis IV detector) and were processed using CrystalClear. The structure of ΔΔIhh-SM was determined by molecular replacement methods, using the program PHASER 41. The search model consisted of residues 1 to 94 and 385 to 439 of ΔI-SM. ΔΔIhh-CM and ΔΔIhh crystallized in the same space group as ΔΔIhh-SM. Therefore, structure refinement for these mutants was started using the ΔΔIhh-SM model, but with a rigid body refinement cycle implemented prior to simulated annealing. Otherwise, refinement of the three structures was performed as for ΔI-SM. Data collection and refinement statistics are listed in Table 2.
Protein Data Bank accession codes
The atomic coordinates and structure factors for I-SM (2IMZ), ΔΔIhh-SM (2IN0), ΔΔIhh-CM (2IN8), and ΔΔIhh (2IN9) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ.
Acknowledgments
We thank Wei Wu and Iveta Kyselova for developing the expression and purification procedures used to prepare the protein samples, John Dansereau for preparing Figures 1, 5, and 7B, and Shekhar Garde and Gil Amitai for helpful discussions and critical reading of the manuscript. We acknowledge use of the X-ray diffraction facilities of the Wadsworth Center’s Macromolecular Crystallography Core and of beamline X12c of the National Synchrotron Light Source, Brookhaven National Laboratory (which is supported by the NIH and the DOE). This work was supported by grants from the NIH (GM44844) and NSF (C1404079). BP was supported by the NIH Biomolecular Science and Engineering training grant GM067545.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Perler FB, Davis EO, Dean GE, Gimble FS, Jack WE, Neff N, Noren CJ, Thorner J, Belfort M. Protein splicing elements: inteins and exteins - a definition of terms and recommended nomenclature. Nucl Acids Res. 1994;22:1125–1127. doi: 10.1093/nar/22.7.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perler FB. InBase, the intein database. Nucl Acids Res. 2002;30:383–384. doi: 10.1093/nar/30.1.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gorbalenya AE. Non-canonical inteins. Nucl Acids Res. 1998;26:1741–1748. doi: 10.1093/nar/26.7.1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaneko T, Nakamura Y, Wolk CP, Kuritz T, Sasamoto S, Watanabe A, Iriguchi M, Ishikawa A, Kawashima K, Kimura T, Kishida Y, Kohara M, Matsumoto M, Matsuno A, Muraki A, Nakazaki N, Shimpo S, Sugimoto M, Takazawa M, Yamada M, Yasuda M, Tabata S. Complete genomic sequence of the filamentous nitrogen-fixing cyanobacterium anabaena sp strain PCC 7120. DNA Res. 2001;8:205–213. doi: 10.1093/dnares/8.5.205. [DOI] [PubMed] [Google Scholar]
- 5.Nakamura Y, Kaneko T, Sato S, Ikeuchi M, Katoh H, Sasamoto S, Watanabe A, Iriguchi M, Kawashima K, Kimura T, Kishida Y, Kiyokawa C, Kohara M, Matsumoto M, Matsuno A, Nakazaki N, Shimpo S, Sugimoto M, Takeuchi C, Yamada M, Tabata S. Complete genome structure of the thermophilic cyanobacterium Thermosynechococcus elongatus BP-1. DNA Res. 2002;9:123–130. doi: 10.1093/dnares/9.4.123. [DOI] [PubMed] [Google Scholar]
- 6.Waters E, Hohn MJ, Ahel I, Graham DE, Adams MD, Barnstead M, Beeson KY, Bibbs L, Bolanos R, Keller M, Kretz K, Lin X, Mathur E, Ni J, Podar M, Richardson T, Sutton GG, Simon M, Soll D, Stetter KO, Short JM, Noordewier M. The genome of Nanoarchaeum equitans: insights into early archaeal evolution and derived parasitism. Proc Natl Acad Sci USA. 2003;100:12984–12988. doi: 10.1073/pnas.1735403100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Derbyshire V, Wood DW, Wu W, Dansereau JT, Dalgaard JZ, Belfort M. Genetic definition of a protein-splicing domain: functional mini-inteins support structure predictions and a model for intein evolution. Proc Natl Acad Sci USA. 1997;94:11466–11471. doi: 10.1073/pnas.94.21.11466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shingledecker K, Jiang SQ, Paulus H. Molecular dissection of the Mycobacterium tuberculosis RecA intein: design of a minimal intein and of a trans-splicing system involving two intein fragments. Gene. 1998;207:187–195. doi: 10.1016/s0378-1119(97)00624-0. [DOI] [PubMed] [Google Scholar]
