

Abstract
Our previous studies demonstrated that sulindac, a non-steroidal anti-inflammatory drug, suppressed intestinal tumor formation in mouse, which is linked to the induction of wild-type p53-activated fragment 1 (p21WAF1, or p21). Here we showed that sulindac also required c-jun N-terminal Kinase 1 (JNK1) to inhibit cell proliferation and induce apoptosis in vitro and in vivo. First, sulindac inhibited cell proliferation and induced apoptosis in colon cancer cell lines HCT116 with wild-type p21 or null p21, which were p21-dependent and were also associated with the induction of p21 and phosphorylation of JNK1. Second, sulindac increased apoptosis in JNK1+/+ and JNK1−/− mouse embryonic fibroblast (MEF) cells, but, the increase of apoptosis in JNK1+/+ cells was more than that in JNK1−/− cells. More interestingly, sulindac significantly inhibited cell proliferation in JNK1+/+ cells, but the inhibition in JNK1−/− cells markedly decreased. Further studies indicated that JNK1 was dramatically induced by sulindac in the JNK1+/+ cells which correlated with the induction of p21. However, the induction of p21 in JNK1−/− cells was less than that in JNK1+/+ cells. Finally, we determined the expression of JNK1 in the intestinal mucosa of Apc+/−, p21+/+ mice, and found that sulindac significantly induced JNK1 phosphorylation, corresponding to the induction of p21, both in mRNA and protein levels. Our data indicates that sulindac-mediated proliferation inhibition and apoptosis induction were not only p21-dependent, but also required JNK1.
Keywords: JNK1, p21, sulindac, proliferation, apoptosis
1. INTRODUCTION
Sulindac is one of nonsteroidal anti-inflammatory drugs that have significant activity in inhibiting colon tumor formation (Clevers, 2006; Kelloff et al., 2006; Szabo, 2006). Sulindac has been shown to be effective in preventing intestinal tumor formation in mouse models that inherited a mutant APC gene (eg. Apc1638+/− mouse) (Lipkin, 1997), in inhibiting tumor formation in mouse models in which an allele of the homologous mouse Apc gene was inactivated by a mutation (ApcMin mice) (Beazer-Barclay et al., 1996) or inactivation of p21 or p27 introduced to Apc1638+/− mice (Apc/p21 and Apc/p27 compound mice), which is linked to induction of p21, leading to the decrease of cell proliferation and increase of cell differentiation and apoptosis in mucosa (Yang et al., 2005a; Yang et al., 2005b; Yang et al., 2001). In our present studies we have found that the induction of p21 by sulindac was linked to JNK1 in vitro and in vivo, and that both JNK1 and p21 were required for sulindac in inhibiting cell proliferation and in inducing apoptosis.
The c-Jun NH2-terminal kinases (JNKs) are identified as members of mitogen-activated protein kinase (MAPK) family and are known to phosphorylate and activate several transcriptional factors, including c-Jun, ATF-2, activation protein-1 (AP-1) and p53 (Chang and Karin, 2001; Davis, 2000; Derijard et al., 1994; Gupta et al., 1996; Hibi et al., 1993; Ip and Davis, 1998; Karin, 1995; Sanchez et al., 1994; Weston and Davis, 2002; Weston et al., 2002; Widmann et al., 1999). Growing evidences have shown that JNK signaling transduction pathway plays important roles in a variety of cellular processes, including cell proliferation, differentiation and apoptosis (Bode and Dong, 2005; Liu and Lin, 2005; Tournier et al., 2000). The JNKs are encoded by three genes, JNK1, JNK2 and JNK3. JNK1 and JNK2 are expressed ubiquitously, but JNK3 expression is limited in the brain and heart (Chang and Karin, 2001; Davis, 2000; Shaulian and Karin, 2002). JNK1 signal transduction pathway is particularly involved in regulating apoptosis (Dong et al., 1998; Kuan et al., 1999; Liu et al., 2004). Our recent studies have shown that JNK1 played a critical role in intestinal tumorigenesis which was associated with p21 expression in vitro and in vivo (Tong et al, unpublished data).
To further elucidate the role of JNK1 and p21 in inhibition of cell proliferation and in induction of cell apoptosis by sulindac, we employed human colon cancer cell lines that are p21- wild-type or null and mouse embryonic fibroblasts that lack the JNK1 gene, and treated with sulindac. Cell proliferation and apoptosis were analyzed, and the alteration of JNK1 and p21 expression was assayed. In addition, p21-related cell cycle regulation genes c-myc, cdk4 and cyclin D1 were also analyzed.
