

Abstract
Prior evidence indicates that bile acids stimulate colon cancer cell proliferation by muscarinic receptor-induced transactivation of epidermal growth factor receptors (EGFR). To explore further the mechanism underlying this action, we tested the hypothesis that bile acids activate a matrix metalloproteinase (MMP) that catalyzes release of an EGFR ligand. Initial studies showed that non-selective MMP inhibitors blocked the actions of deoxycholyltaurine (DCT), thereby indicating a role for MMP-catalyzed release of an EGFR ligand. DCT-induced cell proliferation was reduced by increasing concentrations of EGFR kinase inhibitors, by antibodies to the ligand-binding domain of EGFR, by neutralizing antibodies to heparin binding-EGF-like growth factor (HB-EGF) and by CRM197, an inhibitor of HB-EGF release. These findings and our observations with more selective MMP inhibitors suggested that MMP-7, an enzyme known to release HB-EGF, plays a key role in mediating bile acid-induced H508 colon cancer cell proliferation. We observed that recombinant HB-EGF and MMP-7 mimicked both the signaling and proliferative actions of bile acids. Strikingly, reducing MMP-7 expression with either neutralizing antibody or small interfering RNA attenuated the actions of DCT. MMP-7 expression in H508 cells was confirmed using quantitative reverse transcription PCR. DCT stimulated a greater than 10-fold increase in MMP-7 gene transcription. Co-localization of pro-MMP-7 and pro-HB-EGF at the cell surface (immunofluorescence microscopy) was demonstrated, indicating proximity of the enzyme to its substrate. These findings provide strong evidence that in H508 human colon cancer cells, DCT-induced transactivation of EGFR is mediated by MMP-7-catalyzed release of the EGFR ligand HB-EGF.
Keywords: matrix metalloproteinases, colon cancer cell proliferation, bile acids, epidermal growth factor receptors
1. Introduction
Population and animal studies indicate that fecal secondary bile acids are important co-factors in colon carcinogenesis, particularly with regard to right-sided (cecum and ascending colon) cancer [1–3]. In rodents treated with carcinogens, direct instillation or other means of increasing fecal bile acids augments the development of colon cancer [4–6] (Cheng and Raufman, unpublished observations) and reducing fecal deoxycholic acid concentrations by ursodeoxycholic acid feeding decreases colon tumor formation [7, 8]. Likewise, in humans with inflammatory bowel disease, modulating fecal bile acids with oral ursodeoxycholic acid treatment reduces the risk of colorectal dysplasia and cancer [9]. Nonetheless, the molecular mechanisms underlying the proliferative actions of bile acids on colonic neoplasia have been elusive.
Paradoxically, in colon cancer cell lines, rather than stimulating cell proliferation, secondary bile acids like lithocholic and deoxycholic acids are commonly reported to stimulate apoptosis, [10, 11]. Nonetheless, more recent reports indicate that relatively low concentrations of deoxycholic acid (5 to 50 μM) stimulate radiolabeled thymidine incorporation [12]. These changes were associated with nuclear translocation of β-catenin and activation of cyclin D1, known promoters of colon cancer cell proliferation [12]. Thus, whereas high micromolar concentrations (~ 500 μM) of secondary bile acids are pro-apoptotic [10, 11], low micromolar concentrations (5 to 50 μM) of these agents appear to be pro-proliferative [12]. Moreover, in human colon cancer cells the ability of bile acids to induce proliferation or apoptosis is strongly correlated with their hydrophobicity [13]. That is, equimolar concentrations of conjugated bile acids are less likely to induce apoptosis than their more hydrophobic parent molecules [13, 14]. Recently, we confirmed that low micromolar concentrations of more hydrophilic glycine and taurine conjugates of secondary bile acids stimulate human colon cancer cell proliferation [15, 16]. Furthermore, as measured by activation of caspase-3 [17], incubation for up to 5 days with concentrations of conjugated secondary bile acids that stimulate colon cancer cell proliferation does not result in apoptosis [15, 16].
Proliferative actions of various ligands that interact with G-protein coupled receptors (GPCR) [e.g. M3 muscarinic receptors (M3R)] are dependent on activation of receptor tyrosine kinases (RTK) [e.g. epidermal growth factor receptors (EGFR)]. This phenomenon, commonly designated transactivation, is a required mechanism for the physiological actions of many GPCR ligands. Pai et al. showed that in Caco-2 and HT-29 colon cancer cells prostaglandin E2-induced thymidine incorporation, a marker of cell proliferation, is dependent on Src activation, the release of transforming growth factor-α (TGF-α) and the consequent transactivation of EGFR [18]. EGFR activation is recognized to play a pivotal role in colon carcinogenesis and provides an important therapeutic target [19, 20]. In cell lines derived from human colon cancers, like the H508 cell line that abundantly expresses M3R [21], interaction of acetylcholine (ACh) and conjugated secondary bile acids with M3R results in transactivation of EGFR and activation of pro-proliferative post-receptor signaling [16, 22, 23]. In H508 cells, inhibition of EGFR activation using specific inhibitors or antibodies prevents both ACh- and bile acid-induced colon cancer cell proliferation [16, 23]. Bile acid-induced transactivation of EGFR activates post-receptor signaling cascades that involve phosphorylation of p44/42 mitogen activated protein kinase (p44/42 MAPK, also referred to as ERK 1/2) and p90 ribosomal S kinase (p90RSK), a nuclear response protein downstream of p44/42 MAPK [16, 24]. Blocking EGFR activation and downstream phosphorylation of p44/42 MAPK and p90RSK inhibits the proliferative actions of bile acids [16].
A frequent mechanism underlying EGFR transactivation involves metalloprotease-mediated release of an EGFR ligand [25]. The EGFR ligand family comprises 6 members, including EGF, TGF-α, amphiregulin, heparin-binding EGF-like growth factor (HB-EGF), betacellulin and epiregulin [25, 26]. EGFR ligands are synthesized as pro-ligands that are anchored to cell membranes. A family of secreted and membrane-anchored matrix metalloproteinases (MMP) cleaves pro-EGFR ligands, thereby releasing active ligands from the cell surface [27–29]. Activation of a particular metalloprotease results in release, sometimes referred to as ‘shedding’, of the corresponding EGFR ligand [30]. For example, in human cholangiocytes, bile acids stimulate matrix metalloprotease-3 (MMP-3)-mediated release of TGF-α with consequent phosphorylation (activation) of EGFR [31]. Pretreatment of cholangiocytes with neutralizing antibodies against TGF-α blocks EGFR phosphorylation [31].
Our previous observation that antibodies to the ligand-binding domain of EGFR inhibit the signaling and proliferative actions of bile acids on H508 colon cancer cells implicated the release of an EGFR ligand in this process [16]. This finding stimulated us to elucidate the mechanism of EGFR ligand release and to identify the EGFR ligand responsible for these actions. The initial strategy employed in the present study was to determine the requirement for matrix metalloprotease activation in mediating bile acid-induced transactivation of EGFR. We hypothesized that if the proliferative actions of bile acids depend on activation of a particular matrix metalloprotease, then identification of this agent(s) would provide clues to candidate EGFR ligands that mediate these actions. We examined H508 colon cancer cells because the proliferative actions of bile acids on this colon cancer cell line has been studied extensively and these cells abundantly co-express M3R and EGFR [15, 16, 23]. As demonstrated by the data presented here, using a variety of methods in the H508 human colon cancer cell line, we provide novel evidence that activation of MMP-7 and the release of HB-EGF mediate, at least in part, the proliferative actions of conjugated secondary bile acids.
2. Materials and Methods
2.1. Chemicals and Reagents
The materials used were purchased as follows: PD168393, GM6001, NC-GM6001, MMP inhibitors-II and –III, and MMP-3 inhibitors –II and -IV were from Calbiochem; transforming growth factor-α (TGF-α) and recombinant human MMP-1 from Oncogene; recombinant human MMP-7 from R&D Systems; and FITC- and Tritc-conjugated secondary antibodies and Hoechst stain for immunofluorescence microscopy, CRM197, and deoxycholyltaurine (DCT) were from Sigma. All other chemicals were obtained from Sigma or Fisher.
2.2. Cell Culture
H508 human colon cancer cell lines (ATCC) were grown in RPMI 1640 (GIBCO) supplemented with 10% fetal bovine serum (FBS) (Biowhittaker). Adherent cultures were passaged weekly at subconfluence after trypsinization. Cultures were maintained in incubators at 37°C in an atmosphere of 5% CO2 and 95% air.
2.3. Cytotoxicity Assays
Before proceeding with these studies, we evaluated potential cytotoxic actions of bile acids on H508 cells by trypan blue exclusion (Sigma assay kits). At concentrations used in the following experiments none of these agents altered trypan blue exclusion.
2.4. Antibodies and Immunoblotting
Rabbit polyclonal anti-MAPK, mouse monoclonal antiphospho-MAPK, rabbit polyclonal antiphospho-EGFR and rabbit polyclonal antiphospho-p90RSK were from Cell Signaling (Beverly, MA). TGF-α antibody was from Oncogene; EGF and EGFR antibodies from UBI (Lake Placid, NY); and HB-EGF antibody from Santa Cruz Biotechnology.