- 9.Wu Y, Wang ZX. Comparison of conformational changes and inactivation of soybean lipoxygenase-1 during urea denaturation. Bioch Biophys Acta. 1998;1388:325–336. doi: 10.1016/s0167-4838(98)00182-4. [DOI] [PubMed] [Google Scholar]
- 10.Chong S, Xu MQ. Protein splicing of the Saccharomyces cerevisiae VMA intein without the endonuclease motifs. J Biol Chem. 1997;272:15587–15590. doi: 10.1074/jbc.272.25.15587. [DOI] [PubMed] [Google Scholar]
- 11.Duan X, Gimble FS, Quiocho FA. Crystal structure of PI-SceI, a homing endonuclease with protein splicing activity. Cell. 1997;89:555–564. doi: 10.1016/s0092-8674(00)80237-8. [DOI] [PubMed] [Google Scholar]
- 12.Klabunde T, Sharma S, Telenti A, Jacobs WR, Sacchettini JC. Crystal structure of gyrA intein from Mycobacterium xenopi reveals structural basis of protein splicing. Nat Struct Biol. 1998;5:31–36. doi: 10.1038/nsb0198-31. [DOI] [PubMed] [Google Scholar]
- 13.Ichiyanagi K, Ishino Y, Ariyoshi M, Komori K, Kosuke M. Crystal structure of an archaeal intein-encoded homing endonuclease PI-PfuI. J Mol Biol. 2000;300:889–901. doi: 10.1006/jmbi.2000.3873. [DOI] [PubMed] [Google Scholar]
- 14.Matsamura H, Takahashi H, Inoue T, Yamamoto T, Hashimoto H, Nishioka M, Fujiwara S, Takagi M, Imanaka T, Kai Y. Crystal structure of intein homing endonuclease II encoded in DNA polymerase gene from hyperthermophilic archaeon Thermococcus kodakaraensis strain KOD1. Proteins. 2006;63:711–715. doi: 10.1002/prot.20858. [DOI] [PubMed] [Google Scholar]
- 15.Mizutani R, Anraku Y, Satow Y. Protein splicing of yeast VMA1-derived endonuclease via thiazolidine intermediates. J Synchr Rad. 2004;11:109–112. doi: 10.1107/s0909049503023495. [DOI] [PubMed] [Google Scholar]
- 16.Ding Y, Xu MQ, Ghosh I, Chen X, Ferrandon S, Lesage G, Rao Z. Crystal structure of a mini-intein reveals a conserved catalytic module involved in side chain cyclization of asparagine during protein splicing. J Biol Chem. 2003;278:39133–39142. doi: 10.1074/jbc.M306197200. [DOI] [PubMed] [Google Scholar]
- 17.Sun P, Ye S, Ferrandon S, Evans TCJ, Xu MQ, Rao Z. Crystal structure of an intein from the split dnaE gene of Synechocystis sp PCC6803 reveal the catalytic model without the penultimate histidine and the mechanism of zinc ion inhibition of protein splicing. J Mol Biol. 2005;353:1093–1105. doi: 10.1016/j.jmb.2005.09.039. [DOI] [PubMed] [Google Scholar]
- 18.Wood D, Wu W, Belfort G, Derbyshire V, Belfort M. A genetic system yields self-cleaving inteins for bioseparations. Nat Biotech. 1999;17:889–892. doi: 10.1038/12879. [DOI] [PubMed] [Google Scholar]
- 19.Hiraga K, Derbyshire V, Dansereau JT, Van Roey P, Belfort M. Minimization and stabilization of the Mycobacterium tuberculosis recA intein. J Mol Biol. 2005;354:916–926. doi: 10.1016/j.jmb.2005.09.088. [DOI] [PubMed] [Google Scholar]
- 20.Hall TMT, Porter JA, Young KE, Koonin EV, Beachy PA, Leahy DJ. Crystal structure of a hedgehog autoprocessing domain: conservation of structure, sequence and cleavage mechanism between hedgehog and self-splicing proteins. Cell. 1997;91:85–97. doi: 10.1016/s0092-8674(01)80011-8. [DOI] [PubMed] [Google Scholar]
- 21.Ghosh I, Sun L, Xu MQ. Zinc inhibition of protein trans-splicing and identification of regions essential for splicing and association of a split intein. J Biol Chem. 2001;276:24051–24058. doi: 10.1074/jbc.M011049200. [DOI] [PubMed] [Google Scholar]