2. MATERIALS and METHODS
2.1. Cell lines and cell culture
Human colon cancer cell lines HCT116 that are p21+/+ or p21−/− were kindly provided by Dr. Burt Vogelstein (Johns Hopkins University); the JNK1+/+ and JNK1−/− mouse embryo fibroblast cell lines (MEF cells) were derived from JNK1+/+ and −/− mice (Dong et al., 1998), respectively. HCT116 cells were maintained in McCoy’s 5A medium, while MEF cells were maintained in DMEM. Both medium were supplemented with 10% fetal bovine serum (FBS), 1x antibiotic/antimycotic (100 units/ml streptomycin, 100 units/ml penicillin, and 0.25 μg/ml amphotericin B), 100 μM nonessential amino acids, and 10 mM HEPES buffer solution (all from Invitrogen Corp., Carlsbad, CA). All cells were cultured in 37 °C in 5%CO2. Sulindac (Sigma, St. Louis, MO) was dissolved in dimethyl sulfoxide (DMSO) and diluted in a serials concentration.
2.2. Cell proliferation assay
One hundred μl of cell suspension (1×104 ) cells in either McCoy’s 5A or DMEM medium supplemented with 10% FBS was seeded to each well of 96-well plate the day before the assay. After 18 to 20 h, the medium was removed. 100 μl of full assay medium was added to each well, and simultaneously, sulindac was added to the medium to reach the final concentration of sulindac from 0 to 3.2mM. 100 μl full medium with equal volume of DMSO were added to each well as control. All groups were quadruple. The plates were incubated at 37 °C for 24 h. Then the cell proliferation was determined by MTT [3-(4,5-dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide ] assay (CellTiter 96 Non-Radioactive Cell Proliferation Assay Kit, Promega Corporation, Madison, WI). Briefly, 15μl of dye solution was added to each well, and incubated the plate at 37 °C for 4 h in a humidified and 5% CO2 atmosphere. Added 100μl of stop solution to each well and incubated for 1 h. Finally, the absorbance was read at 570 nm by a MicroPlate Spectrophotometer (Bio-Rad, Hercules, CA). Reference wavelength of 600nm was used to reduce background.
2.3. Analysis of apoptosis
For analysis of apoptosis, cells were seeded in triplicate in 6-well plates. Seeding densities were 6 x 105 cells /well and were calculated such that control cell density approximated 50% confluence at the completion of the experimental period. After being cultured overnight, cells were treated with 1.6 mM of sulindac (Sigma, St. Louis, MO). Post-treatment of 0, 8, 24 and 48 h, the adherent and nonadherent cell populations were harvested, washed in cold PBS, then fixed with 80% ethanol for 8 h at 4 °C, stained with propidium iodide buffer (50 μg/ml propidium iodide, 0.1% sodium citrate, and 0.1% Triton X-100) overnight at 4°C, and 10,000 cells were analyzed for apoptosis using a Becton Dickinson FACScan (Becton Dickinson Immunocytometry Systems, San Jose, CA). The percentage of apoptotic cells was quantified using Cell Quest software (Becton Dickinson, San Jose, CA). Same experiments were triplicated separately.
2.4. Protein extraction and Western blot
Cells were washed twice with ice-cold PBS, then cell lysis buffer (Cell signaling Inc.) was added and the cells were incubated on ice for 15 min. Cells were scraped and transferred to 1.5 ml microcentrifuge tube. Sonicated briefly, total cell lysate was centrifuged at 8,160 g for 10 min at 4 °C. The supernatant was separated in 12% SDS-PAGE and transfer to nitrocellullose membrane. Anti-p21, anti-JNK1, Phosphorylated-JNK1, anti-c-myc, anti-CyclinD1 and anti- CDK4 (Santa Cruz Biotechnology, Santa Cruz, CA) antibodies were used for the probes. Signal was detected by enhanced chemoluminescence techniques (Amersham Life Science, Piscataway, NJ), as described previously (Yang et al., 2005a). The intensities of the signals were quantified by using software BandScan, the quantification (ratio) was normalized with the intensities of β-actin or vinculin.
