

Abstract
Ginkgo biloba (ginkgo) is one of most frequently used botanical dietary supplements. The bioactive constituents include the terpenoid lactones consisting of bilobalide and the ginkgolides A, B, C, and J. A new assay based on high performance liquid chromatography-electrospray tandem mass spectrometry (LC-MS-MS) was developed for the measurement of the terpenoid lactones in ginkgo products such as leaf powder and extracts. Initially, the MS-MS fragmentation pathways of ginkgolides were investigated to identify abundant fragment ions that might be useful for the sensitive and selective detection of ginkgolides and bilobalide during LC-MS-MS. Then, sample preparation and clean-up procedures were streamlined to maximize throughput by taking advantage of the selectivity of LC-MS-MS detection. Analyte recoveries exceeded 90%, the intra-assay and inter-assay relative standard deviations were less than 5%, the relative error was less than 8%, and the limits of detection and quantification were from 3.6–120 fmol and 11–350 fmol, respectively, depending upon the analyte that was injected onto the HPLC column. Therefore, this LC-MS-MS assay facilitated the rapid quantitative analysis of ginkgolides A, B, C, and J, and bilobalide in ginkgo dietary supplements with excellent recovery, reproducibity, accuracy, and sensitivity.
Keywords: bilobalide; Ginkgo biloba; ginkgolide, terpene trilactones; LC-MS-MS
INTRODUCTION
Ginkgo biloba (ginkgo) ranks among the 10 most popular botanical dietary supplements with sales exceeding $ 1 billion US dollars per year [1–2]. Ginkgo has been used in traditional Chinese medicine for many centuries to treat ailments as diverse as asthma, alcohol abuse and bladder inflammation, and currently, ginkgo leaf powders and extracts are being used for the treatment of cerebral and peripheral circulatory disorders [2–4]. Some of the pharmacological activities of ginkgo products that have been reported include antioxidant, anti-ischemic and neuro-protective effects, and cardiovascular, cerebrovascular, and peripheral circulatory benefits [5–9]. Clinical studies have suggested that the consumption of ginkgo extracts can enhance memory and alleviate symptoms of Alzheimer’s disease [10–13]. Although Solomon, et al. [14], reported negative results in a study on possible memory enhancement by ginkgo, the six-week intervention period might have been too short to observe an effect. In addition, the study by Solomon, et al., did not specify the ginkgo product used thereby preventing confirmation by other researchers.
The pharmacologically active constituents of ginkgo include flavonoids and the terpenoid lactones ginkgolides and bilobalide. Ginkgolides A, B, C, and J selectively inhibit platelet aggregating factor [15–16], and bilobalide is believed to function as a neuro-protectant [17]. The structures of these compounds are shown in Figure 1. Based on the biological activities of these particular compounds, commercial ginkgo products are usually standardized to the levels of the terpenoid lactones and flavonoids [18,19].
Figure 1.
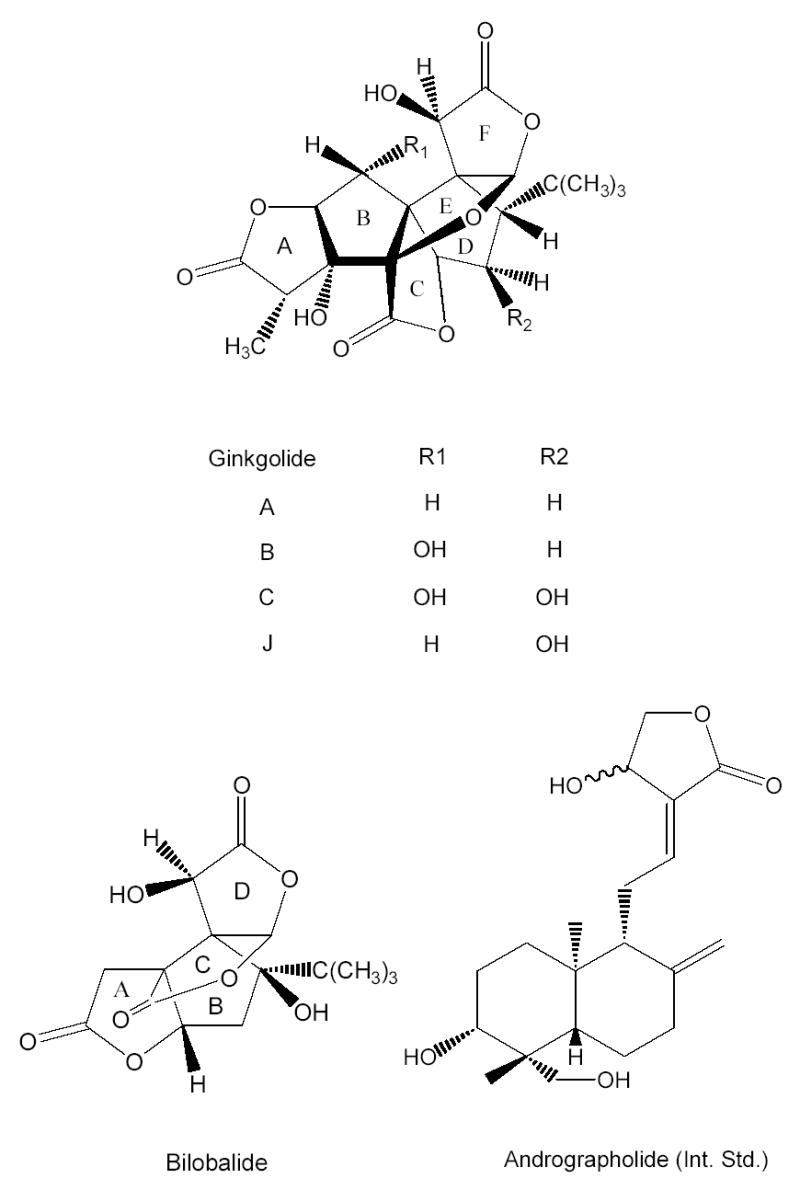
The structures of ginkgolides A, B, C, and J, bilobalide, and andrographolide (internal standard).
