

Abstract
DNA replication of bacmid-derived constructs of the Autographa californica multiple nucleocapsid nucleopolyhedrovirus (AcMNPV) was analyzed by field inversion gel electrophoresis (FIGE) in combination with digestion at a unique Eco81I restriction enzyme site. Three constructs were characterized: a parental bacmid, a bacmid deleted for the alkaline nuclease gene, and a bacmid from which the gp64 gene had been deleted. The latter was employed as a control for comparison with the alkaline nuclease knockout because neither yields infectious virus and their replication is limited to the initially transfected cells. The major difference between DNA replicated by the different constructs was the presence in the alkaline nuclease knockout preparations of high concentrations of relatively small, sub-genome length DNA in preparations not treated with Eco81I. Furthermore, upon Eco81I digestion, the alkaline nuclease knockout bacmid also yielded substantially more sub genome size DNA than the other constructs. Electron microscopic examination of cells transfected with the alkaline nuclease knockout indicated that, in addition to a limited number of normal-appearing electron-dense nucleocapsids, numerous aberrant capsid-like structures were observed indicating a defect in nucleocapsid maturation or in a DNA processing step that is necessary for encapsidation. Because of the documented role of the baculovirus alkaline nuclease and its homologs from other viruses in homologous recombination, these data suggest that DNA recombination may play a major role in the production of baculovirus genomes.
INTRODUCTION
The Baculoviridae is a family of viruses with large, double-stranded, circular DNA genomes of 80 to 180 kb, depending on the strain. The Autographa californica multiple nucleocapsid nucleopolyhedrovirus (AcMNPV) is the type NPV species and is widely used to generate recombinant viruses for expressing heterologous genes. Six viral factors are necessary for replication of plasmids in transient replication assays, including a transactivator of early gene transcription (IE-1), a DNA polymerase, a DNA helicase, a DNA primase (LEF-1), a primase accessory factor (LEF-2), and a single-stranded DNA binding protein (LEF-3) are necessary for replication of plasmids in transient replication assays (Kool et al., 1994; Todd et al., 1995). Although these factors are capable of synthesizing DNA in transfected insect cells, other factors that are likely required for the production of mature genomes are not well characterized. One such factor, called alkaline nuclease (AN) is conserved in all baculovirus genomes sequenced to date. In addition, homologs of AN are widely distributed in eubacteria, archea, and eukaryotes (Aravind et al., 2000). These enzymes participate in the repair of dsDNA breaks and in homologous recombination and are thought to play a vital role in maintaining genome integrity. The best-studied example is the Red system from bacteriophage λ (for review see (Kuzminov, 1999; Poteete, 2001; Stahl, 1998). This system includes the λ exonuclease (Redα), which degrades linear dsDNA from the 5’ ends producing 3’ overhangs, which serve as intermediates in recombination (Little, 1967). During recombination, λ exonuclease interacts with an ssDNA-binding protein (Redβ), which promotes renaturation of complementary strands thereby mediating strand annealing and strand invasion (Cassuto et al., 1971; Li et al., 1998). Comprehensive sequence analyses indicates that the baculovirus AN belong to the same family as the λ exonuclease (Aravind et al., 2000; Bujnicki and Rychlewski, 2001). In addition, the AN of AcMNPV has been shown to interact with the baculovirus ssDNA binding protein (LEF-3) and possesses both a 5’-to-3’ exonuclease activity and structure dependent endonuclease activity (Mikhailov et al., 2003; Mikhailov et al., 2004).
We have previously demonstrated that a baculovirus bacmid construct lacking the alkaline nuclease gene (an) was non-infectious (Okano et al., 2004). However, it was able to replicate DNA after transfection into Sf-9 cells suggesting that the an deletion may cause a defect in the later stages of the replication cycle such as DNA processing. In this report, we describe the characterization of viral DNA that is synthesized by the an deletion mutant and compared it to parental bacmid DNA and a control gp64 deletion mutant by field inversion gel electrophoresis (FIGE). In addition, the structure of capsids produced by the AN knockout were examined by immunoelectron microscopy.
RESULTS
FIGE analysis of viral replication intermediates from infected Sf-9 cells
To characterize baculovirus DNA intermediates produced during replication, we used the FIGE system. We initially focused on analyzing replicated viral DNA in the size range of 50 to 200kb. To accomplish this, FIGE program 4 (Anon., 1993) was selected because under these conditions unit length genomic DNA is resolved. In order to detect unit length viral DNA, we introduced a unique Eco81I restriction site into our bacmid constructs (see Materials and Methods) (Fig. 1). We first examined the DNA from cells infected with budded virus derived from the parental-GUS(Eco81I) bacmid (Fig. 2A) from 0 to 72 h.p.i. and compared it to viral DNA extracted from budded virus (BV). When the DNA was uncut and analyzed by ethidium bromide staining, the major band from infected cells was present at ~250 kb, whereas the DNA from BV migrated at 100 – 150 kb (Fig. 2A). It should be noted that, because bacmid DNA and baculovirus genomes are intact circular DNA molecules, they are likely impaled by agarose fibers during electrophoresis and either do not enter the gel or migrate aberrantly (Cole and Tellez, 2002; Levene and Zimm, 1987). To examine this further, we performed in situ DNA extraction and FIGE analysis on bacterial cells harboring the baculovirus bacmid. Similar to BV DNA, the undigested bacmid DNA was retained in the well, whereas after digestion with Eco81I, it migrated into the gel to the position of unit length genomic DNA (data not shown). Therefore it is not possible to draw conclusions regarding the size of uncut circular DNA.