Phosphorylation of p44/42 MAPK, EGFR and p90RSK was determined by methods described previously [32]. Briefly, cells were subcultured in 6-well plates (106 cells/well). After a 24-hour incubation at 37°C, the cells were serum-starved for an additional 24 hours, washed with PBS, and allowed to recover in PBS for 30 minutes at 37°C before adding test agents. Following a 10-minute incubation with test agents, the reaction was terminated by adding lysis buffer [150 mM NaCl, 10 mM Tris/HCl, 1% (w/v) deoxycholic acid, 1% (v) Nonidet-40, 0.1% (w/v) SDS, 4 mM EDTA, 1 mM Na3VO4, 20 μg/ml leupeptin, 20 μg/ml aprotinin, 250 μg/ml nitrophenylphosphate and 1 mM phenylmethylsulphonyl fluoride, pH 8]. When inhibitors (e.g. GM6001) were used, they were added 30 minutes before test agents. When antibodies (e.g. anti-HB-EGF, anti-TGF-α) were used, they were added 2 hours before test agents. Cell lysates were subjected to SDS-PAGE (10% gel, Invitrogen). Resolved proteins were transferred electrophoretically to nitrocellulose membranes (Micron Separations) and probed with anti-phospho-p44/42 MAPK, anti-phospho-EGFR or anti-phospho-p90RSK. Bound antibody was detected by chemiluminescence (Supersignal kit, Pierce). To verify that equal amounts of protein were added to each lane, the blots used with anti-phospho-p44/42 MAPK were stripped and re-probed with anti-p42.
2.5. Cell Proliferation Assay
Cell proliferation was determined using the validated sulforhodamine B (SRB) colorimetric assay [33]. Cells were seeded in 96-well plates (Corning Glass Works, Corning, NY) at approx. 10% confluence and allowed to attach for 24 hours. The growth medium was removed and fresh medium without FBS and containing the indicated concentration of test agent was added. Cells were incubated for 5 days at 37°C in an atmosphere of 5% CO2 and 95% air without further additions of test agents. After incubation, cells were treated for 30 minutes with 0.4% (w/v) SRB dissolved in 1% acetic acid. Protein-bound dye was extracted with unbuffered 10 mM Tris base. Absorbance was measured at 560 nm using a computer-interfaced, 96-well microtiter plate reader (SpectraMax384).
2.6. Quantitative Real-time PCR
Total cellular RNA was isolated from cells using Trizol reagent (Invitrogen). First strand cDNAs were synthesized using the Superscript III First Strand Synthesis System for RT-PCR (Invitrogen). Primers for RT-PCR were designed using on-line software (www.genscript.com/ssl-bin/app/primer). Real-time PCR was performed using 7900HT Fast System (ABI) with Power SYBR Green master mix (ABI), cDNA was synthesized from 50 ng total RNA with 20 ng of each primer. PCR conditions included 5 min at 95ºC, followed by 35 cycles of 95ºC for 15 sec, 60ºC for 15 sec, 72ºC for 30 sec. PCR data were analyzed using ABI instrument software SDS 2.1. Specificity of the amplifications was confirmed by melting-curve analysis. Relative levels of mRNA expression were calculated according to the standard Ct method. Individual expression values were normalized by comparison with Glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Oligonucleotides used in PCR from different exons of MMP-7 were: forward primer within exon 4, 5’-GGAGATGCTCACTTCGATGA-3’ and reverse primer within exon 5, 5’-ATACCCAAAGAATGGCCAAG-3’. The length of the MMP-7 PCR product is 104 bp. PCR primers used for GAPDH were: forward primer, 5’-CCCCATGGTGTCTGAGCG-3’ and reverse primer, 5’-CGACAGTCAGCCGCATCTT-3’. The length of the GAPDH product is 67 bp.
2.7. Immunofluorescence Microscopy
H508 cells were subcultured in 4-well Lab-Tek II chamber slides (5 x 104 cells/well), and incubated for 24 hours at 37°C. After washing with PBS, PBS/2M NaCl, cells were kept on ice, fixed with cold MeOH for 10 min, treated with 0.1% TX-100 for an additional 10 minutes, and blocked for 30 minutes with PBS/5% serum derived from the same animal species as the secondary antibody. Cells were then incubated at 4°C overnight with the primary antibodies. After incubation, cells were washed in PBS, incubated with secondary FITC- or/and Tritc-conjugated antibodies at room temperature for 30 minutes and washed. Cell nuclei were visualized with Hoechst staining. To determine the cellular and subcellular distribution of pro-MMP-7 and pro-HB-EGF, slides were analyzed using both standard (Nikon Eclipse 80i) and confocal immunofluorescence microscopy (Zeiss LSM 510).
2.8. Small Interfering RNA Transfection
H508 cells were seeded in 96-well plates at approx. 10% confluence and incubated at 37°C for 24 hours. Four pooled small interfering RNA (siRNA) duplex oligos (Dharmacon) targeting human MMP-7 or a nonspecific duplex oligo negative control (fluorescein-conjugated from Cell Signaling) were transfected into H508 cells with transfection reagent (Mirus) according to the manufacturer’s recommendations. Two days following transfection, cells were incubated for an additional 5 days in FBS-free medium containing 50 μM DCT at 37°C in an atmosphere of 5% CO2 and 95% air. Cell proliferation was determined using the SRB assay as described above.
2.9. Statistical Analysis
All figures (e.g. immunoblots) show data representative of at least three independent experiments. All graphs show the mean and standard error of the mean of at least three independent experiments. Statistical calculations were performed using the Student’s two-tailed un-paired t-test assuming normal distribution with equal variance. Statistical significance is given by the number of asterisks (*, P < 0.05; **, P < 0.005) and a P value of less than 0.05 was considered statistically significant.
3. Results
3.1. Dose-response and time-course for the signaling and proliferative actions of deoxycholyltaurine on H508 colon cancer cells
To select appropriate bile acid concentrations and incubation times for the experiments that follow, we examined both the dose-response curves and time-courses for the actions of deoxycholyltaurine (DCT) on p44/42 MAPK phosphorylation (activation) and on cell proliferation. We selected DCT as the test bile acid for these experiments because previous studies indicated that this agent interacts with M3 muscarinic receptors on H508 colon cancer cells, and that this interaction results in transactivation of EGFR, thereby activating post-receptor MAPK signaling and stimulating cell proliferation [16, 23, 34].
As shown in Figure 1A, DCT caused dose-dependent phosphorylation (activation) of p44/42 MAPK that was detectable with 10 μM and was maximal at the highest DCT concentration tested, 300 μM. Based on these findings, to readily detect differences in the degree of protein phosphorylation, we selected 300 μM DCT as the test concentration for experiments involving assays for p44/42 activation. To demonstrate clearly inhibitor effects on p44/42 MAPK phosphorylation, we sometimes used submaximal concentrations of DCT.
Figure 1. Dose-response and time-course for the signaling and proliferative actions of DCT on H508 colon cancer cells.
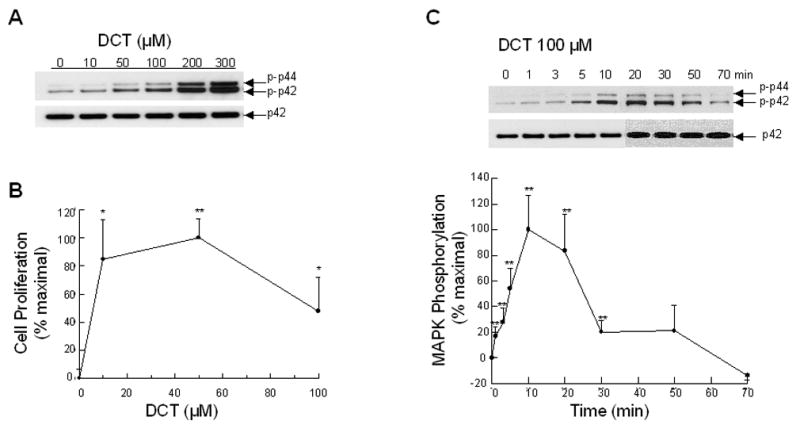
A. Dose-response for DCT-induced p44/42 MAPK phosphorylation. H508 cells were treated with the indicated concentrations of DCT for 10 minutes at 37°C. p44/42 MAPK activity was determined by immunoblotting with antibodies specific for phosphorylated p44/42 MAPK. The quantity of protein added was verified by immunoblotting with antibodies specific for total p42 MAPK. Results are representative of 5 separate experiments. B. Dose-response for DCT-induced cell proliferation. H508 cells were incubated for 5 days at 37°C with the indicated concentrations of DCT. Cell proliferation was determined by the sulforhodamine blue (SRB) colorimetric assay [33]. Results are expressed as mean ± SEM of at least 5 separate experiments. *,**P < 0.05 and 0.005, respectively, vs unstimulated cells. C. Time-course for DCT-induced p44/42 MAPK phosphorylation. H508 cells were treated with 100 μM DCT for 70 minutes at 37°C and p44/42 MAPK activity was determined by immunoblotting at the indicated times with antibodies specific for phosphorylated p44/42 MAPK. The quantity of protein added was verified by immunoblotting with antibodies specific for total p42 MAPK. A representative immunoblot is shown and the graph depicts quantitative densitometric analysis of at least 5 immunoblots. Results are expressed as mean ± SEM. **P < 0.005 vs unstimulated cells.
As shown in Figure 1B, increases in cell proliferation were stimulated over the same range of DCT concentrations that activated p44/42 MAPK. Cell proliferation was maximal with 50 μM DCT and decreased slightly with higher concentrations of the bile acid (Figure 1B). Hence, we used 50 μM DCT as the test concentration for experiments involving cell proliferation. Unless indicated otherwise, the concentrations of inhibitors and antibodies selected for study did not alter basal values for either MAPK phosphorylation or cell proliferation.