To standardize commercial ginkgo products and to carry out quality control studies, assays have been reported based on a wide variety of analytical techniques including HPLC or capillary electrophoresis with UV absorbance detection [20,21], HPLC with refractive index detection [22], HPLC with evaporative light scattering detection (ELSD) [23], gas chromatography (GC) with flame ionization detection [24], GC-mass spectrometry (GC-MS) [25], NMR [26], and liquid chromatography-mass spectrometry (LC-MS) [27–30]. Although ginkgo extracts usually contain considerable quantities of flavonoids which have strong UV absorption, terpenoid lactones lack UV chromophores so that even trace amounts of co-eluting compounds such as the abundant flavonoids might interfere with their detection. Therefore assays based on UV absorption lack sensitivity and selectivity for the quantitative analysis of ginkgolides and bilobalide in complex botanical extracts. Refractive index detection lacks both sensitivity and selectivity and is more suitable for monitoring preparative HPLC separations than for quantitative analysis. ELSD responds to both terpenoid lactones and flavonoids, although the standard curves are non-linear. However, like UV and refractive index detection, ELSD lacks selectivity. As a result, pre-purification of samples and carefully designed HPLC separations are necessary to eliminate interfering substances and to resolve all of the analytes prior to detection. Although both GC with flame ionization detection and GC-MS are highly sensitive analytical techniques, GC-based methods require complicated and time-consuming sample preparation and derivatization. Finally, NMR-based assays of ginkgolides and bilobalide lack sensitivity and are slow compared to LC-MS assays.
Like other HPLC-based assays, LC-MS does not require sample derivatization prior to analysis. Furthermore, the selectivity of mass selective detection minimizes the need for complete chromatographic resolution of analytes and matrix compounds. However, all of the published LC-MS assays of which we are aware are based only on the detection of molecular ion species such as deprotonated molecules, protonated molecules, sodium adducts, or ammonium adducts of ginkgolides and bilobalide. Given the complexity of biological matrices, there is the potential for interference by isobaric ions from other constituents. In the present study, a highly sensitive, accurate, precise, and selective assay based on HPLC-negative ion electrospray tandem mass spectrometry (LC-MS-MS) was developed for the simultaneous quantitative analysis of four ginkgolides and bilobilide in ginkgo extracts.
EXPERIMENTAL
Reagents and Chemicals
Ginkgolides A, B, C, and J, bilobalide, and andrographolide (internal standard) were obtained from a repository of natural products that had been isolated and characterized previously from plant materials in our laboratory. The purity and molecular weight of each standard was verified using LC-MS as described below. The purities exceeded 99.8%. Nine commercially available G. biloba products were purchased at local pharmacies in Chicago, IL, and are described below. All the solvents were HPLC grade or better and were purchased from Fisher Scientific (Hanover Park, IL).
Preparation of Standard Solutions and Samples
The ginkgolide and bilobalide standards (1 mg each) were weighed to the nearest 0.05 mg and dissolved in methanol in a 10-mL volumetric flask to make a stock solution. All working standard solutions for the calibration curves were prepared by diluting the stock solutions using methanol/water (1:1, v/v). Note that all working solutions were stored at −20 °C and warmed to room temperature immediately prior to analysis.
The linear standard curves for bilobalide and each ginkgolide (except ginkgolide J) were constructed using 10 points corresponding to 50, 100, 200, 500, 800, 1000, 2000, 3000, 4000, and 5000 pg of each standard injected on-column. Since the limit of quantitation of ginkgolide J was 150 pg injected on-column, only the highest 8 points were used for the linear standard curve of this analyte. For non-linear polynomial curve fitting, an additional 10 data points were used for the standard curves corresponding to 10,000, 20,000, 30,000, 40,000, 50,000, 60,000, 70,000, 80,000, 90,000, and 100,000 pg (injected on-column) equaling a total of 20 data points per analyte (18 data points for ginkgolide J). Three sets of quality control samples were prepared separately to represent the low, middle, and high ranges of the linear calibration curves (which ranged from approximately 50 to 5000 pg of analyte injected onto the HPLC column). These quality control samples contained 650, 1600 or 4000 pg of each bilobalide or ginkgolide standard.
Three ginkgo tablets or capsules from each commercial ginkgo product were selected, and each ginkgo tablet or the contents of each ginkgo capsule were weighed to the nearest 0.05 mg. The terpenoid lactones from each sample were extracted three times by sonication in 15-mL portions of methanol for 1 h. No other processing of the samples was carried out. Each extract was filtered through Whatman (Clifton, NJ) #1 filter paper into a 250-mL round-bottom flask, and the combined methanol extracts of each sample were evaporated to dryness at 45–50 °C under reduced pressure. The residue of each extract was redissolved in methanol, transferred to a 10-mL volumetric flask, and made up to the volume with methanol. A 10-μL aliquot was removed, diluted 100- or 1000-fold with methanol/water (1:1, v/v), and mixed with the internal standard andrographolide. The final concentration of the internal standard in each aliquot was 0.61 μM (200 pg/μL). After centrifugation, 10-μL aliquots of each supernatant were injected onto the LC-MS-MS for analysis as described below. Each sample was analyzed three times. The recoveries of the ginkgolides and bilobalide were determined by spiking 0.1 or 0.5 mg of each analyte into accurately weighed samples which had already been analyzed for their original ginkgolide and bilobalide content. The sample preparation was identical to that described above for ginkgo dietary supplements.