Fig 1.
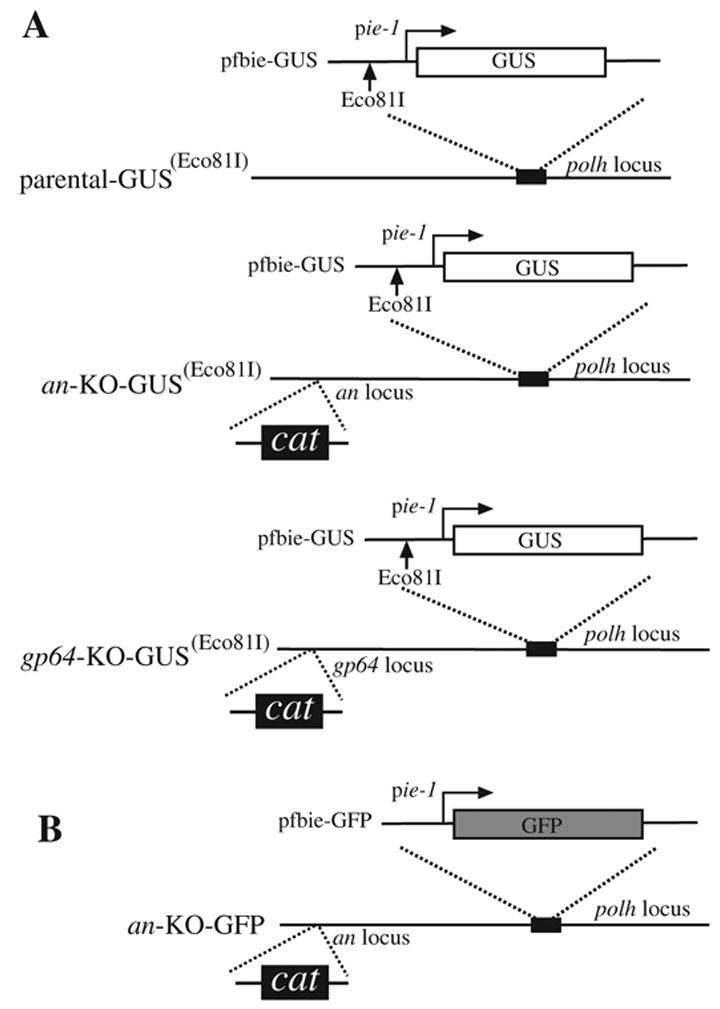
AcMNPV bacmid constructs used in this report and the strategy employed to introduce marker genes and a unique Eco81I restriction site. (A) The transfer plasmid pfbie-GUS containing the GUS reporter gene and the Eco81I restriction site was transposed into a non-modified bacmid (parental), an alkaline nuclease (an), or a gp64 knockout bacmid to generate the constructs parental-GUS(Eco81I), an-KO-GUS(Eco81I), and gp64-KO-GUS(Eco81I), respectively. (B) The transfer plasmid pfbie-GFP was transposed into an an knockout bacmid to generate an-KO-GFP. Transposition occurred at the polyhedrin locus (polh) and was performed in accordance with the bac-to-bac protocol (Invitrogen). The GUS and GFP marker genes are under control of the AcMNPV immediate early 1 (ie-1) promoter. Construction of the an and gp64 null bacmids containing the chloramphenicol resistance gene have been described previously (Okano et al., 2004).
Fig 2.

FIGE analysis of viral DNA replication intermediates from Sf-9 cells infected with the parental-GUS(Eco81I) recombinant virus. Uncut or Eco81I digested total DNA extracts were analyzed after electrophoresis (program 4) by either ethidium bromide staining (A, C, and E) or by Southern blot hybridization with labeled genomic viral DNA as the probe (B, D, and F). Hours post-infection (hpi) are indicated above the panels. M indicates mock infected cells, BV indicated DNA isolated from budded virus, and L (lambda) indicates MidRange PFG DNA size marker I (New England Biolabs). The arrows indicate single unit-size viral DNA and the asterisks indicate viral DNA that migrated larger-than-unit length. Size ranges in kilobases are indicated on the left of the panels.
To further characterize the genomic DNA, cells or virus were embedded in agarose, processed, and digested in situ with Eco81I. As expected, the BV DNA was resolved into a single major band of about 145 kb (the AcMNPV bacmid is larger than wt AcMNPV) (Fig. 2 E,F). In addition, cellular DNA from infected cells was reduced to the 50–100 kb size range and did not hybridize to the viral DNA probe (Fig. 2 C, D), whereas three major species of viral-specific DNA were evident at the later stages of the time-course (Fig. 2 D). These included sub genome size DNA of about 50–100 kb, a genome size band of ~145 kb, and a larger doublet band of >250 kb. A quantity of viral-specific DNA also remained trapped in the well. The FIGE program 4 results in a compression zone in the range of DNA sizes in the >250 kb region and accurate estimates are not possible. FIGE program 5 separates DNA from ~ 50 – 600 kb which would encompass this compressed zone. Using program 5, the >250 kb bands observed in program 4 were not evident indicating that the represent a heterogeneous population (data not shown).