2.5. Quantitative real-time PCR and western blot analysis for mouse intestine
Mouse tissues of small intestine were from our archived frozen tissue of our previous studies (Yang et al., 2001). Total RNA was isolated from the frozen mouse tissues using TRIzol reagent (Invitrogen Life Technology, Carlsbad, CA), as described (Yang et al., 2005a). The quantity of RNA was measured spectrophotometrically. As we described (Yang et al., 2005a), cDNA was synthesized from DNase-treated total RNA using TaqMan Multiscribe Reverse Transcriptase (Applied Biosystems, Inc., Foster City, CA). Quantitative PCR analysis was done using the ABI Prism 7900-HT Sequence Detection System (96-well). The primers for mouse JNK1 were: forward: aatggtttgcca-caaaatcc, reverse: gagtcagctgggaaaagcac. The size of the product is 201 bp. The primers for mouse p21 and β-actin, and amplification conditions for the quantitative real-time PCR and data analysis were as described previously (Yang et al., 2005a). Protein was extracted from the same mouse as RNA isolation. Western blot was performed and analyzed for the alterations of JNK1, phosphorylated JNK1 (p-JNK1) and p21, similar procedure as did in cell lines.
3. RESULTS
3.1. Sulindac inhibited cell proliferation and induced apoptosis in human colon cancer cells, but it was p21-dependent through induction of JNK1
Sulindac has been used for chemoprevention of cancer in vivo and in vitro. Our previous studies have shown that tumor inhibition by sulindac is linked to induction of p21, inhibiting cell proliferation and promoting cell differentiation and apoptosis in intestinal mucosa of mouse (Yang et al., 2005a; Yang et al., 2005b). To further investigate whether this is linked to JNK1, we used human colon cancer lines HCT116 with either p21- wild-type or p21-null, and treated these cells with sulindac.
First, cell proliferation analysis was assayed by MTT test, following manufacturer’s instruction. The same amount of HCT116 p21+/+ or −/− cells were seeded and treated with a series of concentrations of sulindac ranging from 0 to 3.2 mM. We found that sulindac inhibited proliferation of both p21+/+ and p21−/− cells. However, in p21+/+ cells, even in a low concentration of 0.8 mM of sulindac, cell proliferation could be inhibited (figure 1A). The higher the concentration of sulindac, the more inhibition was seen in p21+/+ cells. But, p21−/− cells were resistant to lower concentration of sulindac. Higher concentration of 1.6 mM of sulindac was needed to inhibit cell proliferation, and a narrow efficiency range was observed in these cells. As shown in figure 1A, p21−/− cells were resistant to 3.2 mM of sulindac. This data provides more evidence that sulindac-induced cell inhibition is p21 dependent.