Tandem Mass Spectrometry
All mass spectra were acquired using a Micromass (Manchester, UK) Quattro II triple quadrupole mass spectrometer equipped with an electrospray source and a Waters (Milford, MA) 2690 HPLC system. A 1-μM solution of each compound was infused at 20 μL/min, and either positive or negative ion electrospray mass spectra were acquired. The capillary voltages were 3.3 kV and −3.1 kV for positive and negative ion analysis, respectively. The cone voltage was optimized for each compound and ranged from 50 – 75 V for positive ion electrospray and from 30 – 42 V for negative ion electrospray. MS-MS product ion tandem mass spectra of the deprotonated molecules were acquired using collision-induced dissociation (CID) with argon as the collision gas at 1.1×10−3 Torr. The collision energy was optimized to between 10 and 15 eV for each compound. The most abundant product ion of each compound was selected for use with multiple reaction monitoring (MRM) during LC-MS-MS.
LC-MS-MS
HPLC separations during LC-MS-MS were carried out using a Waters XterraTM C18 reversed phase HPLC column (3.5 μM, 2.1×100 mm). The 15-min gradient separation consisted of 10–70% aqueous methanol at 250 μL/min. Negative ion electrospray was used for ionization at a source temperature of 140 °C and a desolvation temperature of 320 °C. Nitrogen was used as the nebulizing gas at 20 L/hr and as a drying gas at 450 L/hr. CID was carried out as described above for MS-MS.
RESULTS AND DISCUSSION
Tandem Mass Spectrometry of Ginkgo Terpenoid Lactones
During positive ion electrospray mass spectrometry, all four ginkgolides and bilobalide formed abundant [M+Na]+ ions as the base peaks, but no potassium or ammonium adducts were detected. Protonated molecules and complexes of protonated molecules with methanol, [MH+CH3OH]+, were observed at much lower abundances than the sodium adducts. The abundant [M+Na]+ ions were investigated as possible precursors for MRM during LC-MS-MS but were too stable to produce useful product ions during CID in the triple quadrupole mass spectrometer. Furthermore, the relative amounts of [M+Na]+ and [M+H]+ species were variable depending upon the trace amounts of sodium in the sample or mobile phase. Therefore, negative ion electrospray was investigated for use during LC-MS-MS.
Negative ion electrospray produced abundant deprotonated molecules of the terpenoid lactones that formed the base peak of each mass spectrum. In contrast to the multiple adducts observed during positive ion electrospray, no other adducts of the ginkgolides or bilobilide were detected. Some in-source fragmentation was observed corresponding to elimination of one or two carbon monoxide molecules from the [M-H]- precursors. The use of post-column addition of ammonia was investigated as a potential means to enhance the formation of deprotonated molecules. Since lactone hydrolysis occurred instead of enhancement of the deprotonated molecules, post-column addition of ammonia was not pursued further.
The product ion tandem mass spectra of the deprotonated molecules of the ginkgo terpenoid lactones were recorded and are shown in Figure 2. The most favorable fragmentation pathway of the deprotonated ginkgolides was loss of one or two carbon monoxide molecules. In each case, loss of two carbon monoxide molecules, [M-H-2CO]−, was the most abundant fragment pathway (Figure 2). The origins of the neutral losses of carbon monoxide were probably the lactone carbonyl groups of the A and C rings because these structures are highly strained. Although at much lower abundance compared to the neutral losses of carbon monoxide, the ginkgolides also fragmented to eliminate carbon dioxide. The loss of carbon dioxide was more abundant in the tandem mass spectra of ginkgolides A and J than in those of ginkgolides B and C. In addition, a product ion of m/z 319 at low abundance corresponding to the loss of two carbon dioxide neutrals was detected in the tandem mass spectrum of ginkgolide A (Figure 2).
Figure 2.
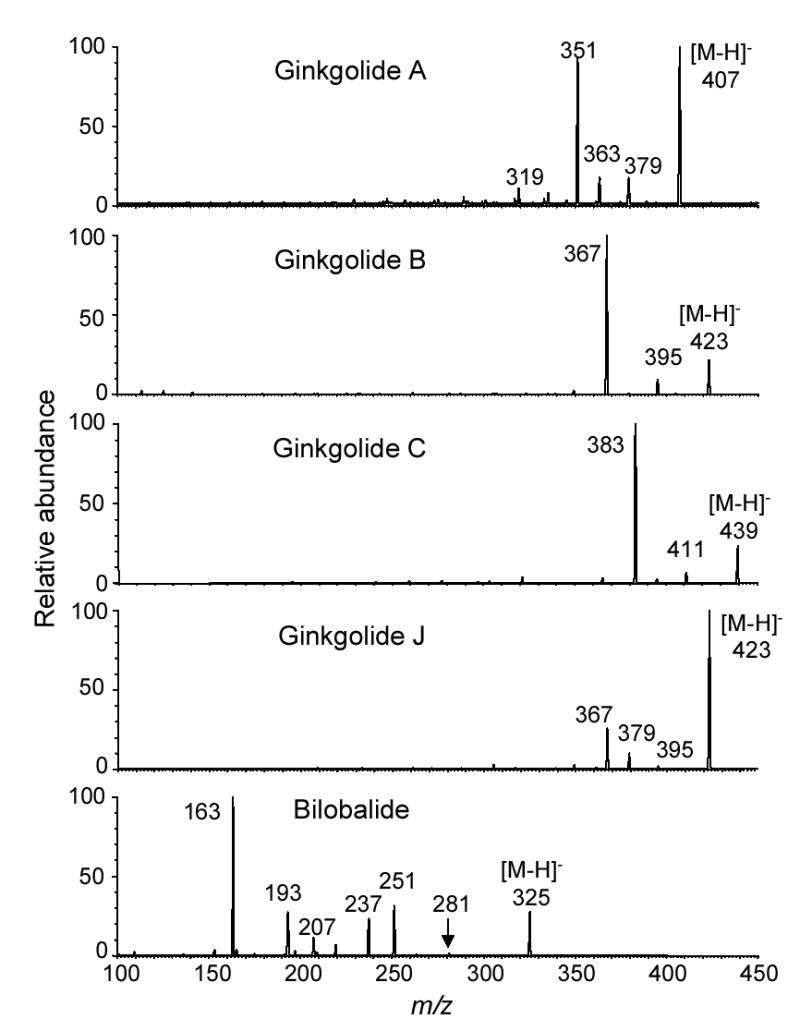
Negative ion electrospray product ion tandem mass spectra of bilobalide and ginkgolides A, B, C, and J.