The trapping of DNA in the wells and the presence of the >250 Kb band suggested that DNA digestion may not be complete. In order to determine the extend of DNA Eco81I digestion, triplicate samples of Sf9 cells at 72 hr after infection with the infectious bacmid construct (Fig. 1) were embedded in agarose, processed as described in the Materials and Methods and then subjected to overnight incubation with or without Eco81I. The DNA was then extracted from the gel and subjected to quantitative PCR using primers that amplified a 500-bp region spanning the Eco81I site. These data indicated that over 98%+/−0.2 of the Eco81I sites were digested (data not shown). Therefore, the DNA bands that we observed were due to complete digestion of the DNA. Despite almost complete digestion, the presence of the DNA in the wells and the compression zone suggested that they might be branched structures that would not migrate as unit length.
FIGE analysis of gp64 and an-knockout transfected cells
In these experiments an AcMNPV bacmid deleted for the gp64 gene was used for a control. Gp64 is the viral envelope fusion protein and is required for the exit (budding) of mature virions from infected cells (Monsma et al., 1996). Therefore the gp64 knockout is not infectious and will not amplify upon transfection. A similar construct has been used by others previously (Lin and Blissard, 2002). FIGE was performed with cells transfected with a gp64 knockout bacmid (gp64-KO-GUS(Eco81I)) at different times post-transfection (Fig. 3A-D). Ethidium bromide stained uncut DNA showed a similar pattern to the parental AcMNPV bacmid infected cells (Fig. 2A) with the majority of the DNA migrating at >250 kb (Fig. 3A) as described previously (Vanarsdall et al., 2006). Similarly, hybridization analysis indicated that from 0 to 24 hr post transfection, the DNA in the >250 kb region is host DNA, but after 24 h.p.t. viral-specific DNA also accumulated at this region (compare Fig. 3B to Fig. 2B). A significant amount of viral-specific DNA was also trapped in the wells similar to parental-GUS Eco81I bacmid infected cells (Fig. 3B). When the DNA from gp64-KO-GUS(Eco81I) transfected cells was digested in situ with DpnI (to digest input bacmid DNA) and Eco81I, three species of DNA were observed similar to DNA from parental infected cells (Fig. 3C and D). These included a major diffuse band of ~50–100 kb, a genome size DNA band of ~145 kb and the higher MW doublet of >250 kb. Some viral-specific DNA is also trapped in the wells. Therefore, these results indicate that replication intermediates generated by parental bacmid infected and gp64-KO-GUS(Eco81I) transfected cells are similar.
Fig 3.
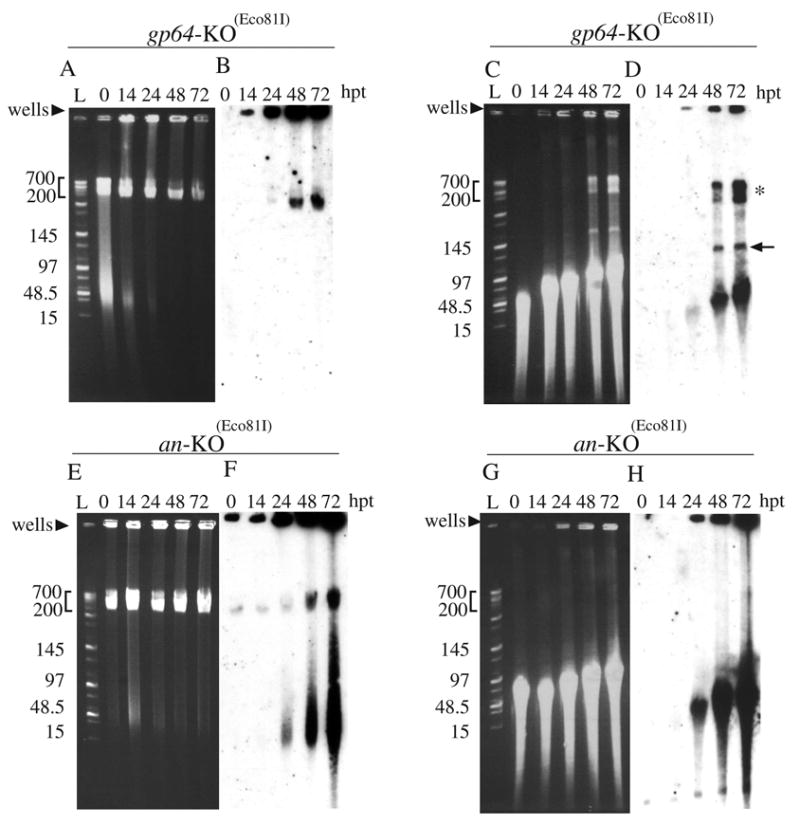
FIGE analysis of viral DNA replication intermediates from gp64 or an knockout transfected Sf-9 cells. DNA was analyzed after electrophoresis (program 4) by either ethidium bromide staining (A, C, E and G) or by Southern blot hybridization with labeled genomic viral DNA as the probe (B, D, F and H). The bacmid constructs used for transfection and the time cells were harvested post-transfection (h.p.t.) are indicated above the panels. L indicates MidRange PFG DNA size marker I (New England Biolabs). The DNA in panels A, B, E, and F were uncut and the DNA in panels C, D, G, and H were digested with Eco81I. The arrows indicate single unit-size viral DNA and the asterisk indicates viral DNA that migrated larger-than-unit length. Size ranges in kilobases are indicated on the left of the panels.