Figure 1.
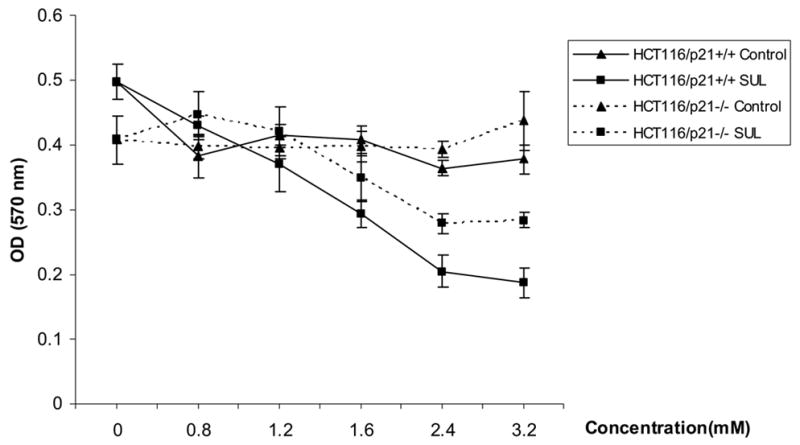

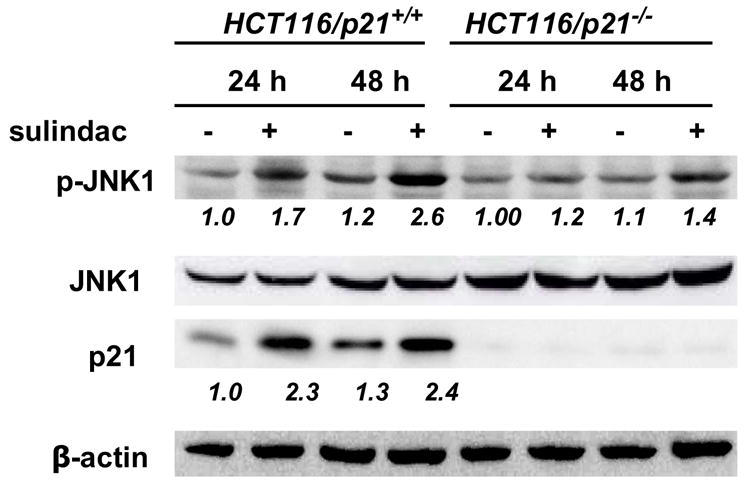
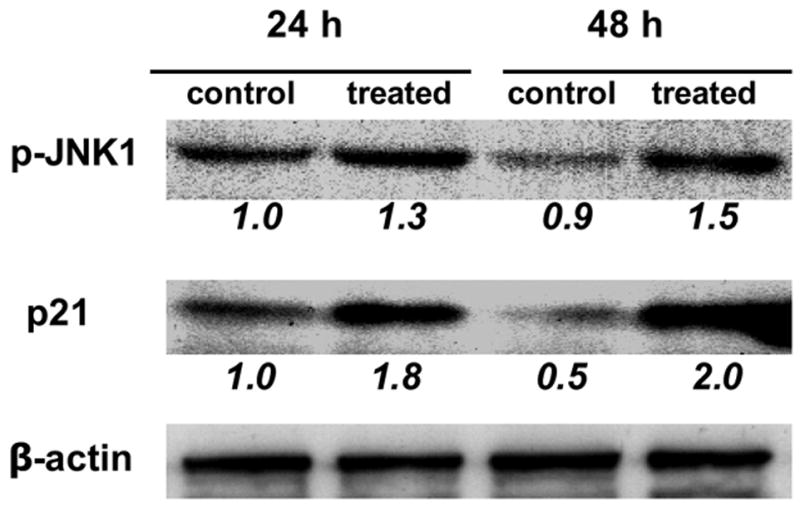
1A, Sulindac inhibited the colon cancer cells proliferation, and p21+/+ cells were more sensitive than p21−/− cells. (SUL: sulindac). 1B and 1C, Sulindac dramatically induced apoptosis in HCT116/p21+/+ cells, corresponding to the induction of JNK1 phosphorylation and p21WAF1 expression. However, the induction of apoptosis in HCT116/p21−/− cells was significantly less than HCT116/p21+/+ cells, correlated with less induction of JNK1-phosphorylation. 1D, Sulindac induced JNK1 phosphorylation and p21 expression in other colon cancer line SW620. The numbers underneath the bands indicate the quantification of signal normalized with the intensities of β-actin.
Since both p21+/+ and −/− cells were sensitive to sulindac at concentration of 1.6 mM, we picked this concentration for time course studies. Total cell population was collected post treatment of 0, 8, 24 and 48 h, for apoptosis analysis. As shown in figure 1B, sulindac induced apoptosis in both cell lines after 24 and 48 h. However, sulindac-induced apoptosis in p21+/+ cells was significantly more than that in p21−/− cells at the same time point.
To further elucidate whether the induction of apoptosis was associated with the alterations of JNK1 and p21, protein was extracted from the cells to determine p21 and JNK1 expression. As we documented previously (Yang et al., 2005a; Yang et al., 2001) and shown in figure 1C, p21 was significantly induced by sulindac in p21+/+ cells, no p21 was detectable, nor inducible in p21−/− cells. More interestingly, the phosphorylation of JNK1, an active isoform of JNK1, was significantly induced in p21+/+ cells, but was only slightly induced in p21−/− cells (figure 1C). Similar results were seen in another human colon cancer cell line SW620 (figure 1D), in which p21 was wild-type, where both p21 and JNK1 were induced by sulindac after the treatment of 24 or 48 h. Therefore, the absence of p21 caused less JNK1 phosphorylation, induced less apoptosis.
To investigate whether sulindac regulated cell cycle regulation genes, we detected the expression of c-myc and its target gene, cdk4, and cyclin D1 as well. We found that in contrast to the induction of p21 and JNK1, sulindac suppressed c-myc, cdk4 and cyclin D1 expression in both cell lines, and the longer treatment was given, the stronger suppression was seen after quantifying the signals (figure 2), partially contributing to the proliferation inhibition by sulindac in both cell lines.
Figure 2.