Since ginkgolides contain a rigid carbon skeleton consisting of six fused 5-membered rings but bilobalide has a more flexible structure containing only four 5-membered rings, the tandem mass spectra of these species are significantly different. For example instead of eliminating carbon monoxide, deprotonated bilobalide at m/z 325 fragmented during MS-MS with CID to eliminate one, two, or three molecules of carbon dioxide, which were detected at m/z 281 (trace), 237, and 193, respectively (Figure 2). Loss of two or three carbon dioxide molecules was more favorable than elimination of only one molecule. The formation of the ion of m/z 251 was due to loss of both a tert-butyl group and a hydroxyl group, and the additional elimination of one and two carbon dioxide groups from the other rings resulted in the formation of the ions of m/z 207 and the base peak of m/z 163, respectively.
LC-MS-MS of Ginkgolides, Bilobalide and Ginkgo Extracts
Based on the negative ion tandem mass spectra of the deprotonated molecules of the ginkgolides and bilobalide shown in Figure 2, the most abundant fragment ions were selected for use during quantitative analysis using LC-MS-MS with MRM. The precursor ions in each case were the deprotonated molecules. For the ginkgolides, the most abundant product ions corresponded to the elimination of two carbon monoxide molecules from the deprotonated molecules. The precursor ion pairs used for MRM were m/z 407→351, 423→367, 439→383, and 423→367, for ginkgolides A, B, C, and J, respectively. In the case of bilobalide, the precursor/product ion pair was m/z 325→163.
Unlike previously published LC-MS assays of ginkgolides and bilobalide, an internal standard was added to each extract immediately before analysis using LC-MS-MS. The internal standard was used to control for any sample injection variation and instrument response changes that might occur during the course of multiple analyses. The botanical natural product andrographolide, a terpene lactone from Andrographis paniculata, was selected as the internal standard since it is similar in molecular weight and contains some of the same functional groups as bilobalide and the ginkgolides. Like the ginkgolides and bilobalide, andrographolide formed an abundant deprotonated molecule during negative ion electrospray, and like bilobalide, the deprotonated molecule fragmented during CID to eliminate carbon dioxide. The abundant product ion corresponding to elimination of both carbon dioxide and water from the deprotonated molecule of andrographolide was used during MRM. This ion pair for MRM of andrographolide was m/z 349→287.
The LC-MS-MS analysis of a standard mixture of bilobalide and four ginkgolides is shown in Figure 3. Bilobalide eluted first at 7.6 min followed by ginkgolides J, C, A, and B at 9.0, 9.3, 11.2 and 11.5 min, respectively. Baseline separation was obtained for each of these terpenoid lactones in less than 12 min. The internal standard andrographolide eluted last at 13.9 min. Therefore, the entire separation was completed in approximately 14 min, which is as fast as any of the previously published HPLC-based assays of these compounds. To the best of our knowledge, the fastest HPLC method for these compounds that was published previously was that of Jensen, et al., (29) who reported the partial separation of bilobalide and four ginkgolides in 14 min.
Figure 3.
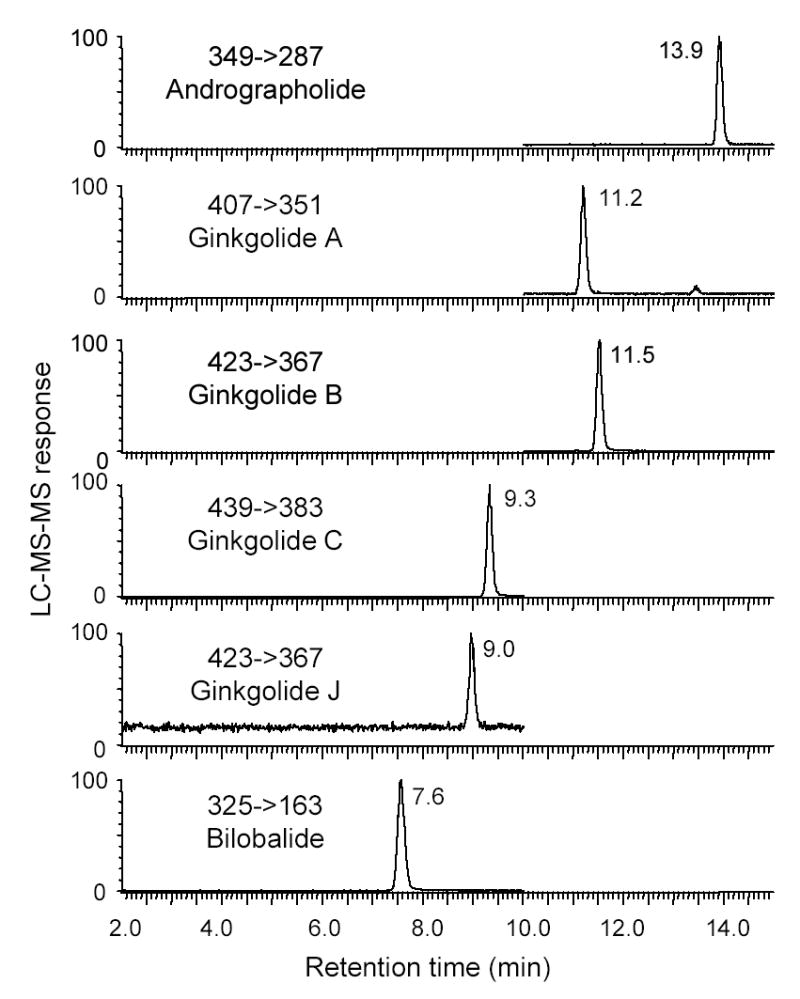
Negative ion electrospray LC-MS-MS analysis of a mixture of ginkgolides A, B, C, J, bilobalide, and andrographolide (internal standard). Multiple reaction monitoring (MRM) was used to measure the most abundant product ion of the deprotonated molecule of each analyte following collision-induced dissociation (CID).