We previously demonstrated that, although the alkaline nuclease knockout bacmid was able replicate DNA, no infectious virus were produced (Okano et al., 2004). Therefore, we examined the DNA that was replicated by this mutant in more detail using FIGE analysis. When DNA from an-KO-GUS(Eco81I) transfected cells was not digested with DpnI and Eco81I, no discernable difference could be seen from the ethidium bromide stained gel when compared to gp64-KO-GUS(Eco81I) transfected cells (compare Fig. 3A and 3E). However, Southern blot analysis of uncut DNA from an-KO-GUS(Eco81I) transfected cells revealed a high molar ratio of relatively small sub genome-size DNA fragments (50–100kb) that first appeared by 24 h.p.t. and continued to accumulate for the remainder of the time-course (Fig. 3F). No DNA of this size was present in blots from either parental infected or gp64-KO-GUS(Eco81I) transfected cells when the DNA remained uncut (Fig. 2B and 3B, respectively). Furthermore, digestion with DpnI and Eco81I yielded predominantly small DNA fragments that appeared to be concentrated mostly in the 50kb to 100 kb size range, but did extend higher and overlapped genome size DNA (Fig. 3H). In addition, the high MW doublet band of >250 kb was not evident. If these bands are branched structures produced by recombination, this could explain the lack of these structures in the DNA produced by alkaline nuclease knockout bacmid (see Discussion). Lastly, when the FIGE profile of DNA from cells infected with the AN repaired virus (Okano et al., 2004) was examined, it showed a similar pattern as that of the wt (data not shown). Therefore, the fragmented viral DNA profile produced by the an-KO bacmid (Fig. 3 F, H) is due to the deletion of the an gene.
Comparison of DNA replication by the alkaline nuclease and gp64 knockout bacmids by quantitative PCR
To further investigate DNA synthesis by the an knockout, we compared it to the gp64 knockout using real time PCR. Although we previously demonstrated that the an knockout is able to replicate DNA in transfected cells, the level was lower than that from wt infected cells which was likely due to spread of the infection to surrounding cells by the wt virus (Okano et al., 2004). In contrast, with the an and gp64 knockout constructs, BV would not spread and infect surrounding cells thereby allowing for a more accurate comparison. Analysis by real time PCR of DNA from cells transfected with these two constructs demonstrated that they synthesized similar levels of DNA over the time-course (Fig. 4).
Fig. 4.

Real-time PCR analysis of viral DNA. At the designated time-point, total DNA was isolated from Sf-9 cells transfected with the indicated bacmid construct, digested with the restriction enzyme DpnI to eliminate input bacmid DNA, and analyzed by real-time PCR using SYBR green I. Values are displayed as the average from transfections performed in triplicate with error bars indicating standard deviations.
Immunoelectron microscopy of cells transfected with the an knockout bacmid
To determine if the knockout of an affects nucleocapsid production, electron microscopy was performed on sections from cells transfected with the an knockout bacmid and immuno-stained with a monoclonal antibody against the AcMNPV major capsid protein VP39. Images of cells transfected with the gp64-KO bacmid construct under these conditions showed positive immuno-staining of electron dense rod-shaped nucleocapsids typical of a baculovirus infection (Fig. 5A). Analysis of alkaline nuclease knockout bacmid transfected cells also revealed the presence of a limited number of electron-dense, rod-shaped capsids indicating that the mutant has the ability to generate a low number of normal-appearing nucleocapsids (Fig. 5 B). However, most of the capsid-like structures that we observed were aberrant, elongated structures that were electron-translucent and were not observed in cells transfected with the gp64 knockout bacmid (Fig. 5C and D). Therefore these data indicate that a bacmid lacking alkaline nuclease is defective in producing normal levels of mature nucleocapsids which may be a reflection of the apparent defect in DNA processing.
Fig. 5.

Immunogold staining of thin sections generated from gp64-KO or an-KO-GFP transfected cells at 72 h.p.t. (A) Image of a gp64-KO transfected cell showing normal looking viral nucleocapsids containing an electron-dense core. (B) Image of an an-KO-GFP transfected cell also showing normal looking electron-dense nucleocapsids. (c–d) Images of an-KO-GFP transfected cells showing aberrant rod-shaped capsid structures lacking an electron dense core. For all samples, sections were stained with the primary monoclonal antibody to the AcMNPV p39 major capsid protein as undiluted tissue culture supernatant. The secondary 10 nm gold-conjugated antibody was used at 1:50 dilution. The bar equals 0.25 μm.
DISCUSSION
FIGE analysis of DNA
In this report, we describe the characterization of viral-specific DNA that is synthesized during the baculovirus replication cycle. A large proportion of the replicative intermediates isolated from infected cells and genomic DNA isolated from BV were trapped in the wells of an agarose gel when analyzed by FIGE. Concordant with previous reports, this phenomena may be an inherent feature of this type of analysis due to the fact that large circular DNA may become impaled on the free ends of gel fibers preventing its migration under electrophoresis (Cole and Tellez, 2002). In addition, it is thought that herpes simplex virus 1 (HSV-1) replicative intermediates, which are also retained in wells, may be complex branched structures, possibly derived by strand invasion, or by nascent DNA synthesized from replication origins present on DNA being replicated (Severini et al., 1996; Zhang et al., 1994) [reviewed in (Boehmer and Lehman, 1997)]. Only after digesting DNA with a single cutting restriction enzyme, did we observe monomeric DNA molecules from parental bacmid infected cells. Therefore the retention of undigested DNA may be caused by circular and/or complex branched structures. To ensure that the DNA was completely digested under the in situ conditions that we employed, we used quantitative PCR with primers flanking the Eco81I site to compare the DNA with and without Eco81I digestion. We determined that over 98% of the DNA had been digested indicating that our results were not caused by incomplete digestion by the enzyme.