Previous extensive work with conjugated secondary bile acids indicates that DCT-induced H508 colon cancer cell proliferation is maximal after 5 days incubation [15, 16]. Hence, we selected 5-day incubations to study changes in cell proliferation. In contrast, activation of post-receptor signaling cascades is rapid. As shown in Figure 1C, DCT-induced activation of p44/42 MAPK was detected within 1 to 2 minutes and was maximal at 10 minutes. The level of p44/42 MAPK phosphorylation declined after 10 minutes and returned to baseline by 70 minutes (Fig. 1C). Based on these results, to study MAPK activation we examined 10-minute incubations with test agents.
3.2. Matrix metalloproteinase inhibitors block DCT-induced p44/42 MAPK phosphorylation
A common mechanism for release of EGFR ligands, involves activation of secreted or cell membrane-anchored MMP’s [30]. We used non-selective and selective MMP inhibitors as tools to determine whether the proliferative actions of bile acids are dependent on release of an EGFR ligand. GM6001, a non-selective hydroxamate MMP inhibitor [35], blocked DCT-induced p44/42 MAPK phosphorylation (Fig. 2A). In contrast, NC GM6001, an inert control agent, did not alter the actions of DCT (Figure 2A). To determine the functional correlate of inhibiting MMP action with GM6001, we examined the effect of the inhibitor and control agent on DCT-induced cell proliferation. As show in Figure 2B, the addition of GM6001, but not NC GM6001, caused a significant reduction in DCT-induced H508 cell proliferation.
Figure 2. Actions of matrix metalloproteinase (MMP) inhibitors on bile acid-induced MAPK phosphorylation and stimulation of H508 cell proliferation.
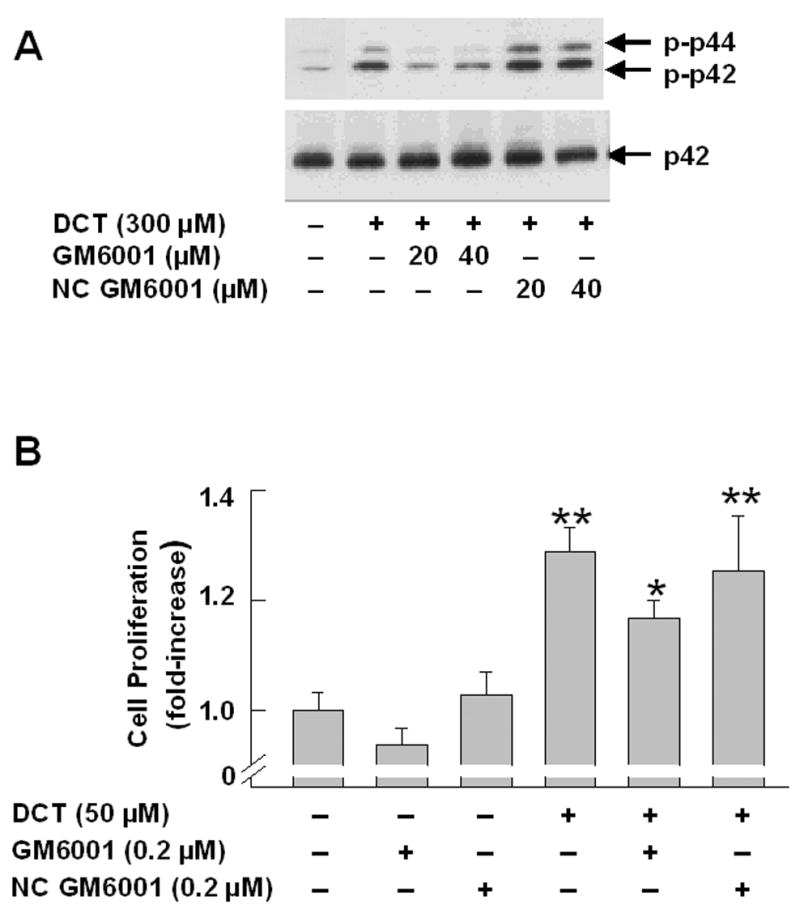
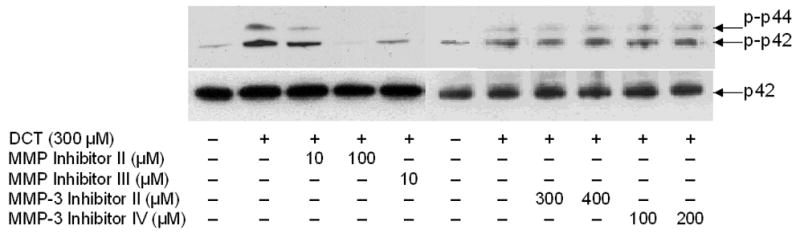
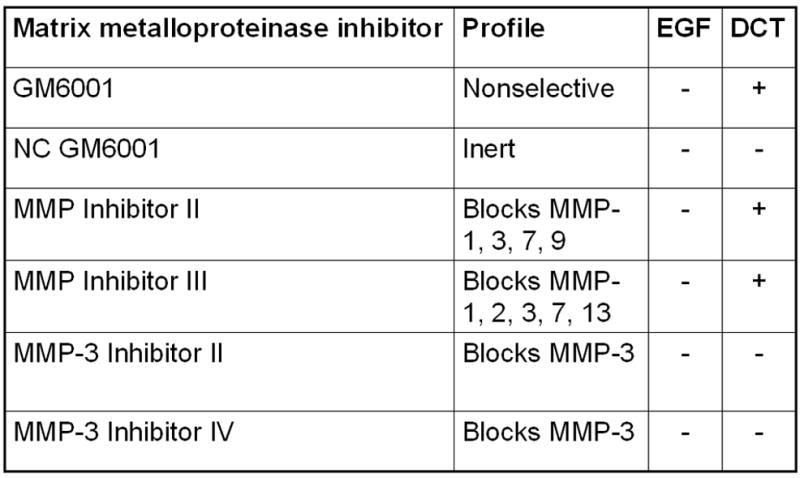
A. MAPK phosphorylation. H508 cells were treated with the indicated concentration of DCT for 10 minutes at 37°C, alone or with two concentrations of GM6001 and NC GM6001. p44/42 MAPK activity was determined by immunoblotting with antibodies specific for phosphorylated p44/42 MAPK. The quantity of protein added was verified by immunoblotting with antibodies specific for total p42 MAPK. Results are representative of 3 separate experiments. B. Cell proliferation. H508 cells were incubated with DCT, alone and with GM6001 or NC GM6001 for 5 days at 37°C. Cell proliferation was determined by the sulforhodamine blue (SRB) colorimetric assay [33]. Results are expressed as mean ± SEM of at least 3 experiments. *P < 0.05 vs cells stimulated with DCT alone. **P < 0.005 vs unstimulated cells. C. Actions of MMP Inhibitors II and III and MMP-3 Inhibitors II and IV on H508 cell proliferation. H508 cells were treated with 300 μM DCT for 10 minutes at 37°C, alone or with the indicated concentrations of MMP inhibitors II and III, and MMP-3 inhibitors II and IV. The quantity of protein added was verified by immunoblotting with antibodies specific for total p42 MAPK. Results are representative of 3 separate experiments. D. Profiles and actions of MMP inhibitors on EGF- and bile acid-induced phosphorylation of p44/42 MAPK in H508 human colon cancer cells. - indicates the inhibitor had no effect; + indicates inhibition by the inhibitor.
To define more clearly which MMP mediates the actions of DCT, we examined the actions of more selective MMP inhibitors. MMP inhibitors -II and –III both blocked the actions of DCT (Fig. 2C). Within the MMP family, the spectrum of MMP inhibition by GM6001, MMP inhibitor-II, and MMP inhibitor-III, limits the likely candidates to MMP-1, -3 and –7 [30] (Fig. 2D). We used MMP-3 inhibitors -II and -IV to evaluate the possibility that MMP-3, which catalyzes release of TGF-α, mediates the actions of DCT. Addition of these MMP-3 inhibitors did not alter DCT-induced p44/42 MAPK phosphorylation (Fig. 2C). To exclude non-specific effects of the MMP inhibitors, control experiments showed that none of these agents altered EGF-induced p44/42 MAPK phosphorylation (not shown). These control experiments indicate that MMP inhibitors block release of EGFR ligands but do not block EGFR activation per se (that is, EGF stimulates p44/42 MAPK phosphorylation in the absence or presence of MMP inhibitors).
The activity profile for the MMP inhibitors used in these studies limits candidate MMP’s mediating the actions of bile acids to MMP-1 and MMP-7 (Fig. 2D). A selective MMP inhibitor that can distinguish between MMP-1 and MMP-7 activity is not currently available. Overall, these data provide additional evidence in support of the hypothesis that bile acids stimulate the release from H508 cells of a ligand that binds to EGFR and activates post-receptor signaling. Moreover, the inhibitory actions of GM6001 and MMP inhibitors -II and -III on DCT-induced signaling support the hypothesis that bile acids activate EGFR by indirect mechanisms. That is, one would not expect MMP inhibitors to block cell proliferation if this was a consequence of direct interaction of DCT with EGFR. Narrowing candidate MMP’s that mediate bile acid actions to MMP-1 and/or MMP-7 limits the relevant reported non-structural extracellular matrix component substrates for these enzymes to TGF-α and HB-EGF [30, 36].
3.3. HB-EGF release and interaction with EGFR plays a role in mediating bile acid-induced p44/42 MAPK phosphorylation and proliferation of H508 colon cancer cells
Based on the results of our experiments with MMP inhibitors, to identify the EGFR ligand that mediates DCT-induced transactivation of EGFR we used antibodies to three common EGFR ligands: HB-EGF, EGF, and TGF-α (Fig. 3). TGF-α is the most common EGFR ligand in the intestines and is reported to mediate carbachol- and bile acid-induced EGFR transactivation in T84 colon cancer cells [29, 37] and in a cholangiocyte cell line [31], respectively.