LC-MS-MS calibration curves were prepared using bilobalide and ginkgolide standards. The data for each analyte could be fit to straight line with a good correlation coefficient (R2) extending over only two orders of magnitude (see Table 1). For example, the correlation coefficient for ginkgolide A was 0.9972 for 50–5000 pg injected on-column. However, these same data fit much better to a curve described by a second-order polynomial. In the case of ginkgolide A, this non-linear second-order polynomial fit produced a correlation coefficient (R2) of 1.000 for ginkgolide A over more than three orders of magnitude, 50–100,000 pg. The correlation coefficients for linear and second-order polynomial non-linear fits of the calibration curve data for all five analytes are shown in Table 1. The linear standard curves were used for the quantitative analysis of bilobalide and ginkgolides in the ginkgo commericial products (see results below), but the use of the non-linear standard curves produced similar results.
Table 1.
Limits of Detection (LOD), Limits of Quantification (LOQ) and Correlation Coefficients for the LC-MS-MS Calibration Curves of Ginkgolides and Bilobalide
| Correlation coefficient | ||||
|---|---|---|---|---|
| Compound | LODa fmol (pg) on-column | LOQb fmol (pg) on-column | R2 (linear) | R2 (2nd-order polynomial) |
| Ginkgolide A | 35 (15) | 118 (50) | 0.9972 | 1.0000 |
| Ginkgolide B | 3.7 (1.3) | 11 (4) | 0.9956 | 0.9996 |
| Ginkgolide C | 3.6 (1.6) | 11 (5) | 0.9950 | 0.9995 |
| Ginkgolide J | 120 (50) | 350 (150) | 0.9971 | 0.9998 |
| Bilobalide | 4.9 (1.6) | 15 (5) | 0.9995 | 1.0000 |
S/N = 3
S/N = 10
The limits of detection (LOD), defined as a signal-to-noise ratio of 3:1, ranged from 3.6 fmol (1.6 pg) injected on-column for ginkgolide C to 120 fmol (50 pg) for ginkgolide J. Defining the limit of quantification (LOQ) as a signal-to-noise ratio of 10:1, the LOQ values for these compounds ranged from 11 fmol (4–5 pg) injected on-column for ginkgolides B and C to 350 fmol (150 pg) for ginkgolide J. The LOD and LOQ values for all five analytes are summarized in Table 1. Compared to the LC-MS assay reported by Jensen, et al. [29], which to our knowledge is the most sensitive LC-MS method reported previously, our LOD values for GB, GC, and BB are approximately 10-fold more sensitive (15-fold, 12-fold, and 6.2-fold, respectively) and our LOD values for GA and GJ are in the same range. Jensen, et al. did not report values for LOQ for comparison.
The recovery of each terpenoid lactone from the G. biloba products was determined by spiking 0.1 and 0.5 mg of each standard compound into three sets of an accurately weighed G. biloba product (Nature’s Resource® Ginkgo Biloba). Extraction and LC-MS-MS analysis of the spiked samples were carried out as described in the Experimental section. The recovery data are summarized in Table 2 and ranged from 90.0–99.4% at the 0.1 mg level to 92.6–102.2% at the 0.5 mg level. The relative standard deviations for the recoveries of the ginkgolides and bilobalide were 1.8–5.5% (see Table 2). Therefore, the extraction procedure was shown to give excellent recovery of each of the five analytes.
Table 2.
Recoveries of Ginkolides and Bilobalide from G. biloba Leaf Powdera
| Compound | 0.1 mg spike | 0.5 mg spike | ||
|---|---|---|---|---|
| Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | |
| Ginkgolide A | 96.1b | 2.2 | 102.2 | 3.8 |
| Ginkgolide B | 99.4 | 3.8 | 93.5 | 5.5 |
| Ginkgolide C | 90.0 | 3.3 | 92.6 | 1.8 |
| Ginkgolide J | 91.8 | 3.5 | 93.3 | 3.6 |
| Bilobalide | 90.1 | 4.1 | 93.2 | 4.4 |
Nature’s Resource® Ginkgo Biloba
n = 3 for each measurement
The precision and accuracy of this LC-MS-MS assay were evaluated by analyzing three sets of standards representing the low, middle, and high concentrations of the standard curve. In addition to intra-day variation, analyses were carried out on three separate days to evaluate inter-day variation. These data are shown in Table 3. In summary, the relative standard deviations for the intra-day and inter-day measurements of each ginkgolide and bilobalide were less than 5%, and the relative errors for these intra-day and inter-day measurements were less than 8%. Therefore, this LC-MS-MS assay was shown to provide both high precision and high accuracy for the quantitative analysis of four ginkgolides and bilobalide. To the best of our knowledge, previous LC-MS assays for the analysis of ginkgolides and bilobalide did not include the use of an internal standard and did not report the relative errors. The precision and accuracy of LC-MS or LC-MS-MS assays would be expected to decrease without an internal standard.