In order to resolve full length genomic DNA after digestion with the single cutting enzyme Eco81I, the parameters defined by program 4 of the field inversion apparatus were most optimal. Under these conditions, however, in addition to unit length genomic DNA, a compressed population of DNA molecules of >250 kb were also produced. However, using a different FIGE program (program 5) which is suitable for separating DNA in the size range of 50 to 600 kb, the migration pattern became more diffuse indicating that this DNA is likely heterogeneous in size and not a discrete species of a specific length (data not shown). This has led us to suggest that the migration of this high MW DNA may be the result of branched structures produced by multiple initiation of DNA replication on the same molecule or by recombination by strand invasion.
We have previously described a bacmid knocked out for vlf-1 and found that it was similar to the an knockout in that it lacked infectivity and was able to synthesize DNA at similar to gp64 knockout levels (Vanarsdall et al., 2004; Vanarsdall et al., 2006). However, in contrast to the an knockout, FIGE analysis demonstrated that the pattern of DNA synthesized by the vlf-1 knockout was indistinguishable from the gp64 knockout virus. In contrast, the DNA produced by the an knockout appeared to contain a large and heterogeneous population of low MW species of about 50 to 100 kb that were not present in the gp64 knockout control or parental bacmid infected cells. Furthermore, digestion with Eco81I yielded predominantly DNA smaller than genome length and the higher MW (>250 kb) bands were not evident. These data indicated that, although ‘wt’ levels of DNA can be synthesized by this mutant, much of the DNA is of sub-genome size.
A role for recombination in baculovirus DNA replication?
Although the production of sub genome size DNA by the alkaline nuclease knockout could have been caused by a requirement for alkaline nuclease for the processivity of the DNA polymerase complex, there is no evidence for the baculovirus alkaline nuclease or its homologs playing such a role. In contrast, the baculovirus alkaline nuclease is a member of a family of enzymes that are involved in homologous recombination, and the fact that we have demonstrated that the AcMNPV AN also has enzymatic properties characteristic of these proteins (Mikhailov et al., 2003), the presence of these small DNA fragments are consistent with the an knockout virus being defective in homologous recombination. The reason that AcMNPV may employ homologous recombination for genome production is not clear. It may be required because the production of subgenome size DNA may be an intrinsic feature of AcMNPV DNA replication, but this has not been documented. It could also be a response to apoptosis. Although the ability of AcMNPV to block apoptosis is well-documented (Clem et al., 1991), the blockage may not be complete and a recombination system may assist in repairing partially degraded viral DNA.
The fragmented DNA produced by the bacmid lacking an could also result from a defect in packaging. If the DNA produced by this mutant is not packaged properly, it may not be protected from nucleases in the cell. However, with a vlf-1 knockout mutant that we have investigated, although the DNA does not appear to be packaged, it is not fragmented (Vanarsdall et al., 2006)
In addition to participating in the production of replication intermediates, alkaline nuclease could have other effects on virus replication. Electron microscopic examination of the nucleocapsids produced by the alkaline nuclease indicates that most of them are aberrant in structure suggesting that the DNA produced by the alkaline nuclease knockout cannot be packaged properly. Furthermore, homologous recombination may be a final event in the production of circular genomes.
Recently plant mitochondrial and chloroplast DNA has been characterized. In both instances, evidence for recombination-dependent replication and the presence of complex branched structures were reported (Bendich, 2004; Manchekar M and Related Articles, 2006). The maize chloroplast (cp) genome is similar in size to the baculovirus bacmid genomes that we have investigated (~146 kb) and appears to map as two isomers caused by inversion between an inverted repeat region, however much of the DNA appears to be present in a complex structure(Oldenburg and Bendich, 2004b). When run on a pulsed-field gel, much of the cp DNA was retained in the well. Visualization of this DNA by ethidium bromide staining and fluorescence microscopy under electrophoretic or fluid force revealed that the well-bound DNA consists of multiple linear DNA fibers extending ‘medusa-like’ from a central core (Oldenburg and Bendich, 2004a; Oldenburg and Bendich, 2004b). Upon treatment with a restriction enzyme with a single site in the cp genome, the long DNA fibers were released, but a core with short fibers comprising 28% of the DNA remained in the well. The nature of the core structure remains to be determined.
A role of alkaline nuclease in other viral systems
The importance of alkaline nuclease has been documented in other viral systems. Mutant herpes simplex virus with knockouts of their alkaline nuclease gene have also been investigated. Similar to our observations for the baculovirus alkaline nuclease knockout (an-KO-GUS(Eco81I)), the HSV-1 an mutant is capable of synthesizing viral DNA (Shao et al., 1993), but the DNA is non infectious when transfected into susceptible cells (Porter and Stow, 2004). In addition, HSV-1 AN mutants show a high frequency of abortive packaging and few mature complete capsids (Shao et al., 1993). Similarly, the baculovirus alkaline nuclease knockout that we have characterized produced numerous aberrant capsids. It is difficult to compare FIGE analyses of DNA produced by baculovirus and HSV-1 alkaline nuclease mutants because mature baculovirus genomes are circular rather than linear and therefore cannot be readily analyzed by FIGE without restriction endonuclease digestion. In addition, the capsid structures differ (rod-shaped vs icosahedral) which might influence the packaging process. However, in one study when HSV DNA was isolated from wells and examined by electron microscopy, it was found that DNA from the alkaline nuclease knockout appeared to be much shorter than that from wt virus suggesting that it was more fragile than wt DNA (Goldstein and Weller, 1998).