Figure 3. Effect of adding neutralizing EGFR ligand antibodies on DCT-induced p44/42 MAPK phosphorylation and H508 colon cancer cell proliferation.
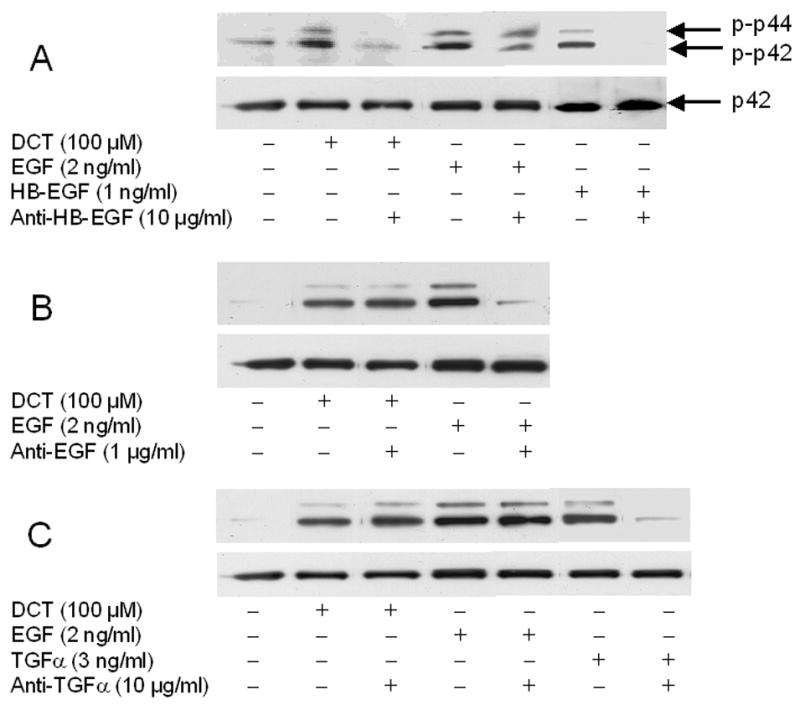
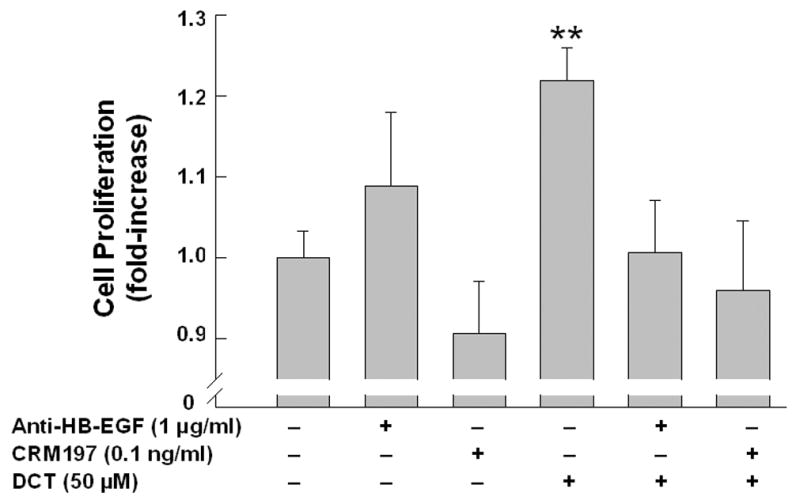
A–C. Effect of adding HB-EGF, EGF, and TGF-α antibodies on p44/42 MAPK phosphorylation. H508 cells were treated with DCT, EGF, HB-EGF and TGF-α for 10 minutes at 37°C, alone or with the EGFR ligand antibodies shown. p44/42 MAPK activity was determined by immunoblotting with antibodies specific for phosphorylated MAPK. The quantity of protein added was verified by immunoblotting with antibodies specific for total p42 MAPK. Results are representative of 3 separate experiments. D. Effect of adding HB-EGF antibodies and CRM197 on cell proliferation. H508 cells were incubated with DCT, alone and with antibody to HB-EGF or with CRM197 (0.1 ng/ml) for 5 days at 37°C. Cellular proliferation was determined by the sulforhodamine blue (SRB) colorimetric assay [33]. Results are mean ± SEM of 3 to 5 experiments. **P < 0.005 vs unstimulated cells.
Strikingly, in H508 colon cancer cells, neutralizing antibody to HB-EGF abolished the actions of DCT and HB-EGF, but not those of EGF (Fig. 3A). Neutralizing antibody to EGF reduced the actions of EGF but not those of DCT (Fig. 3B), whereas neutralizing TGF-α antibody reduced the actions of TGF-α but did not alter the actions of DCT or EGF (Fig. 3C). Collectively, the MMP inhibitor profile (Fig. 2) and the results with neutralizing antibodies to EGFR ligands (Fig. 3) indicated that HB-EGF was a promising candidate for the EGFR ligand that mediates the proliferative actions of DCT.
As shown in Figure 3D, both neutralizing antibody to HB-EGF, and a non-toxic mutant form of diphtheria toxin that prevents HB-EGF release, CRM197, attenuated the proliferative actions of DCT. Control experiments with both anti-EGF and anti-TGFα antibodies showed that these agents inhibited EGF- and TGFα-induced cell proliferation (p < 0.05 and < 0.005), respectively, but did not alter DCT-induced cell proliferation (not shown). Likewise, anti-EGF antibody did not block the proliferative effects of TGFα and anti-TGFα antibody did not block the proliferative effects of EGF. Overall, these data support the hypothesis that, in H508 colon cancer cells, MMP-catalyzed release of the EGFR ligand HB-EGF, but not EGF or TGF-α, plays a role in mediating bile acid-induced cell signaling and proliferation.
3.4. Recombinant HB-EGF-induced activation of p44/42 MAPK is inhibited by EGFR and MEK inhibitors and by antibody to the ligand-binding domain EGFR
To confirm that HB-EGF activates EGFR and post-receptor signaling in H508 cells, we examined the actions of increasing concentrations of the EGFR ligand on p44/42 MAPK phosphorylation (Fig. 4A). p44/42 MAPK phosphorylation was detected with 0.2 and maximal with 20 ng/ml HB-EGF (Fig. 4A). As expected for the actions of an EGFR ligand, an antibody to the ligand-binding domain of EGFR, an inhibitor of EGFR activation (PD168393), and an inhibitor of MEK (MAPK kinase) (PD98059) all inhibited HB-EGF-induced p44/42 MAPK phosphorylation (Fig. 4B). Regarding effects on cell proliferation, addition of either EGFR inhibitors or antibody to the ligand binding domain of EGFR attenuated HB-EGF-induced cell proliferation (p < 0.005 for both) (not shown).
Figure 4. Actions of recombinant HB-EGF on p44/42 MAPK phosphorylation in H508 colon cancer cells.
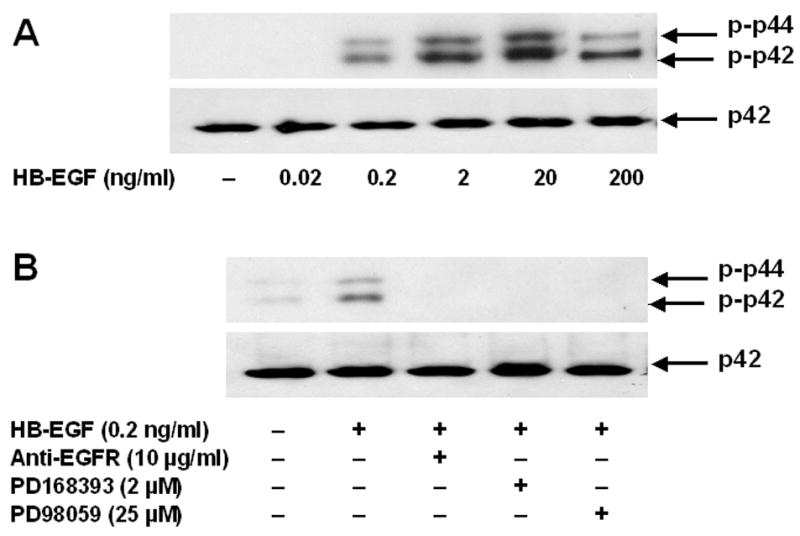
A. Actions of increasing concentration of HB-EGF on p44/42 MAPK phosphorylation. H508 cells were treated with increasing concentrations of HB-EGF for 10 minutes at 37°C. p44/42 MAPK activity was determined by immunoblotting with antibodies specific for phosphorylated MAPK. The quantity of protein added was verified by immunoblotting with antibodies specific for total p42 MAPK. Results are representative of 3 separate experiments. B. Antibodies to the ligand-binding domain of EGFR, and EGFR (PD168393) and MEK (PD98059) inhibitors block HB-EGF-induced p44/42 MAPK phosphorylation. H508 cells were treated with the indicated concentration of HB-EGF for 10 minutes at 37°C, alone or in the presence of the indicated concentrations of antibodies and inhibitors. p44/42 MAPK activity was determined by immunoblotting with antibodies specific for phosphorylated MAPK. The quantity of protein added was verified by immunoblotting with antibodies specific for total p42 MAPK. Results are representative of 3 separate experiments.