Table 3.
Precision and Accuracy of LC-MS-MS Analysis of Ginkgolides and Bilobalide
| Day-1
|
Day-2
|
Day-3
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | Actual amount (pg) | Measured amount (pg) | RSDa (%) | REb (%) | Measured amount (pg) | RSD (%) | RE (%) | Measured amount (pg) | RSD (%) | RE (%) | Inter-day RSD (%) |
| Ginkgolide A | 650 | 689 | 2.82 | 6.00 | 696 | 3.76 | 7.07 | 693 | 2.27 | 6.58 | 0.51 |
| 1600 | 1659 | 4.4 | 3.69 | 1573 | 0.61 | −1.68 | 1658 | 3.88 | 3.65 | 3.03 | |
| 4000 | 3817 | 1.86 | −4.57 | 3990 | 4.46 | −0.24 | 3906 | 1.08 | −2.35 | 3.17 | |
| Ginkgolide B | 650 | 699 | 3.13 | 7.62 | 695 | 3.34 | 6.95 | 694 | 1.03 | 6.76 | 0.38 |
| 1600 | 1689 | 1.87 | 5.58 | 1648 | 2.34 | 2.97 | 1644 | 4.54 | 2.75 | 1.50 | |
| 4000 | 3754 | 2.58 | −6.16 | 4000 | 2.27 | 0.00 | 3888 | 3.00 | −2.80 | 2.22 | |
| Ginkgolide C | 650 | 676 | 2.58 | 3.94 | 702 | 2.60 | 8.08 | 638 | 2.76 | −1.84 | 4.79 |
| 1600 | 1671 | 3.74 | 4.44 | 1608 | 2.11 | 0.51 | 1650 | 2.34 | 3.14 | 1.95 | |
| 4000 | 3808 | 2.97 | −4.80 | 4036 | 4.46 | 0.91 | 3849 | 2.47 | −3.76 | 3.12 | |
| Ginkgolide J | 650 | 673 | 2.85 | 3.60 | 637 | 1.45 | −2.03 | 701 | 4.98 | 7.81 | 4.79 |
| 1600 | 1646 | 3.63 | 2.85 | 1622 | 3.98 | 1.37 | 1728 | 4.63 | 8.01 | 3.34 | |
| 4000 | 3734 | 1.01 | −6.65 | 4018 | 3.46 | 0.36 | 3954 | 2.57 | −1.14 | 3.78 | |
| Bilobalide | 650 | 667 | 2.40 | 2.67 | 696 | 3.93 | 7.07 | 631 | 4.17 | −2.85 | 4.90 |
| 1600 | 1591 | 4.47 | −0.53 | 1588 | 4.97 | −0.69 | 1713 | 4.46 | 7.08 | 4.37 | |
| 4000 | 3813 | 2.34 | −4.68 | 3976 | 3.00 | −0.60 | 3964 | 0.83 | −0.91 | 2.32 | |
RSD = relative standard deviation (n = 3)
RE = relative error
The levels of bilobalide and four ginkgolides were measured in nine commercially available G. biloba products using this new LC-MS-MS assay. To illustrate the appearance of the original data, LC-MS-MS chromatograms for the analysis of Nature’s Resource® Ginkgo Biloba are shown in Figure 4. The results of all of the LC-MS-MS analyses are summarized in Table 4. All four ginkgolides and bilobalide were detected in each of the dietary supplements indicating that they contained G. biloba as labeled. Bilobalide and ginkgolide A were the most abundant species, followed in decreasing order by ginkgolides B, C and J. Six of the nine commercial products declared their total terpenoid lactone contents (as percent weight) on the labels, and these values are compared with our measured values in Table 4. Our data indicate that the terpenoid lactone contents of three of the commercial G. biloba products were higher (7–22%) and three were lower (6–24%) than the values determined using LC-MS-MS. These variations are probably the result of the use of different analytical methods by the manufacturer (with different levels of accuracy and precision) and/or variation between the samples tested in our laboratory and those tested by the manufacturer.
Figure 4.