Replication of baculovirus genomes
It has previously been suggested that baculovirus genome DNA is replicated by a rolling circle type mechanism (Oppenheimer and Volkman, 1997). When we examined DNA by partial digestion at 10, 20 and 72 hr p.i. DNA with Eco81I followed by FIGE using either program 4 or program 5 we found no evidence for concatemeric DNA being produced (data not shown). However, even if concatemeric DNA fragments were observed, it would not rule out that they had been produced by DNA recombination or a combination of recombination and rolling circle replication. Our observations are similar to those for the characterization of HSV-1 replication intermediates which were found to be less than two genomes in length (Deshmane et al., 1995).
The use of baculoviruses as expression vectors has long been exploited because of their facility to incorporate genetic material via homologous recombination. The conservation of alkaline nuclease in all baculovirus genomes that have been sequenced suggests that recombination may be an essential feature of baculovirus genome replication. The research reported here, suggests that alkaline nuclease is essential for the production of DNA that can be processed into mature genomes. The exact mechanism for genome replication and the details of the replication intermediates remains to be determined.
MATERIALS AND METHODS
Cell culture
Sf9 cells were cultured in SF900II serum-free medium (Invitrogen) with penicillin G (50 units/ml), streptomycin (50 μg/ml; Whittaker Bioproducts), and fungizone (amphotericin B; 375 ng/ml) (Invitrogen) as previously described (Li and Rohrmann, 2000).
Bacmid contructs; introduction of a unique Eco81I site and egfp
The an and gp64 null parental bacmids has been described previously (Okano et al., 2004) and (Vanarsdall et al., 2006), respectively. To allow identification of genome length DNA, a unique Eco81I restriction site was introduced into an unmodified control, an, and gp64 knockout bacmids to generate the constructs parental-GUS(Eco81I), an-KO-GUS(Eco81I, and gp64-KO-GUS(Eco81I, respectively (Fig. 1A). To accomplish this, the transfer vector pfbIEGUS (Okano et al., 2004) was modified by digesting with XhoI followed by ligation with the self-annealed oligonucleotide TCGACCCTAAGGG containing the Eco81I restriction sequence (CCTNAGG). To permit monitory of transfection efficiency for immuno electron microscopy, GFP expressing bacmids were produced. The transfer plasmid pfbie-GFP (Vanarsdall et al., 2006) containing a GFP reporter gene was transposed into an an knockout bacmid to generate an-KO-GFP (Fig. 1B). Transposition was performed in accordance with the Bac-to-Bac protocol (Invitrogen).
DNA transfection, viral infection, field inversion gel electrophoresis and hybridization
Bacmid DNA was purified from 0.5 liter cultures using the Large-Construct purification kit (Qiagen) according to the manufacturer’s instructions. Two μg of purified DNA was used for transfecting Sf-9 cells (0.9×106/ml) seeded in a six-well plate via a cationic liposome method (Campbell, 1995). Infections were performed at an MOI of about 5 as described previously (28). For FIGE analysis, cells were harvested with a rubber policeman and collected by low speed centrifugation (1000 rpm, 5 min), washed twice with phosphate-buffered saline (PBS) and mixed with 1% low-melting agarose (FMC Seaplaque) to a final cell density of 3 × 106 cells/200 μl. The mixture was poured into a plug mold (BioRad), resulting in 20 × 9 × 1.5 mm agarose plugs that were then cut into 8 to 10 pieces. For DNA purification, the agarose plugs were treated with 10 mM Tris-HCl pH 8.0, 100mM EDTA, 1% N-lauroyl sarcosine, 200 μg/ml Proteinase K at 50 ºC over night (about 1 ml for 8–10 agarose plugs). Plugs were then washed 5–8 times (about 1.5 ml/wash with rotation for 15 min) with 10 mM Tris-HCl at 4º C and then stored at 4 ºC. For DNA digestion, the proteinase-treated plugs were incubated in 100 μl of Eco81I reaction buffer containing 30U Eco81I and 20U DpnI (both from Fermantas) and incubated at 37ºC overnight. The reaction was terminated by the addition of 20 μl of 0.5 M EDTA. Agarose plugs were placed into the wells of 1% Pulsed Field Certified Agarose gel (Bio-Rad) in 0.5 × TBE buffer (45mM Tris-borate, pH 8.0, 1 mM EDTA) and separated by field inversion gel electrophoresis (FIGE) using a MJ Research PPI-200 programmable pulse inverter. Two FIGE programs were used (Anon., 1993): to separate DNA in the 15 to 200 kb range we used Program 4 (A; initial reverse time: 0.05 min, B; reverse increment: 0.01 min, C; initial forward time: 0.15 min, D; forward increment: 0.03 min, E; number of steps: 81, F; reverse increment: 0.001 min, G; forward increment: 0.003); 8V/cm for 17 hr at 4 ºC. To separate DNA in the 50 to 600 kb range we used Program 5 (A; 0.1 min, B; 0.01 min, C; 0.3 min, D; 0.03 min, E; 45, F; 0.01 min, G; 0.03 min); 9V/cm for 17 hr at 4ºC. MidRange PFG marker I and Yeast chromosome PFG markers (New England Biolabs) were used as DNA size markers. For Southern blot analysis, the agarose gel was soaked in 0.25M HCl for 15 min followed by 0.4M NaOH for 15 min. The DNA was then transferred to a nylon membrane and hybridized with viral genomic DNA digested with EcoRI and HindIII that was labeled using the Alk-Phos Direct Labeling and Detection system (Amersham) as described previously (Okano et al., 2004).