- 22.Mills KV, Paulus H. Reversible inhibition of protein splicing by zinc ion. J Biol Chem. 2001;276:10832–10838. doi: 10.1074/jbc.M011149200. [DOI] [PubMed] [Google Scholar]
- 23.Nichols NM, Benner JS, Martin DD, Evans TCJ. Zinc ion effects on individual Ssp DnaE intein splicing steps: regulating pathway progression. Biochemistry. 2003;42:5301–5311. doi: 10.1021/bi020679e. [DOI] [PubMed] [Google Scholar]
- 24.Shemella P, Pereira B, Zhang Y, Van Roey P, Belfort G, Garde S, Nayak SK. Mechanism for Intein C-terminal Cleavage: A proposal from quantum mechanical calculations. Biophys J. 2006 doi: 10.1529/biophysj.106.092049. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu MQ, Comb DG, Paulus M, Noren CJ, Shao Y, Perler FB. Protein splicing: an analysis of the branched intermediate and its resolution by succinimide formation. EMBO J. 1994;23:5517–5522. doi: 10.1002/j.1460-2075.1994.tb06888.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu MQ, Perler FB. The mechanism of protein splicing and its modulation by mutation. EMBO J. 1996;15:5146–5153. [PMC free article] [PubMed] [Google Scholar]
- 27.Paulus H. Protein splicing and related forms of protein autoprocessing. Ann Rev Biochem. 2000;69:447–496. doi: 10.1146/annurev.biochem.69.1.447. [DOI] [PubMed] [Google Scholar]
- 28.Shao Y, Xu MQ, Paulus H. Protein splicing: characterization of the aminosuccinimide residue at the carboxyl terminus of the excised intervening sequence. Biochemistry. 1995;34:10844–10850. doi: 10.1021/bi00034a017. [DOI] [PubMed] [Google Scholar]
- 29.Mills KV, Paulus H. Biochemical mechanisms of intein-mediated protein splicing. In: Belfort M, Derbyshire V, Stoddard BL, Wood DW, editors. Homing Endonucleases and Inteins. Springer-Verlag; 2005. pp. 233–255. [Google Scholar]
- 30.Woo HA, Chae HZ, Hwang AC, Yang KS, Kang AW, Kim K, Rhee AG. Reversing the inactivation of peroxiredoxins caused by cysteine sulfinic acid formation. Science. 2003;300:653–656. doi: 10.1126/science.1080273. [DOI] [PubMed] [Google Scholar]
- 31.Sharma GN, Nickel C, Rahlfs S, Fischer M, Becker K, Karplus PA. Crystal structure of a novel Plasmodium falciparum 1-Cys peroxiredoxin. J Mol Biol. 2005;346:1021–1034. doi: 10.1016/j.jmb.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 32.Katayama Y, Hashimoto K, Nakayama H, Mino H, Nojiri M, Ono T-a, Nyunoya H, Yohda M, Takio K, Odaka M. Thiocyanate hydrolase is a cobalt-containing metalloenzyme with a cysteine-sulfinic acid ligand. J Am Chem Soc. 2006;128:728–729. doi: 10.1021/ja057010q. [DOI] [PubMed] [Google Scholar]
- 33.Werner E, Wende W, Pingoud A, Heinemann U. High resolution crystal structure of domain I of the Saccharomyces cerevisiae homing endonuclease PI-SceI. Nucl Acids Res. 2002;30:3962–3971. doi: 10.1093/nar/gkf523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Poland BW, Xu MQ, Quiocho FA. Structural insights into the protein splicing mechanism of PI-SceI. J Biol Chem. 2000;275:16408–16413. doi: 10.1074/jbc.275.22.16408. [DOI] [PubMed] [Google Scholar]
- 35.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Meth Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 36.Terwilliger TC, Berendzen J. Automated structure solution for MIR and MAD. Acta Crystallogr. 1999;D55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr. 2000;D56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 39.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for the building of protein models in electron-density maps and the location of errors in these maps. Acta Crystallogr. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 40.Laskowski RA, McArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:282–291. [Google Scholar]
- 41.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Raed RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr. 2005;D61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]