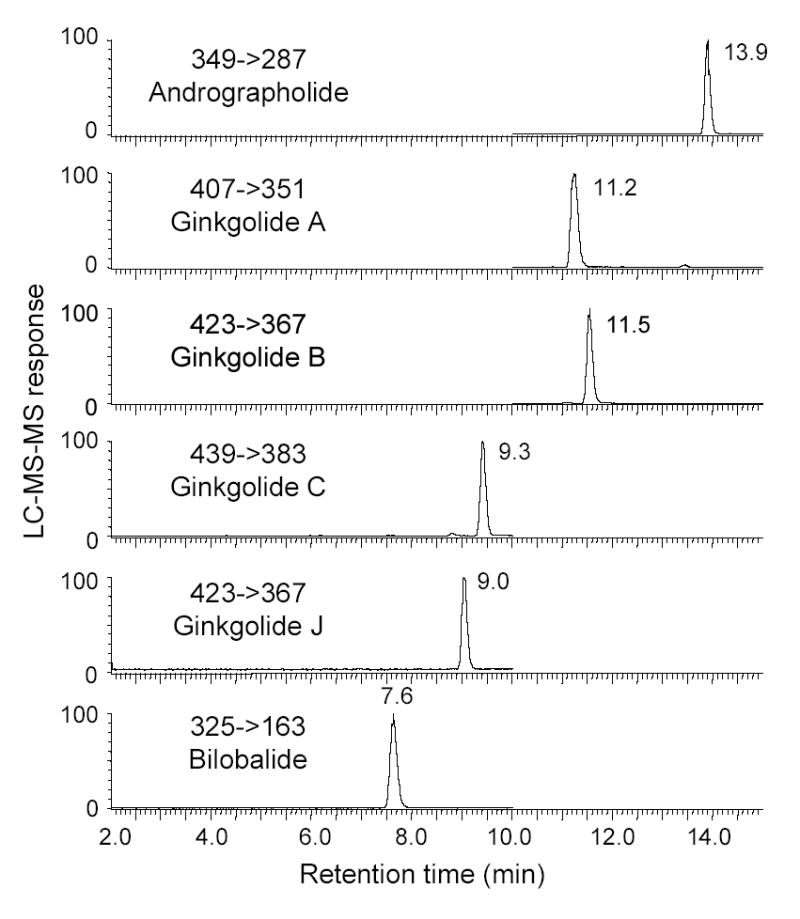
Negative ion electrospray LC-MS-MS MRM analysis of bilobalide and ginkgolides A, B, C, and J extracted from the commercial G. biloba dietary supplement Nature’s Resource® Ginkgo Biloba.
Table 4.
Terpenoid Lactones (wt%) in G. biloba Dietary Supplements
| Total terpenoid lactones
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Commercial Product | Lot # | GA %a | GB % | GC % | GJ % | BB % | Measured | Declared on label |
| Nature’s Resource®Ginkgo Biloba | LB11565N | 0.20 ± 0.01 | 0.09 ± 0.01 | 0.11 ± 0.01 | 0.03 ± 0.00 | 0.19 ± 0.01 | 0.62 | 0.66 |
| Finest Ginkgo-MemoTM Concentrate | JJ10559 | 0.41 ± 0.02 | 0.12 ± 0.01 | 0.08 ± 0.00 | 0.07 ± 0.00 | 0.54 ± 0.01 | 1.22 | - |
| Ginkoba® | FD0534A | 0.10 ± 0.01 | 0.15 ± 0.00 | 0.03 ± 0.00 | 0.05 ± 0.00 | 0.24 ± 0.01 | 0.57 | - |
| Finest Natural Ginkgo Biloba | 6764-3 | 0.32 ± 0.02 | 0.10 ± 0.00 | 0.05 ± 0.00 | 0.05 ± 0.00 | 0.20 ± 0.00 | 0.72 | 0.86 |
| Nature’s RewardTM Ginkogin | CG912-682B | 0.04 ± 0.00 | 0.03 ± 0.00 | 0.02 ± 0.00 | 0.02 ± 0.00 | 0.03 ± 0.00 | 0.19 | 0.25 |
| Natrol®Ginkgo Biloba | 941583 | 0.27 ± 0.00 | 0.24 ± 0.00 | 0.10 ± 0.00 | 0.04 ± 0.00 | 0.44 ± 0.01 | 1.09 | - |
| Sundown® Ginkgo Biloba | 991534 | 0.12 ± 0.00 | 0.09 ± 0.00 | 0.02 ± 0.00 | 0.02 ± 0.00 | 0.14 ± 0.01 | 0.39 | 0.32 |
| Sundown® Ginkgo Biloba xtra® | 278024 | 0.27 ± 0.01 | 0.26 ± 0.00 | 0.07 ± 0.00 | 0.02 ± 0.00 | 0.33 ± 0.00 | 0.95 | 0.84 |
| Sundown® Ginkgo Biloba | 243240 | 0.29 ± 0.01 | 0.27 ± 0.01 | 0.09 ± 0.00 | 0.02 ± 0.00 | 0.39 ± 0.02 | 1.06 | 0.99 |
Three different samples of each commercial product were used per determination (n=3), and the extract of each sample was measured three times.
CONCLUSIONS
A quantitative assay was developed based on LC-MS-MS for the analysis of the pharmacologically active terpenoid lactones in G. biloba dietary supplements. Using this assay bilobalide and ginkgolides A, B, C, and J could be measured with high precision, accuracy, and sensitivity. The presence of flavonoids, which are another group of pharmacologically active compounds in ginkgo products, did not interfere with this assay. Therefore, sample preparation could be streamlined and HPLC separation time could be kept to a minimum. To the best of our knowledge, this assay is unsurpassed in terms of sensitivity, selectivity, and speed for the measurement of terpenoid lactones in ginkgo products.
Acknowledgments
This research was supported by grant P50 AT00155 provided jointly by the Office of Dietary Supplements (ODS), the National Center for Complementary and Alternative Medicine (NCCAM), the Office for Research on Women’s Health (ORWH), and the National Institute of General Medicine (NIGMS) of the National Institutes of Health (NIH). The contents of this paper are solely the responsibility of the authors and do not necessarily represent the official views of the ODS, NCCAM, ORWH, NIGMS, or NIH.