Analysis of in situ digestion of agarose embedded DNA
In order to determine the ability of Eco81I to digest DNA in situ, triplicate samples of LMT agarose embedded Sf-9 cells at 72 hpi were processed as described above. After deproteinization, one set of samples was subjected to overnight digestions with 30U of Eco81I while the other set was incubated in digestion buffer under identical conditions without restriction enzyme. After overnight digestion, one-half of the sample was removed, melted at 60C for 20 min, phenol extracted and analyzed by Q-PCR with primers that amplify a 500 nt fragment across the Eco81I restriction site. The primers used were Ac126710 (5’-CAATGGACCCGACAAATTCT-3’) and SV40polA (5’-ATGTCGACAGATACATTGATGAGTTTGG-3’) that amplify a 500 bp fragment spanning the Eco81I restriction site. The remaining agarose was inserted into a 1% slab gel and electrophoresed under standard FIGE conditions to characterize the DNA profile.
Q-PCR replication assay and immunoelectron microscopy
These procedures were carried out as previously described (Vanarsdall et al., 2005).
Acknowledgments
We thank Victor Mikhailov for his advice throughout this project and for his review of this manuscript and Loy Volkman for the monoclonal antibody to AcMNPV vp39. This research was supported by National Institutes of Health Grant GM060404 (to G. F. R).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anon . The MJ Research PPI-200 Programmable Power Inverter. 1993. [Google Scholar]
- Aravind L, Makarova K, Koonin E. SURVEY AND SUMMARY: holliday junction resolvases and related nucleases: identification of new families, phyletic distribution and evolutionary trajectories. Nucleic Acids Res. 2000;28(18):3417–32. doi: 10.1093/nar/28.18.3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendich AJ. Circular chloroplast chromosomes: the grand illusion. Plant Cell. 2004;16:1661–6. doi: 10.1105/tpc.160771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehmer PE, Lehman IR. Herpes simplex virus DNA replication. Annu Rev Biochem. 1997;66:347–84. doi: 10.1146/annurev.biochem.66.1.347. [DOI] [PubMed] [Google Scholar]
- Bujnicki J, Rychlewski L. The herpesvirus alkaline exonuclease belongs to the restriction endonuclease PD-(D/E)XK superfamily: insight from molecular modeling and phylogenetic analysis. Virus Genes. 2001;22(2):219–30. doi: 10.1023/a:1008131810233. [DOI] [PubMed] [Google Scholar]
- Campbell MJ. Lipofection reagents prepared by a simple ethanol injection technique. BioTechniques. 1995;18:1027–1032. [PubMed] [Google Scholar]
- Cassuto E, Lash T, Sriprakash K, Radding C. Role of exonuclease and beta protein of phage lambda in genetic recombination. V Recombination of lambda DNA in vitro. Proc Natl Acad Sci USA. 1971;68(7):1639–43. doi: 10.1073/pnas.68.7.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clem RJ, Fechheimer M, Miller LK. Prevention of apoptosis by a baculovirus gene during infection of insect cells. Science. 1991;254:1388–1390. doi: 10.1126/science.1962198. [DOI] [PubMed] [Google Scholar]
- Cole KD, Tellez CM. Separation of large circular DNA by electrophoresis in agarose gels. Biotechnol Prog. 2002;18(1):82–7. doi: 10.1021/bp010135o. [DOI] [PubMed] [Google Scholar]
- Deshmane SL, Raengsakulrach B, Berson JF, Fraser NW. The replicating intermediates of herpes simplex virus type 1 DNA are relatively short. J Neurovirol. 1995;1:165–76. doi: 10.3109/13550289509113963. [DOI] [PubMed] [Google Scholar]
- Goldstein JN, Weller SK. In vitro processing of herpes simplex virus type 1 DNA replication intermediates by the viral alkaline nuclease, UL12. J Virol. 1998;72(11):8772–81. doi: 10.1128/jvi.72.11.8772-8781.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kool M, Ahrens C, Goldbach RW, Rohrmann GF, Vlak JM. Identification of genes involved in DNA replication of the Autographa californica baculovirus. Proc Natl Acad Sci USA. 1994;91:11212–11216. doi: 10.1073/pnas.91.23.11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzminov A. Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda. Microbiol Mol Biol Rev. 1999;63(4):751–813. doi: 10.1128/mmbr.63.4.751-813.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levene SD, Zimm BH. Separations of open-circular DNA using pulsed-field electrophoresis. Proc Natl Acad Sci U S A. 1987;84(12):4054–7. doi: 10.1073/pnas.84.12.4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Rohrmann Characterization of a baculovirus alkaline nuclease. J Virol. 2000;74:6401–6407. doi: 10.1128/jvi.74.14.6401-6407.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Karakousis G, Chiu S, Reddy G, Radding C. The beta protein of phage lambda promotes strand exchange. J Mol Biol. 1998;276(4):733–44. doi: 10.1006/jmbi.1997.1572. [DOI] [PubMed] [Google Scholar]