3.5. Recombinant MMP-7 mimics the actions of bile acids and antibody to MMP-7 blocks these actions
Based on the spectrum of action of MMP inhibitors used for these experiments (Fig. 2), we concluded that activation of either MMP-1 or MMP-7 may play a role in mediating the actions of DCT. Previous reports indicate that MMP-7 (also known as matrilysin; EC 3.4.24.23) catalyzes release of HB-EGF [36]. Moreover, in clinical studies, expression of MMP-7 increases progressively with more advanced grades of colonic epithelial dysplasia and cancer [38–40] and, in a rodent model, the formation of intestinal tumors is reduced in Min mice that do not express MMP-7 [41]. Hence, experimental data support a role for MMP-7 in facilitating colon carcinogenesis. Our data suggest that bile acid-induced activation of the enzyme with consequent release of the EGFR ligand HB-EGF represents the molecular mechanism underlying these observations.
To resolve further the role of matrix metalloproteinases in mediating the observed actions of bile acids, we examined the effect of adding exogenous human recombinant MMP-1 and MMP-7 to H508 cells. As shown in Figure 5A, MMP-7 but not MMP-1 (both 0.1 μg/ml) stimulated p44/42 MAPK phosphorylation. Adding an EGFR inhibitor (PD168393), neutralizing antibodies to HB-EGF or antibodies directed to the ligand-binding domain of EGFR blocked the actions of MMP-7 (Fig. 5A).
Figure 5. MMP-7 mimics the actions of bile acids on H508 colon cancer cells.
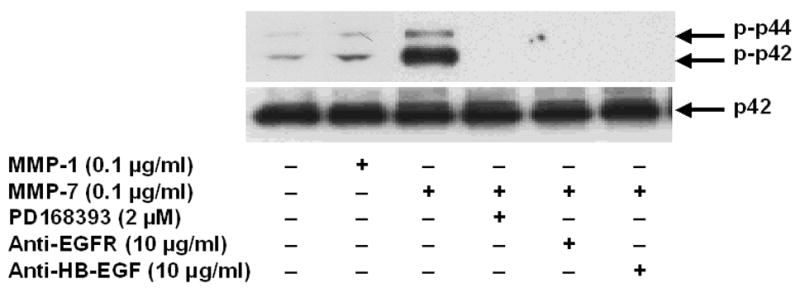
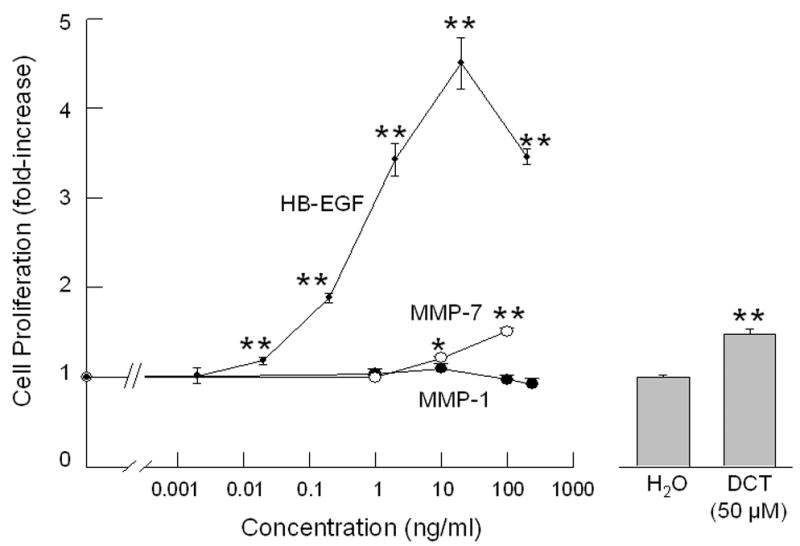
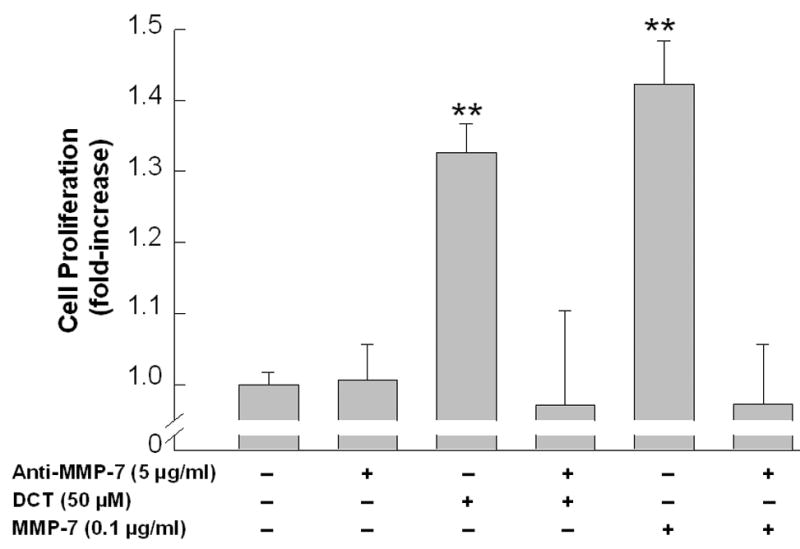
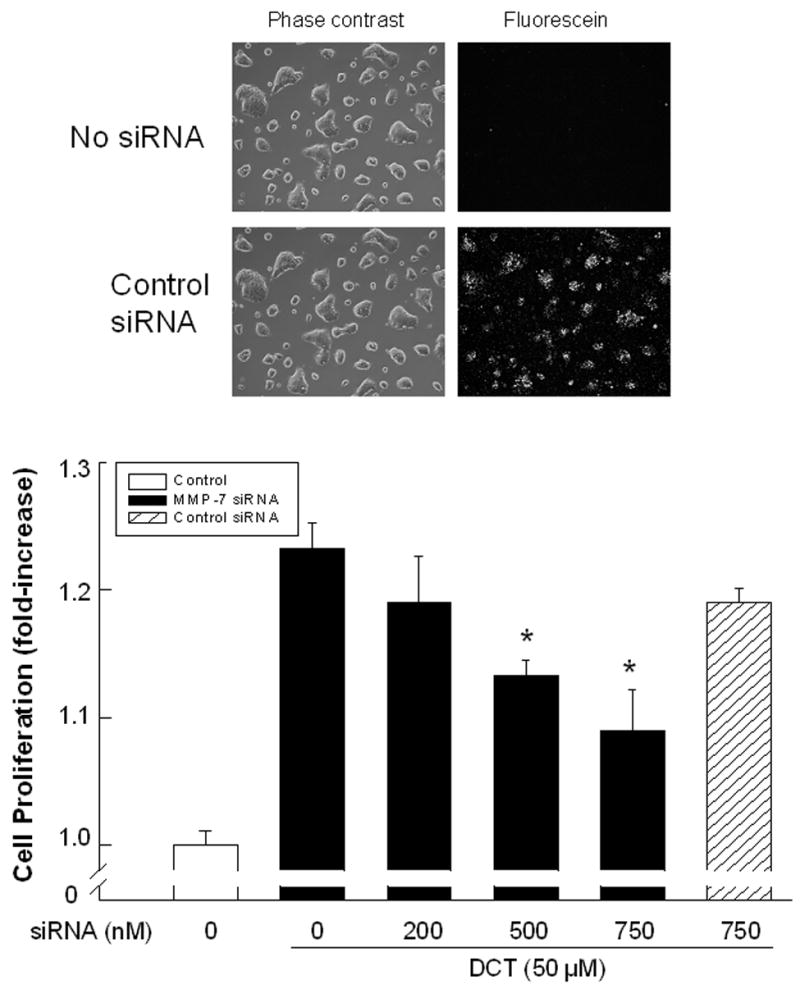
A. Effect of MMP-1 and MMP-7, alone or in the presence of an EGFR inhibitor and antibodies to EGFR and HB-EGF, on p44/42 MAPK phosphorylation. H508 cells were treated with MMP-7 (0.1 μg/ml) and MMP-1 (0.1 μg/ml), alone or in the presence of an EGFR inhibitor (PD168393, 2 μM), an anti-EGFR antibody (10 μg/ml) and an anti-HB-EGF neutralizing antibody (10 μg/ml), for 10 minutes at 37°C. p44/42 MAPK activity was determined by immunoblotting with antibodies specific for phosphorylated MAPK. The quantity of protein added was verified by immunoblotting with antibodies specific for total p42 MAPK. Results are representative of 3 separate experiments. B. Comparison of the efficacy of increasing concentrations of HB-EGF, MMP-7 and MMP-1, with that of DCT for stimulating H508 cell proliferation. Cells were incubated with increasing concentrations of MMP-7, MMP-1, and HB-EGF for 5 days at 37°C and stimulation of cell proliferation was compared to that observed with water and DCT. Results are mean ± SEM of 3 experiments. *,**P < 0.05 and 0.005, respectively vs unstimulated cells. The vertical axis (cell proliferation) is the same for the line drawing (left panel) and bar graph (right panel). C. MMP-7 antibodies inhibit the proliferative actions of MMP-7 and DCT on H508 colon cancer cells. Cells were incubated with DCT and MMP-7, alone and in the presence of antibody to MMP-7, for 5 days at 37°C. Cellular proliferation was determined by the sulforhodamine blue (SRB) colorimetric assay [33]. Results are mean ± SEM of 3 experiments. **P < 0.005 vs unstimulated cells. D. Transfection of cells with MMP-7 siRNA inhibits the proliferative actions of DCT. H508 cells were transfected with siRNA duplex oligos targeting human MMP-7 or a fluorescein-tagged nonspecific duplex oligo negative control. Two days following transfection, cells were incubated for an additional 5 days in FBS-free medium containing 50 μM DCT. The inset at the top shows cellular accumulation of fluorescent control siRNA. Cellular proliferation was determined by the sulforhodamine blue (SRB) colorimetric assay [33]. Results are mean ± SEM of 3 experiments. *P < 0.05 vs cells incubated with DCT alone.