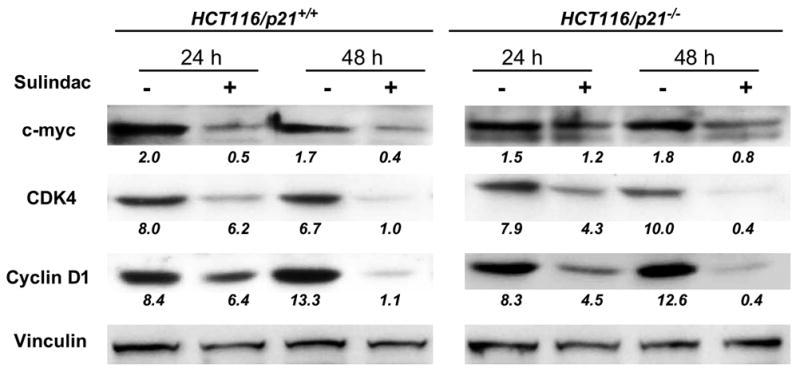
Sulindac suppressed c-myc, CDK4 and cyclin D1 expression in human colon cancer cell lines HCT116 which are p21+/+ or −/− after the treatment of 24 and 48 h. Vinculin was used as loading control. The numbers underneath the bands indicated the quantification of signal normalized with the intensities of vinculin.
3.2. Sulindac inhibited cell proliferation and induced apoptosis in mouse embryonic fibroblasts, which required JNK1
Since sulindac-induced p21 expression was linked to JNK1, we determined whether JNK1 is required for sulindac in inhibiting cell proliferation and promoting apoptosis. Therefore, JNK1-null mouse embryonic fibroblasts (MEF cells) were derived from JNK1 knockout mouse (Black 6 background). As a control, JNK1+/+ MEF cells were derived from normal C57Bl/6J mouse. Both cell lines were treated with a series concentration for proliferation assay, similar to experiments done in human colon cancer cell lines.
To our surprise, JNK1−/− MEF cells were resistant to sulindac at series concentration of 0.8, 1.2 mM, and even at a higher concentration of 1.6 mM that was the usually used concentration in vitro. JNK1−/− MEF cells responded to sulindac at 2.4 mM, and quickly became resistant at 3.2 mM (figure 3A). Thus, sulindac could not inhibit cell proliferation in the cells lacking JNK1 except at a higher concentration than usual. In contrast, JNK1+/+ MEF cells were sensitive to sulindac from low concentrations to high concentrations (figure 3A). Our results indicated that JNK1 was also required for sulindac in inhibiting cell proliferation.
Figure 3.

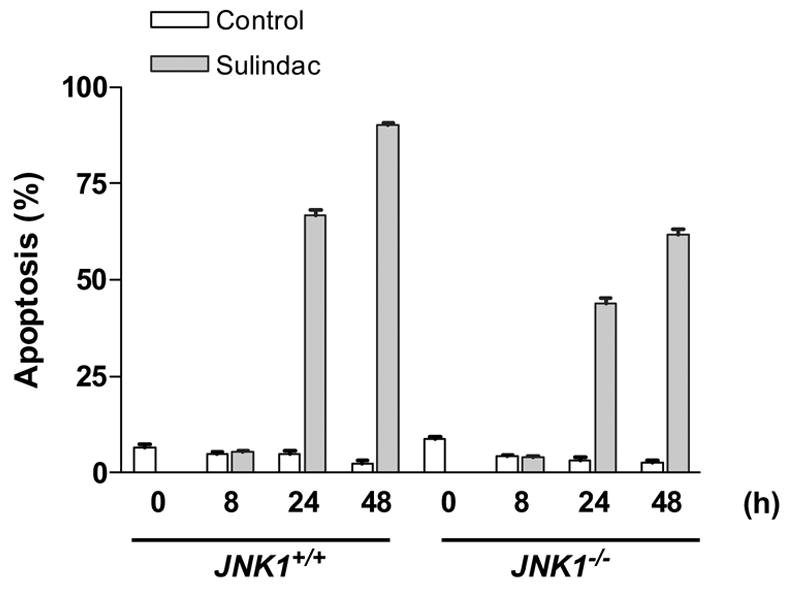

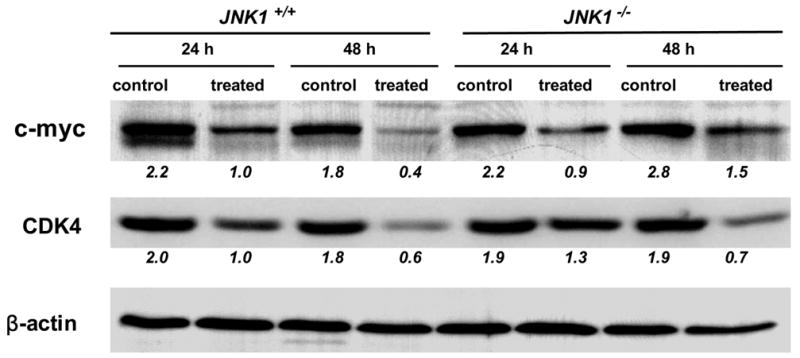
3A, Sulindac inhibited the mouse embryonic fibroblasts proliferation. JNK1+/+ MEF cells were sensitive to sulindac even at a low concentration, but the cells lacked JNK1 (JNK1−/− MEF cells) were resistant to sulindac; 3B and 3C, Sulindac induced apoptosis in JNK1+/+ and −/− mouse embryonic fibroblasts (MEF cells), which was associated with induction of JNK1 and p21. 3D, Sulindac dramatically suppressed c-myc and CDK4 expression in JNK1 MEF cells after treatment of 24 and 48 h. The numbers underneath the bands indicated the quantification of signal normalized with the intensities of β-actin.