- Lin G, Blissard GW. Analysis of an Autographa californica nucleopolyhedrovirus lef-11 knockout: LEF-11 is essential for viral DNA replication. J Virol. 2002;76(6):2770–9. doi: 10.1128/JVI.76.6.2770-2779.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little J. An exonuclease induced by bacteriophage lambda. II Nature of the enzymatic reaction. J Biol Chem. 1967;242(4):679–86. [PubMed] [Google Scholar]
- Manchekar M SGK, Song D Khazi F, McLean SL Nielsen BL, Related Articles L. DNA recombination activity in soybean mitochondria. J Mol Biol. 2006;356:288–99. doi: 10.1016/j.jmb.2005.11.070. [DOI] [PubMed] [Google Scholar]
- Mikhailov V, Okano K, Rohrmann G. Baculovirus Alkaline Nuclease Possesses a 5’->3’ Exonuclease Activity and Associates with the DNA-Binding Protein LEF-3. J Virol. 2003;77(4):2436–2444. doi: 10.1128/JVI.77.4.2436-2444.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhailov V, Okano K, Rohrmann G. Specificity of the Endonuclease Activity of the Baculovirus Alkaline Nuclease for Single-stranded DNA. J Biol Chem. 2004;279(15):14734–14745. doi: 10.1074/jbc.M311658200. [DOI] [PubMed] [Google Scholar]
- Monsma SA, Oomens AGP, Blissard GW. The gp64 envelope fusion protein is an essential baculovirus protein required for cell-to-cell transmission of infection. J Virol. 1996;70:4607–4616. doi: 10.1128/jvi.70.7.4607-4616.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano K, Vanarsdall AL, Rohrmann GF. Characterization of a baculovirus lacking the Alkaline Nuclease Gene. J Virol. 2004;78:10650–10656. doi: 10.1128/JVI.78.19.10650-10656.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldenburg DJ, Bendich AJ. Changes in the structure of DNA molecules and the amount of DNA per plastid during chloroplast development in maize. J Mol Biol. 2004a;344:1311–30. doi: 10.1016/j.jmb.2004.10.001. [DOI] [PubMed] [Google Scholar]
- Oldenburg DJ, Bendich AJ. Most chloroplast DNA of maize seedlings in linear molecules with defined ends and branched forms. J Mol Biol. 2004b;335:953–70. doi: 10.1016/j.jmb.2003.11.020. [DOI] [PubMed] [Google Scholar]
- Oppenheimer DI, Volkman LE. Evidence for rolling circle replication of Autographa californica M nucleopolyhedrovirus genomic DNA. Arch Virol. 1997;142:2107–2113. doi: 10.1007/s007050050229. [DOI] [PubMed] [Google Scholar]
- Porter I, Stow N. Virus particles produced by the herpes simplex virus type 1 alkaline nuclease null mutant ambUL12 contain abnormal genomes. J Gen Virol. 2004;85 doi: 10.1099/vir.0.19657-0. [DOI] [PubMed] [Google Scholar]
- Poteete A. What makes the bacteriophage lambda Red system useful for genetic engineering: molecular mechanism and biological function. FEMS Microbiol Lett. 2001;201(1):9–14. doi: 10.1111/j.1574-6968.2001.tb10725.x. [DOI] [PubMed] [Google Scholar]
- Severini A, Scraba DG, Tyrrell DLJ. Branched structures in the intracellular DNA of herpes simplex virus type I. J Virol. 1996;70:3169–3175. doi: 10.1128/jvi.70.5.3169-3175.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao L, Rapp LM, Weller SK. Herpes simplex virus 1 alkaline nuclease is required for efficient egress of capsids from the nucleus. Virology. 1993;196(1):146–62. doi: 10.1006/viro.1993.1463. [DOI] [PubMed] [Google Scholar]
- Stahl F. Recombination in phage lambda: one geneticist’s historical perspective. Gene. 1998;223:95–102. doi: 10.1016/s0378-1119(98)00246-7. [DOI] [PubMed] [Google Scholar]
- Todd JW, Passarelli AL, Miller LK. Eighteen baculovirus genes, including lef-11, p35, 39K, and p47, support late gene expression. J Virol. 1995;69:968–974. doi: 10.1128/jvi.69.2.968-974.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanarsdall AL, Okano K, Rohrmann GF. Characterization of a baculovirus with a deletion of vlf-1. Virology. 2004;326(1):191–201. doi: 10.1016/j.virol.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Vanarsdall AL, Okano K, Rohrmann GF. Characterization of the replication of a baculovirus mutant lacking the DNA polymerase gene. Virology. 2005;331(1):175–180. doi: 10.1016/j.virol.2004.10.024. [DOI] [PubMed] [Google Scholar]
- Vanarsdall AL, Okano K, Rohrmann GF. Characterization of the role of VLF-1 in baculovirus capsid structure and DNA processing. J Virol. 2006;80:1724–1733. doi: 10.1128/JVI.80.4.1724-1733.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Efstathiou S, Simmons A. Identification of novel herpes simplex virus replicative intermediates by field inversion gel electrophoresis: implications for viral DNA amplification strategies. Virology. 1994;202(2):530–9. doi: 10.1006/viro.1994.1375. [DOI] [PubMed] [Google Scholar]