As shown in Figure 5B, the addition of increasing concentrations of MMP-7 and HB-EGF, but not of MMP-1, stimulated H508 cell proliferation. Over the range of concentrations tested, HB-EGF was both more potent and more efficacious than MMP-7. Moreover, as shown in Figure 5B, the magnitude of MMP-7- and HB-EGF-induced cell proliferation overlapped with that observed with a maximally efficacious concentration of DCT. The increase in H508 cell proliferation observed with 50 μM DCT was achieved with approximately 100 ng/ml MMP-7 or 0.2 ng/ml HB-EGF. These findings support the hypothesis that the actions of DCT are mediated by MMP-7-catalyzed release of HB-EGF with consequent autocrine stimulation of EGFR. Furthermore, the findings in Figures 5A and 5B clearly exclude the possibility that MMP-1 plays any role in these actions.
To test further the hypothesis that activation of MMP-7 mediates the proliferative actions of DCT, we examined the effects of reducing MMP-7 expression with neutralizing antibody and siRNA. As shown in Figure 5C, MMP-7 antibody blocked both MMP-7- and DCT-induced H508 cell proliferation. Likewise, as shown in Figure 5D, transfecting cells with increasing concentrations of MMP-7, but not control, siRNA progressively inhibited DCT-induced H508 cell proliferation. Fluorescein imaging with labeled control siRNA verified the efficiency of transfection (Fig. 5D top). These data provide strong evidence that MMP-7 mediates the proliferative actions of the bile acid.
3.6. PCR confirms MMP-7 expression and induction by DCT, and immunofluorescence microscopy reveals plasma membrane co-localization of pro-HB-EGF and pro-MMP-7 expression in H508 colon cancer cells
To confirm that H508 human colon cancer cells express both pro-HB-EGF and pro-MMP-7 and to determine the subcellular localization of these molecules, we used quantitative reverse transcription-PCR and immunofluorescence microscopy. As shown in Figure 6A, in H508 cells, PCR confirmed both that MMP-7 is expressed and that DCT induces time-dependent MMP-7 gene transcription. GAPDH gene transcription is shown as a control. As shown in Figure 6B, adding 100 μM DCT stimulates a > 10-fold induction of MMP-7 gene transcription (compared to the GAPDH control at each time point). This effect is maximal at 4–6 hrs and MMP-7 mRNA levels return to near basal values by 24 hrs. For immunohistochemistry, cells were incubated with goat anti-human pro-HB-EGF antibody and mouse anti-human pro-MMP-7 antibody. This was followed by a second incubation with FITC-conjugated rabbit anti-goat antibody and Tritc-conjugated goat anti-mouse antibody. Control cells incubated with only the second antibody did not reveal cellular immunofluorescence (not shown). As shown in Figure 6C, phase contrast microscopy of H508 cells revealed many pleomorphic cells with multiple nuclei and an increased nuclear to cytoplasmic ratio typical of cancer cells. Cell treatment with both the primary antibody and the second antibody conjugated to the fluoroprobe revealed that human H508 cells express both pro-HB-EGF (green) and pro-MMP-7 (red). Moreover, Hoechst nuclear staining along with a merged image of both pro-HB-EGF and pro-MMP-7 immunostaining revealed cytoplasmic co-localization of pro-HB-EGF and pro-MMP-7 (Fig. 6C, extreme right panel).
Figure 6. Expression of MMP-7 and plasma membrane co-localization of pro-HB-EGF and pro-MMP-7 in H508 human colon cancer cells.
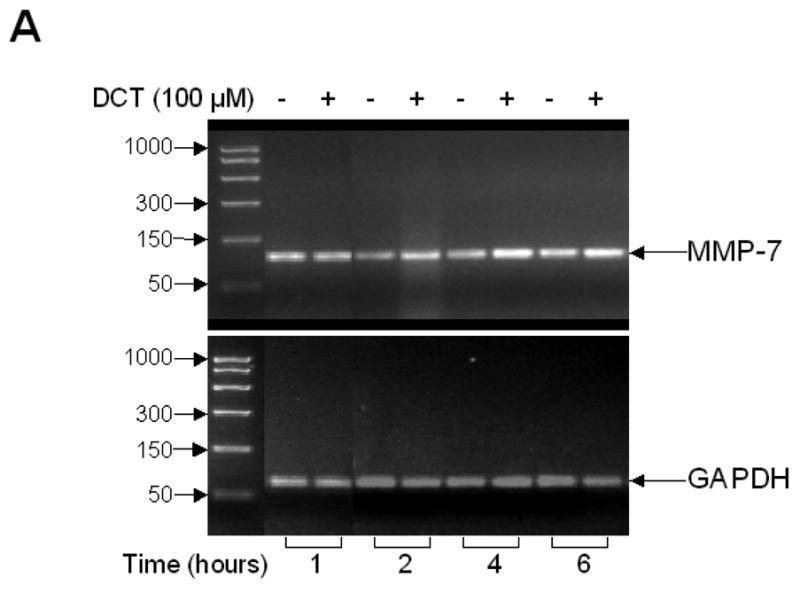
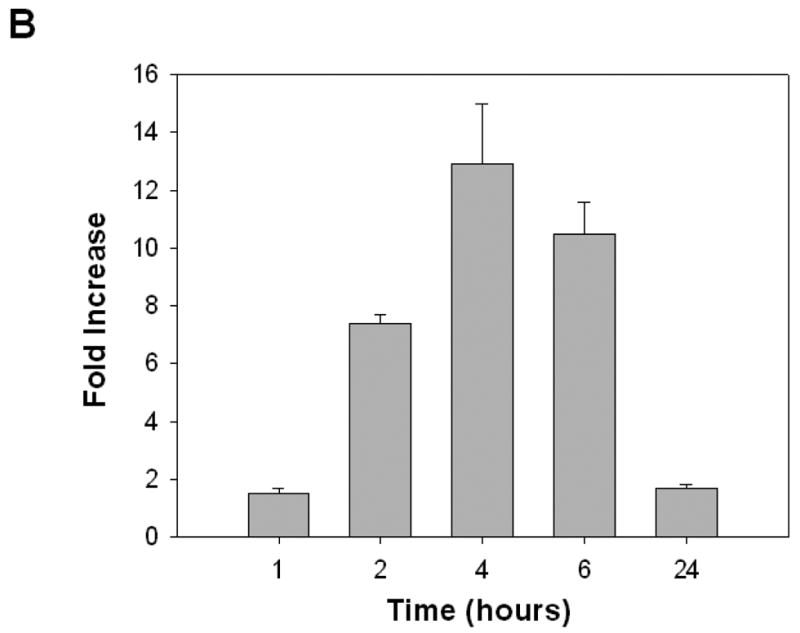
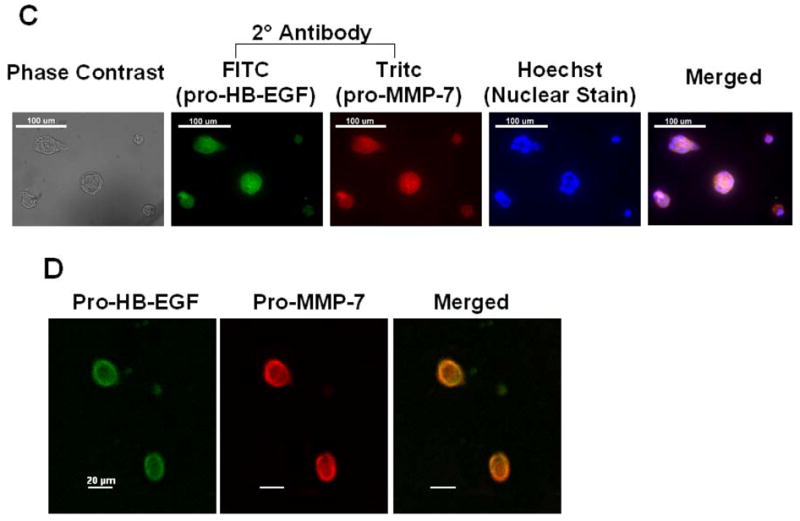
A. Agarose gel (2%) of PCR products from quantitative reverse transcription-PCR in H508 cells confirms MMP-7 gene transcription and induction by DCT. H508 cells were cultured in serum-free media for 24 hrs prior to DCT (100 μM) treatment. Total RNA was isolated following harvest of cells at the indicated time points. DNA size markers (base pairs) are indicated at left with arrows. Arrows at right indicate PCR product for MMP-7 and the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) control. B. DCT induces MMP-7 mRNA expression in H508 colon cancer cells. The level of MMP-7 mRNA was determined by quantitative real-time PCR as described in Materials and methods. Results are mean ± SEM of 3 experiments. C. Immunofluorescence microscopy. Left panel: Phase contrast microscopy of H508 colon cancer cells. Immunofluorescence localization in H508 colon cancer cells using pro-HB-EGF (FITC, green fluorescence) and pro-MMP-7 (Tritc, red fluorescence) antibodies is shown. Cell nuclei are stained with Hoechst (blue). Extreme right panel: Merged images of pro-HB-EGF, pro-MMP-7 and cell nuclei. Cytoplasmic co-localization of pro-HB-EGF and pro-MMP-7 is shown by light orange color. Scale bar = 100 μm. D. Confocal microscopy. Immunofluorescence staining of pro-HB-EGF (left, green fluorescence) and pro-MMP-7 (middle, red fluorescence) demonstrates cell surface co-localization (right, light orange fluorescence). Scale bar = 20 μm.