To determine the role of JNK1 in sulindac-induced apoptosis, we treated both JNK1−/− and JNK1+/+ MEF cells with 1.6 mM of sulindac at different time course. We found that sulindac induced MEF cells apoptosis of MEF cells in both cell lines after 24 and 48 h treatment, but the induction of apoptosis in JNK1−/− MEF cells were much less than that in JNK1+/+ MEF cells (figure 3B). Even p21 protein was induced by sulindac in the both cell lines as documented previously and shown in figure 3C, JNK1 was also significantly induced by sulindac in JNK1+/+ MEF cells after treatment of 24 and 48 h (figure 3C). However, JNK1 was not detected and was not induced post treatment in JNK1−/− MEF cells (data not shown). Due to the short half-life of JNK1 protein, the more death of cells (in terms of more than 90% of apoptosis), the less detectable JNK1 protein in JNK1+/+ MEF cells after 48 h treatment with sulindac. In addition, the baseline of p21 was lower in JNK1−/− MEF cells and the induction of p21 was much less than that in JNK1+/+ MEF cells by sulindac. Thus, JNK1 might be synergistic to p21 in regulating apoptosis through the interaction of p21 and JNK1 signal transduction pathway.
In a complementary determination, we found that similar as in human colon cancer cells, c-myc and cdk4 were remarkably suppressed by sulindac in both JNK1+/+ and JNK1−/− MEF cells (figure 3D).
3.3. Sulindac induced phosphorylation of JNK1 and expression of p21 in intestine of mice
To determine the importance of JNK1 for sulindac in vivo, mRNA and protein were extract from mouse small intestine. As reported previously, Apc+/−, p21+/+ compound mice were fed sulindac (0.2%) supplemented diet for 9 months (Yang et al., 2001). Sulindac inhibited intestinal tumor formation in p21 wild-type mice, but abrogated inhibition in p21−/− mice, suggesting that p21 was essential for sulindac in vivo. As shown in figure 4A, JNK1 mRNA was significantly increased in the intestine of mice with sulindac supplemented diet, compared to the mice with AIN-76A diet, assayed by quantitative real-time PCR; corresponding to the increase of p21mRNA (figure 4B). Even though total JNK1 was not induced, phosphorylated JNK1 was significantly induced in the intestine of mice fed sulindac supplemented diet in comparison with the mice fed AIN-76A diet (4C). This was consistent with the findings seen in cultured human colon cancer cell lines HCT116 and SW620, suggesting, again, that both JNK1 and p21WAF1 were required for sulindac in tumor inhibition.
Figure 4.
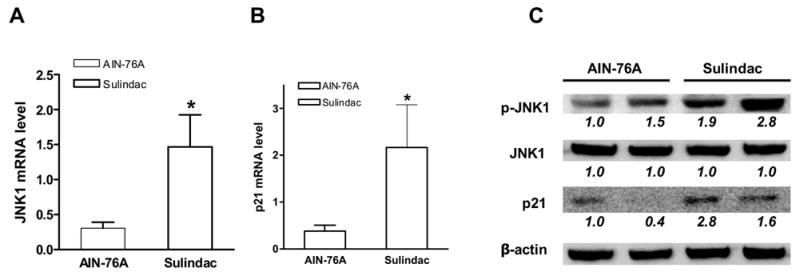
Sulindac induced expression of p21 and phosphorylation of JNK1 in normal mucosa of the small intestine of Apc/p21 compound mice. A, JNK1 mRNA level was induced by sulindac; β-actin was used as internal control. B, p21 mRNA level was increased by sulindac; (*, p< 0.05 compared to AIN-76A). C, phosphorylation of JNK1 and p21 protein was induced by sulindac; β-actin was used as loading control. The numbers underneath the bands indicate the quantification of signal normalized with the intensities of β-actin.