References
- 1.Mar C, Bent S. Western J Med. 1999;171:168–171. [PMC free article] [PubMed] [Google Scholar]
- 2.van Beek TA. Ginkgo biloba. Harwood Academic Publishers; Australia: 2000. [Google Scholar]
- 3.Perry LM. Medicinal Plants of East and Southeast Asia: Attributed Properties and Uses. MIT Press; Cambridge, MA: 1984. [Google Scholar]
- 4.Deng Q. Drug Perspect. 1988;1:57–58. [Google Scholar]
- 5.Diamond BJ, Schiflett SC, Feiwel N, Matheis Noskin O, Richard JA, Schonenberger NE. Arch Phys Med Rehabil. 2000;81:668–678. doi: 10.1016/s0003-9993(00)90052-2. [DOI] [PubMed] [Google Scholar]
- 6.McKenna DJ, Jones K, Hughes K. Altern Ther Health Med. 2001 7;70–86:88–90. [PubMed] [Google Scholar]
- 7.Kleijnen J, Knipschild P. Brti J Clin Pharmacol. 1992;34:352–358. doi: 10.1111/j.1365-2125.1992.tb05642.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeFeudies FV. Ginkgo biloba Extract (Egb 761): Pharmacological Activities and Clinical Applications. Elsevier; Paris, France: 1991. [Google Scholar]
- 9.Fugh-Berman A, Cott JM. Psychosom Med. 1999;61:712–728. doi: 10.1097/00006842-199909000-00012. [DOI] [PubMed] [Google Scholar]
- 10.Kanowski S, Herrmann WM, Stephen K, Wierich W, Horr R. Pharmacopsychiatry. 1996;29:47–56. doi: 10.1055/s-2007-979544. [DOI] [PubMed] [Google Scholar]
- 11.Le Bars P, Katz MM, Berman N, Itli TM, Freedman AM, Schatzberg AF. JAMA. 1997;278:1327–1332. doi: 10.1001/jama.278.16.1327. [DOI] [PubMed] [Google Scholar]
- 12.Maurer K, Dierks RI, Frolich L. J Psychiat Res. 1997;31:645–655. doi: 10.1016/s0022-3956(97)00022-8. [DOI] [PubMed] [Google Scholar]
- 13.Luo Y. J Alzheimers Dis. 2001;3:401–407. doi: 10.3233/jad-2001-3407. [DOI] [PubMed] [Google Scholar]
- 14.Solomon PR, Adams F, Silver A, Zimmer J, DeVeaux R. JAMA. 2002;288:835–40. doi: 10.1001/jama.288.7.835. [DOI] [PubMed] [Google Scholar]
- 15.Smith PF, MacLennan K, Darlington CL. J Ethnopharmacol. 1996;50:131–139. doi: 10.1016/0378-8741(96)01379-7. [DOI] [PubMed] [Google Scholar]
- 16.Li CL, Wong YY. Planta Med. 1997;63:563. doi: 10.1055/s-2006-957768. [DOI] [PubMed] [Google Scholar]
- 17.Bruno C, Cuppini R, Sartini S, Cecchini T, Ambrogini P, Bombardelli E. Planta Med. 1993;59:302–307. doi: 10.1055/s-2006-959686. [DOI] [PubMed] [Google Scholar]
- 18.Sticher O. Planta Med. 1993;59:2–11. doi: 10.1055/s-2006-959593. [DOI] [PubMed] [Google Scholar]
- 19.Camponovo FF, Soldati F. In: Medicinal and Aromatic Plants - Industrial Profiles, Vol. 12, 245–265. van Beek TA, editor. Harwood; Amsterdam, The Netherlands: 2000. [Google Scholar]
- 20.Pietta P, Mauri PL, Rava A. J Pharm Biomed Anal. 1992;10:1077–1079. doi: 10.1016/0731-7085(91)80123-q. [DOI] [PubMed] [Google Scholar]
- 21.Oehrle SA. J Liq Chromatogr. 1995;18:2855–2859. [Google Scholar]
- 22.van Beek TA, Scheeren HA, Rantio T, Melger WC, Lelyveld GP. J Chromatogr. 1991;543:373–387. [Google Scholar]
- 23.Li W, Fitzloff JF. J Pharm Biomed Anal. 2002;30:67–75. doi: 10.1016/s0731-7085(02)00201-7. [DOI] [PubMed] [Google Scholar]
- 24.Lolla E, Paletti A, Peterlongo F. Fitoterapia. 1998;69:513–519. [Google Scholar]
- 25.Biber A, Koch E. Planta Med. 1999;65:192–193. doi: 10.1055/s-2006-960467. [DOI] [PubMed] [Google Scholar]
- 26.van Beek TA, van Veldhuizen A, Leyveld GP, Piron I, Lankhorst PP. Phytochem Anal. 1993;4:261–268. [Google Scholar]
- 27.Mauri P, Migliazza B, Pietta P. J Mass Spectrom. 1999;34:1361–1367. doi: 10.1002/(SICI)1096-9888(199912)34:12<1361::AID-JMS895>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 28.Li X-F, Ma M, Scherban K, Tam YK. Analyst. 2002;127:641–646. doi: 10.1039/b200849a. [DOI] [PubMed] [Google Scholar]
- 29.Jensen AG, Ndjoko K, Wolfender J-L, Hostettmann K, Camponovo FF, Soldati F. Phytochem Anal. 2002;13:31–38. doi: 10.1002/pca.614. [DOI] [PubMed] [Google Scholar]
- 30.Mauri P, Simonetti P, Gardana C, Morazzoni PB, Bombardelli E, Pietta P. Rapid Commun Mass Spectrom. 2001;15:929–934. doi: 10.1002/rcm.316. [DOI] [PubMed] [Google Scholar]
- 31.van Beek TA, Piron I. Phytochem Anal. 1993;4:109–114. [Google Scholar]
- 32.Steinke B, Muller B, Wagner H. Planta Med. 1993;59:155–160. doi: 10.1055/s-2006-959633. [DOI] [PubMed] [Google Scholar]