To determine the subcellular localization of HB-EGF (green) and pro-MMP-7 (red), we used confocal microscopy. As shown in Figure 6D, confocal immunofluourescence studies showed a prominent co-localization of pro-HB-EGF and pro-MMP-7 (light orange) at the cell surface, presumably corresponding to the plasma membrane. These experiments confirmed both the expression and plasma membrane co-localization of pro-HB-EGF and pro-MMP-7 in H508 human colon cancer cells.
4. Discussion
To implicate activation of a particular MMP in the promotion of colon cancer cell proliferation, several criteria must be achieved; the candidate MMP must be expressed in the cell being studied at a pericellular location, at the right time and concentration, and it must be activated or inhibited appropriately. Previously, we presented evidence for the functional interaction of conjugated secondary bile acids with muscarinic receptors and the resulting transactivation of EGFR [15, 16, 34]. Novel evidence favoring important roles for MMP-7 and HB-EGF in mediating the proliferative actions of bile acids include the following observations: the pattern of activity of MMP inhibitors is consistent with a role for MMP-7; recombinant MMP-7 mimics both the signaling and proliferative actions of HB-EGF and DCT; PCR confirms MMP-7 expression and induction by DCT; immunofluorescence microscopy demonstrates pro-MMP-7 localization at the cell surface in association with the pro-HB-EGF substrate; neutralizing HB-EGF antibody inhibits the effects of DCT; neutralizing MMP-7 antibody inhibits both MMP-7- and DCT-induced cell proliferation; and finally, cell transfection with siRNA to reduce MMP-7 expression inhibits DCT-induced cell proliferation.
Upregulation of EGFR expression and activation plays a pivotal role in colon cancer cell proliferation (reviewed in [25]). Like other mammalian EGFR ligands, HB-EGF is synthesized as type I transmembrane protein (pro-ligand) [29]. EGFR pro-ligand cleavage is dependent on the actions of endopeptidases, primarily matrix metalloproteases, which regulate release of the active proliferative factor [29]. Active matrix metalloproteases are secreted or activated as part of a membrane-anchored pro-ligand/enzyme complex [29, 36], and target a variety of cellular and extracellular protein substrates, including EGFR pro-ligands [30]. Collectively, the data presented herein provide compelling evidence that bile acid-induced proliferation of colon cancer cells is mediated by MMP-7-catalyzed release of HB-EGF, followed by activation of EGFR and downstream signaling cascades. The latter events alter gene transcription and result in cellular proliferation [29, 42].
The matrix metalloprotease family comprises at least 22 members that are grouped by structure and cellular location (reviewed in [30, 43]). Of 8 structural groups, 5 are secreted into the extracellular space and 3 are tethered to the cell membrane by covalent linkages [30, 43]. MMP-7 (matrilysin; EC3.4.24.23), a secreted ‘minimal-domain’ MMP that is associated with the cell surface, contains three major structural components; an amino-terminal sequence that directs it to the endoplasmic reticulum, a sequence containing a zinc-interacting thiol group that maintains MMP-7 as an inactive zymogen, and a zinc-binding catalytic domain [30]. As confirmed in the present study by immunofluorescence microscopy, cell surface association increases MMP-7 stability and proximity to potential plasma membrane substrates (e.g. pro-HB-EGF) [30, 36, 44].
MMP-7-mediated cleavage of pro-HB-EGF (206 amino acids) and release of HB-EGF (86 amino acids), is reported in several cancer cell types [30, 36]. Studies conducted more than a decade ago, reported MMP-7 expression in approximately 80% of human colon cancers [38] and in the CaR-1 human rectal carcinoma cell line [45]. Moreover, in a murine cancer model, MMP-7 gene knockout reduces intestinal tumor formation [41]. In the present report, we contribute the novel observations that H508 human colon cancer cells express MMP-7, that a bile acid induces a substantial increase in MMP-7 expression, and that MMP-7-catalyzed release of HB-EGF plays a key role in mediating the proliferative actions of DCT.
Experiments, beyond the scope of the present communication, are needed in human colon cancer cells to validate the role of MMP-7 in mediating bile acid signaling and to place it in the context of other enzymes that can release EGFR ligands. Demonstrating expression and bioactivity of MMP-7 in colon cancer cells is necessary, but not sufficient to prove that this is the only enzyme that mediates bile acid-induced transactivation of EGFR. Although our results using MMP-7 neutralizing antibody and siRNA provide strong evidence that the enzyme plays an important role in mediating the actions of bile acids, enzymes in the metzincin superfamily of metalloproteases, designated ‘disintegrin and metalloproteinase’ domain metalloproteases (ADAMs), can also catalyze EGFR ligand release [43]. Current data on the expression of ADAM metalloproteases in normal or neoplastic colonic epithelium are limited, and few selective inhibitors of their actions are available. Hence, the possibility that other metalloproteases besides MMP-7 play a role in bile acid-induced HB-EGF release is currently being explored in our laboratory. It is also conceivable that in a given cancer cell line more than one metalloprotease mediates these actions or that different colon cancer cell lines express different bile acid-responsive metalloproteases. A multiplicity of EGFR pro-ligand releasing enzymes could provide additional regulatory control of bile acid-induced cell proliferation. As we showed for MMP-7, beyond demonstrating expression of a particular metalloprotease(s), it is necessary to confirm that the actions of bile acids (e.g. activation of EGFR signaling and stimulation of cell proliferation) are attenuated by pharmacological inhibition, and by molecular approaches that knockout and/or knockdown the enzyme(s).
Our work now focuses on identifying additional MMPs and/or ADAMs that are expressed in H508 colon cancer cells. Future studies will elucidate the precise mechanisms underlying bile acid-induced metalloprotease activation and the role of bile acid interaction with muscarinic receptors in initiating this process [16, 23]. Based on studies from our laboratory and others, we conclude that conjugated secondary bile acids function as growth factors, thereby contributing to a favorable microenvironment that promotes the survival and proliferation of colon cancer cells. Blocking the proliferative actions of bile acids may retard colon cancer growth and, possibly, tumor invasion and spread. The clinical and therapeutic implications of our work include the possibility of achieving these goals by targeting multiple steps leading from metalloprotease activation to release of HB-EGF and activation of EGFR signaling.
Acknowledgments
Grant support: this work was supported by the Office of Research and Development, Medical Research Service, Department of Veterans Affairs (JPR) and by NIH grants CA107345 (JPR) and T32 DK067872 (GX).
Abbreviations
- GPCR
G-protein coupled receptor
- M3R
M3 muscarinic receptor
- ACh
acetylcholine
- RTK
receptor tyrosine kinase
- EGFR
epidermal growth factor receptor
- TGF-α
transforming growth factor-α
- MAPK
mitogen activated protein kinase
- p90RSK
p90 ribosomal S kinase
- HB-EGF
heparin binding-epidermal growth factor-like growth factor
- MMP
matrix metalloproteinase
- siRNA
small interfering RNA
- DCT
deoxycholyltaurine
- ADAM
a disintegrin and metalloprotease
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hill MJ. Bile acids and colorectal cancer: hypothesis. Eur J Cancer Prev. 1991;1:69–74. doi: 10.1097/00008469-199110002-00012. [DOI] [PubMed] [Google Scholar]
- 2.Reddy BS, Wynder EL. Metabolic epidemiology of colon cancer. Fecal bile acids and neutral sterols in colon cancer patients and patients with adenomatous polyps. Cancer. 1977;39:2533–9. doi: 10.1002/1097-0142(197706)39:6<2533::aid-cncr2820390634>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 3.Glinghammar B, Rafter J. Carcinogenesis in the colon: interaction between luminal factors and genetic factors. Eur J Cancer Prev. 1999;8:S87–S94. [PubMed] [Google Scholar]
- 4.Narisawa T, Magadia NE, Weisburger JH, Wynder EL. Promoting effect of bile acids on colon carcinogenesis after intrarectal instillation of N-methyl-N'-nitro-N-nitrosoguanidine in rats. J Natl Cancer Instit. 1974;53:1093–7. doi: 10.1093/jnci/53.4.1093. [DOI] [PubMed] [Google Scholar]
- 5.Reddy BS, Narasawa T, Weisburger JH, Wynder EL. Promoting effect of sodium deoxycholate on colon adenocarcinomas in germfree rats. J Natl Cancer Instit. 1976;56:441–2. doi: 10.1093/jnci/56.2.441. [DOI] [PubMed] [Google Scholar]
- 6.Nagengast FM, Grubben MJAL, van Munster IP. Role of bile acids in colorectal carcinogenesis. Eur J Cancer. 1995;31A:1067–70. doi: 10.1016/0959-8049(95)00216-6. [DOI] [PubMed] [Google Scholar]