4. DISCUSSION
It has been well established that JNK1 plays important roles in triggering apoptosis in response to cellular stresses such as UV irradiation and cytokines. However, in these studies, we found that JNK1 is also important in the response to sulindac in chemoprevention through the pathways of inhibition of cell proliferation and induction of apoptosis in vitro and in vivo, in which p21 was synergistic to JNK1. Our data indicated that sulindac-mediated proliferation inhibition and apoptosis induction were not only p21-dependent, but also required JNK1. The induction of p21 by sulindac was linked to JNK1 expression and JNK1 phosphorylation. Loss of p21 caused less induction of apoptosis (figure 1B) and less induction of phosphorylation of JNK1 (figure 1C); loss of JNK1 resulted in cell resistance to sulindac and less induction of apoptosis and p21 (figure 3), by sulindac. Actually, our recent studies (Tong, et al, unpublished data) and previous report (Patel et al., 1998) have shown that JNK1 physically binds to p21WAF1, indicating that interaction of JNK1 and p21 plays a crucial role in inhibiting cell proliferation and inducing apoptosis by sulindac. The functional and physical interaction between JNK1 and p21 suggested at least some of their functions in the response to sulindac are through a complex that includes the both molecules. Therefore, the molecular mechanisms of the interaction of JNK1 signal transduction pathway and p21WAF/c-myc/cdk4 pathway in chemoprevention by sulindac need to be further investigated.
Taken together, our data demonstrated that both JNK1 and p21WAF1 were required for sulindac in the inhibition of cell proliferation and in the induction of apoptosis by sulindac, providing important clinical information and shedding light in developing strategies for chemoprevention and chemotherapy of cancers.
Acknowledgments
We would like to thank Dr. Bert Vogelstein (Johns Hopkins University, Baltimore, MD) for kindly providing colon cancer cell lines HCT116/p21+/+ and HCT116/p21−/−.
Footnotes
Grant support: This work was supported in part by National Cancer Institute grants R01 CA112081 (to W. Yang), R01 CA96605, U54 CA100926 (to LH. Augenlicht), and American Institute for Cancer Research grant 05A121 (to W. Yang).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Beazer-Barclay Y, Levy DB, Moser AR, Dove WF, Hamilton SR, Vogelstein B, Kinzler KW. Sulindac suppresses tumorigenesis in the Min mouse. Carcinogenesis. 1996;17:1757–1760. doi: 10.1093/carcin/17.8.1757. [DOI] [PubMed] [Google Scholar]
- Bode AM, Dong Z. Signal transduction pathways in cancer development and as targets for cancer prevention. Prog Nucleic Acid Res Mol Biol. 2005;79:237–297. doi: 10.1016/S0079-6603(04)79005-4. [DOI] [PubMed] [Google Scholar]
- Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- Clevers H. Colon cancer--understanding how NSAIDs work. N Engl J Med. 2006;354:761–763. doi: 10.1056/NEJMcibr055457. [DOI] [PubMed] [Google Scholar]
- Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- Derijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T, Karin M, Davis RJ. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- Dong C, Yang DD, Wysk M, Whitmarsh AJ, Davis RJ, Flavell RA. Defective T cell differentiation in the absence of Jnk1. Science. 1998;282:2092–2095. doi: 10.1126/science.282.5396.2092. [DOI] [PubMed] [Google Scholar]
- Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HK, Derijard B, Davis RJ. Selective interaction of JNK protein kinase isoforms with transcription factors. Embo J. 1996;15:2760–2770. [PMC free article] [PubMed] [Google Scholar]
- Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993;7:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- Ip YT, Davis RJ. Signal transduction by the c-Jun N-terminal kinase (JNK)--from inflammation to development. Curr Opin Cell Biol. 1998;10:205–219. doi: 10.1016/s0955-0674(98)80143-9. [DOI] [PubMed] [Google Scholar]
- Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- Kelloff GJ, Lippman SM, Dannenberg AJ, Sigman CC, Pearce HL, Reid BJ, Szabo E, Jordan VC, Spitz MR, Mills GB, Papadimitrakopoulou VA, Lotan R, Aggarwal BB, Bresalier RS, Kim J, Arun B, Lu KH, Thomas ME, Rhodes HE, Brewer MA, Follen M, Shin DM, Parnes HL, Siegfried JM, Evans AA, Blot WJ, Chow WH, Blount PL, Maley CC, Wang KK, Lam S, Lee JJ, Dubinett SM, Engstrom PF, Meyskens FL, Jr, O'Shaughnessy J, Hawk ET, Levin B, Nelson WG, Hong WK. Progress in chemoprevention drug development: the promise of molecular biomarkers for prevention of intraepithelial neoplasia and cancer--a plan to move forward. Clin Cancer Res. 2006;12:3661–3697. doi: 10.1158/1078-0432.CCR-06-1104. [DOI] [PubMed] [Google Scholar]
- Kuan CY, Yang DD, Samanta Roy DR, Davis RJ, Rakic P, Flavell RA. The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron. 1999;22:667–676. doi: 10.1016/s0896-6273(00)80727-8. [DOI] [PubMed] [Google Scholar]
- Lipkin M. New rodent models for studies of chemopreventive agents. J Cell Biochem Suppl. 1997:28–29. 144–147. [PubMed] [Google Scholar]
- Liu J, Lin A. Role of JNK activation in apoptosis: a double-edged sword. Cell Res. 2005;15:36–42. doi: 10.1038/sj.cr.7290262. [DOI] [PubMed] [Google Scholar]
- Liu J, Minemoto Y, Lin A. c-Jun N-terminal protein kinase 1 (JNK1), but not JNK2, is essential for tumor necrosis factor alpha-induced c-Jun kinase activation and apoptosis. Mol Cell Biol. 2004;24:10844–10856. doi: 10.1128/MCB.24.24.10844-10856.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel R, Bartosch B, Blank JL. p21WAF1 is dynamically associated with JNK in human T-lymphocytes during cell cycle progression. J Cell Sci. 1998;111 ( Pt 15):2247–2255. doi: 10.1242/jcs.111.15.2247. [DOI] [PubMed] [Google Scholar]
- Sanchez I, Hughes RT, Mayer BJ, Yee K, Woodgett JR, Avruch J, Kyriakis JM, Zon LI. Role of SAPK/ERK kinase-1 in the stress-activated pathway regulating transcription factor c-Jun. Nature. 1994;372:794–798. doi: 10.1038/372794a0. [DOI] [PubMed] [Google Scholar]
- Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol. 2002;4:E131–136. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- Szabo E. Selecting targets for cancer prevention: where do we go from here? Nat Rev Cancer. 2006;6:867–874. doi: 10.1038/nrc2008. [DOI] [PubMed] [Google Scholar]
- Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA, Davis RJ. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science. 2000;288:870–874. doi: 10.1126/science.288.5467.870. [DOI] [PubMed] [Google Scholar]
- Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Genet Dev. 2002;12:14–21. doi: 10.1016/s0959-437x(01)00258-1. [DOI] [PubMed] [Google Scholar]
- Weston CR, Lambright DG, Davis RJ. Signal transduction. MAP kinase signaling specificity. Science. 2002;296:2345–2347. doi: 10.1126/science.1073344. [DOI] [PubMed] [Google Scholar]
- Widmann C, Gibson S, Jarpe MB, Johnson GL. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol Rev. 1999;79:143–180. doi: 10.1152/physrev.1999.79.1.143. [DOI] [PubMed] [Google Scholar]
- Yang W, Bancroft L, Augenlicht LH. Methylation in the p21(WAF1/cip1) promoter of Apc(+/−), p21(+/−) mice and lack of response to sulindac. Oncogene. 2005a;24:2104–2109. doi: 10.1038/sj.onc.1208444. [DOI] [PubMed] [Google Scholar]
- Yang W, Bancroft L, Liang J, Zhuang M, Augenlicht LH. p27kip1 in intestinal tumorigenesis and chemoprevention in the mouse. Cancer Res. 2005b;65:9363–9368. doi: 10.1158/0008-5472.CAN-05-2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Velcich A, Mariadason J, Nicholas C, Corner G, Houston M, Edelmann W, Kucherlapati R, Holt PR, Augenlicht LH. p21(WAF1/cip1) is an important determinant of intestinal cell response to sulindac in vitro and in vivo. Cancer Res. 2001;61:6297–6302. [PubMed] [Google Scholar]