- 7.Earnest DL, Holubec H, Wali RK, Jolley CS, Bissonette M, Bhattacharyya AK, et al. Chemoprevention of azoxymethane-induced colonic carcinogenesis by supplemental dietary ursodeoxycholic acid. Cancer Res. 1994;54:5071–4. [PubMed] [Google Scholar]
- 8.Narisawa T, Fukaura Y, Terada K, Sekiguchi H. Prevention of N-methylnitrosourea-induced colon tumorigenesis by ursodeoxycholic acid in F344 rats. Jpn J Cancer Res. 1998;89:1009–13. doi: 10.1111/j.1349-7006.1998.tb00489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pardi DS, Loftus EV, Jr, Kremers WK, Keach J, Lindor KD. Ursodeoxycholic acid as a chemopreventive agent in patients with ulcerative colitis and primary sclerosing cholangitis. Gastroenterology. 2003;124:889–93. doi: 10.1053/gast.2003.50156. [DOI] [PubMed] [Google Scholar]
- 10.Martinez JD, Stratagoules ED, LaRue JM, Powell AA, Gause PR, Craven MT, et al. Different bile acids exhibit distinct biological effects: the tumor promoter deoxycholic acid induces apoptosis and the chemopreventive agent ursodeoxycholic acid inhibits cell proliferation. Nutr Cancer. 1998;31:111–8. doi: 10.1080/01635589809514689. [DOI] [PubMed] [Google Scholar]
- 11.Qiao D, Stratagouleas ED, Martinez JD. Activation and role of mitogen-activated protein kinases in deoxycholic acid-induced apoptosis. Carcinogenesis. 2001;22:35–41. doi: 10.1093/carcin/22.1.35. [DOI] [PubMed] [Google Scholar]
- 12.Pai R, Tarnawski AS, Tran T. Deoxycholic acid activates beta-catenin signaling pathway and increases colon cell cancer growth and invasiveness. Mol Biol Cell. 2004;15:2156–63. doi: 10.1091/mbc.E03-12-0894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Powell AA, LaRue JM, Batta AK, Martinez JD. Bile acid hydrophobicity is correlated with induction of apoptosis and/or growth arrest in HCT116 cells. Biochem J. 2001;356:481–6. doi: 10.1042/0264-6021:3560481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martinez-Diez MC, Serrano MA, Monte MJ, Marin JJ. Comparison of the effects of bile acids on cell viability and DNA synthesis by rat hepatocytes in primary culture. Biochim Biophys Acta. 2000;1500:153–60. doi: 10.1016/s0925-4439(99)00099-x. [DOI] [PubMed] [Google Scholar]
- 15.Cheng K, Chen Y, Zimniak P, Raufman J, Xiao Y, Frucht H. Functional interaction of lithocholic acid conjugates with M3 muscarinic receptors on a human colon cancer cell line. Biochim Biophys Acta. 2002;1588:48–55. doi: 10.1016/s0925-4439(02)00115-1. [DOI] [PubMed] [Google Scholar]
- 16.Cheng K, Raufman JP. Bile acid-induced proliferation of a human colon cancer cell line is mediated by transactivation of epidermal growth factor receptors. Biochem Pharmacol. 2005;70:1035–47. doi: 10.1016/j.bcp.2005.07.023. [DOI] [PubMed] [Google Scholar]
- 17.Lakhani SA, Masud A, Kuida K, Porter GA, Jr, Booth CJ, Mehal WZ, et al. Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science. 2006;311:847–51. doi: 10.1126/science.1115035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pai R, Soreghan B, Szabo IL, Pavelka M, Baatar D, Tarnawski AS. Prostaglandin E2 transactivates EGF receptor: a novel mechanism for promoting colon cancer growth and gastrointestinal hypertrophy. Nat Med. 2002;8:289–93. doi: 10.1038/nm0302-289. [DOI] [PubMed] [Google Scholar]
- 19.Roberts RB, Min L, Washington MK, Olsen SJ, Settle SH, Coffey RJ, et al. Importance of epidermal growth factor receptor signaling in establishment of adenomas and maintenance of carcinomas during intestinal tumorigenesis. Proc Natl Acad Sci U S A. 2002;99:1521–6. doi: 10.1073/pnas.032678499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fischer OM, Hart S, Gschwind A, Ullrich A. EGFR signal transactivation in cancer cells. Biochem Soc Trans. 2003;31:1203–8. doi: 10.1042/bst0311203. [DOI] [PubMed] [Google Scholar]
- 21.Frucht H, Jensen RT, Dexter D, Yang W-L, Xiao Y. Human colon cancer cell proliferation mediated by the M3 muscarinic cholinergic receptor. Clin Cancer Res. 1999;5:2532–9. [PubMed] [Google Scholar]
- 22.Ukegawa JI, Takeuchi Y, Kusayanagi S, Mitamura K. Growth-promoting effect of muscarinic acetylcholine receptors in colon cancer cells. J Cancer Res Clin Oncol. 2003;129:272–8. doi: 10.1007/s00432-003-0433-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng K, Zimniak P, Raufman JP. Transactivation of the epidermal growth factor receptor mediates cholinergic agonist-induced proliferation of H508 human colon cancer cells. Cancer Res. 2003;63:6744–50. [PubMed] [Google Scholar]
- 24.Frodin M, Gammeltoft S. Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Mol Cell Endocrinol. 1999;151:65–77. doi: 10.1016/s0303-7207(99)00061-1. [DOI] [PubMed] [Google Scholar]
- 25.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–37. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 26.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–25. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 27.Dong J, Opresko LK, Dempsey PJ, Lauffenburger DA, Coffey RJ, Wiley HS. Metalloprotease-mediated ligand release regulates autocrine signaling through the epidermal growth factor receptor. Proc Natl Acad Sci U S A. 1999;96:6235–40. doi: 10.1073/pnas.96.11.6235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, et al. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature. 1999;402:884–8. doi: 10.1038/47260. [DOI] [PubMed] [Google Scholar]
- 29.Harris RC, Chung E, Coffey RJ. EGF receptor ligands. Exp Cell Res. 2003;284:2–13. doi: 10.1016/s0014-4827(02)00105-2. [DOI] [PubMed] [Google Scholar]
- 30.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–74. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 31.Werneburg NW, Yoon JH, Higuchi H, Gores GJ. Bile acids activate EGF receptor via a TGF-alpha-dependent mechanism in human cholangiocyte cell lines. Am J Physiol Gastrointest Liver Physiol. 2003;285:G31–6. doi: 10.1152/ajpgi.00536.2002. [DOI] [PubMed] [Google Scholar]
- 32.Schmidt M, Huwe SM, Fasselt B, Homann D, Rumenapp U, Sandmann J, et al. Mechanisms of phospholipase D stimulation by m3 muscarinic acetylcholine receptors. Evidence for involvement of tyrosine phosphorylation. Eur J Biochem. 1994;225:667–75. doi: 10.1111/j.1432-1033.1994.00667.x. [DOI] [PubMed] [Google Scholar]
- 33.Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst. 1990;82:1107–12. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 34.Raufman JP, Chen Y, Cheng K, Compadre C, Compadre L, Zimniak P. Selective interaction of bile acids with muscarinic receptors: a case of molecular mimicry. Eur J Pharmacol. 2002;457:77–84. doi: 10.1016/s0014-2999(02)02690-0. [DOI] [PubMed] [Google Scholar]
- 35.Grobelny D, Poncz L, Galardy RE. Inhibition of human skin fibroblast collagenase, thermolysin, and Pseudomonas aeruginosa elastase by peptide hydroxamic acids. Biochemistry. 1992;31:7152–4. doi: 10.1021/bi00146a017. [DOI] [PubMed] [Google Scholar]
- 36.Yu WH, Woessner JF, Jr, McNeish JD, Stamenkovic I. CD44 anchors the assembly of matrilysin/MMP-7 with heparin-binding epidermal growth factor precursor and ErbB4 and regulates female reproductive organ remodeling. Genes Dev. 2002;16:307–23. doi: 10.1101/gad.925702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCole DF, Keely SJ, Coffey RJ, Barrett KE. Transactivation of the epidermal growth factor receptor in colonic epithelial cells by carbachol requires extracellular release of transforming growth factor-alpha. J Biol Chem. 2002;277:42603–12. doi: 10.1074/jbc.M206487200. [DOI] [PubMed] [Google Scholar]
- 38.Newell KJ, Witty JP, Rodgers WH, Matrisian LM. Expression and localization of matrix-degrading metalloproteinases during colorectal tumorigenesis. Mol Carcinog. 1994;10:199–206. doi: 10.1002/mc.2940100404. [DOI] [PubMed] [Google Scholar]
- 39.Newell KJ, Matrisian LM, Driman DK. Matrilysin (matrix metalloproteinase-7) expression in ulcerative colitis-related tumorigenesis. Mol Carcinog. 2002;34:59–63. doi: 10.1002/mc.10049. [DOI] [PubMed] [Google Scholar]
- 40.Takeuchi N, Ichikawa Y, Ishikawa T, Momiyama N, Hasegawa S, Nagashima Y, et al. Matrilysin gene expression in sporadic and familial colorectal adenomas. Mol Carcinog. 1997;19:225–9. [PubMed] [Google Scholar]
- 41.Wilson CL, Heppner KJ, Labosky PA, Hogan BL, Matrisian LM. Intestinal tumorigenesis is suppressed in mice lacking the metalloproteinase matrilysin. Proc Natl Acad Sci U S A. 1997;94:1402–7. doi: 10.1073/pnas.94.4.1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jorissen RN, Walker F, Pouliot N, Garrett TP, Ward CW, Burgess AW. Epidermal growth factor receptor: mechanisms of activation and signalling. Exp Cell Res. 2003;284:31–53. doi: 10.1016/s0014-4827(02)00098-8. [DOI] [PubMed] [Google Scholar]
- 43.Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu WH, Woessner JF., Jr Heparan sulfate proteoglycans as extracellular docking molecules for matrilysin (matrix metalloproteinase 7) J Biol Chem. 2000;275:4183–91. doi: 10.1074/jbc.275.6.4183. [DOI] [PubMed] [Google Scholar]
- 45.Imai K, Yokohama Y, Nakanishi I, Ohuchi E, Fujii Y, Nakai N, et al. Matrix metalloproteinase 7 (matrilysin) from human rectal carcinoma cells. Activation of the precursor, interaction with other matrix metalloproteinases and enzymic properties. J Biol Chem. 1995;270:6691–7. doi: 10.1074/jbc.270.12.6691. [DOI] [PubMed] [Google Scholar]