

Abstract
Purpose
To relate clinical issues to the clinical manifestations of prostate cancers across disease states using the eligibility and outcome criteria defined by Response Evaluation Criteria in Solid Tumors (RECIST).
Experimental Design
The manifestations of prostate cancer that characterize localized, recurrent, and metastatic disease were considered using the eligibility criteria for trials defined by RECIST. To do so, we analyzed the sites, size, and distribution of lesions in patients enrolled on contemporary Institutional Review Board ^ approved trials for progressive castrate and noncastrate metastatic disease. Prostate-specific antigen (PSA) levels were also assessed. RECIST-defined outcome measures for tumor regression were then applied to the metastatic patient cohorts, and separately to the states of a rising PSA (noncastrate and castrate) and localized disease.
Results
Only 43.5% of men with castrate metastatic and16% of noncastrate metastatic disease had measurable target lesions >2 cmin size. Overall, 84.4% of the target lesions were lymphnodes, of which 67.7% were ≥2 cm in the long axis. There are no target lesions in patients in the states of a rising PSA and localized disease, making them ineligible for trials under these criteria. PSA-based eligibility and outcomes under RECIST conflict with established reporting standards for the states of a rising PSA and castrate metastatic disease. The clinical manifestations of prostate cancer across multiple disease states are not addressed adequately using the eligibility criteria and outcomes measures defined by RECIST. Important treatment effects are not described.
Conclusions
Trial eligibility and end points based solely on tumor regression are not applicable to the majority of the clinical manifestations of prostate cancers representing all clinical states. Treatment effects can be described more precisely if eligibility criteria are adapted to the clinical question being addressed and clinical state under study, focusing on the duration of benefit defined biochemically, radiographically, and/or clinically.
An objective of a phase II trial is to assess the effect of a treatment on a given manifestation of a disease or a clinical scenario. Another objective is to determine how long the effect lasts. Whether the effect should be based on measures of tumor shrinkage or other parameters is controversial, although it is unusual for compounds to be approved for use on the basis of tumor regression alone (1). For prostate cancer clinical trials, it has long been recognized that reliable phase II end points are lacking (2). This is because measurable tumor masses that can be assessed objectively for changes in size following an intervention occur infrequently. It is difficult to determine a favorable outcome in bone; the most common site of prostate cancer spread; and the correlation between a given post therapy change in prostate-specific antigen (PSA) and true clinical benefit has not been fully defined (3).
The explosion of knowledge of the targets and pathways associated with prostate cancer progression has led to the evaluation of a range of therapies beyond the traditional cytotoxic agents. Drugs that inhibit cell signaling, proapoptotic therapies, inhibitors of specific components of the metastatic process, and a range of biological therapies are now in our armamentarium. Many are designed to slow tumor growth without necessarily killing cells and have the potential to reduce prostate cancer–specific morbidity and mortality in the absence of objective tumor shrinkage. Evaluating these types of drugs using clinical trial end points solely based on tumor regression may lead to discarding potentially useful therapies (4). These considerations raise the question of whether the primary end point of phase II investigations in prostate cancer should be tumor regression, or whether other outcomes might be more informative (1).
Historically, between 5% and 20% of prostate cancer patients enrolled on trials for castration-resistant disease had measurable tumors. However, even when such lesions are present, they are few in number, small in size, and may have a distinct biological makeup relative to osseous disease in the same patient (5–8). In this report, we examine a contemporary data set of patients representing various disease states enrolled on Institutional Review Board–approved protocols at Memorial Sloan-Kettering Cancer Center (MSKCC) to determine whether present-day clinical trial end points are more applicable than those used (and ultimately discarded) in the past.
To do so, we related the specific manifestations of prostate cancer by site of disease and by clinical state (9) to clinical trial end points based on tumor regression as defined by Response Evaluation Criteria in Solid Tumors (RECIST; ref. 10). The clinical states represent common scenarios encountered in routine clinical practice, where an intervention might be offered to control or to eliminate a disease manifestation that is present at a particular time or to prevent a clinical event from occurring in the future (9). The states include newly diagnosed patients with localized disease for whom the treatment objective is to eliminate the tumor surgically or with radiation therapy; patients with an increasing PSA (before or after androgen ablation) after local therapy, where the objective is to control the disease and to prevent metastases; and patients with metastatic disease (before or after androgen ablation), where the objective is to palliate and prevent symptoms and to prolong life (Fig. 1). Starting with an evaluation of the distribution and size of lesions in patients enrolled on trials for castrate and noncastrate metastatic disease, we show that significant treatment effects are not described adequately. We also show how the therapeutic effects of any agent, regardless of mechanism, can be assessed more appropriately using short-term measures of success or failure when specific disease manifestations are present, and by using definitions of progression that determine the duration of treatment effects. Acceptance of the treatment will then depend on the adverse effects encountered relative to the degree and duration of benefit observed. Most important is that outcomes are tailored to the specific indication for the treatment and the point in the illness at which it is being utilized.
Fig. 1.
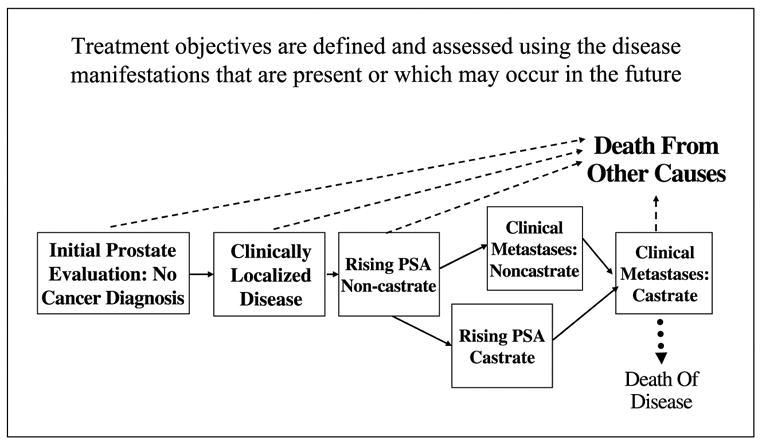
Prostate cancer clinical states: Short-term objectives are to eliminate, relieve, or control manifestations that are present. Longer-term objectives are to prevent or delay them from occurring or reoccurring in the future. Modified from ref. (9) and reprinted with permission from Elsevier.
Materials and Methods
To examine the manifestations of progressing metastatic prostate cancers that are detectable on an imaging study, the sites and size of lesions from patients enrolled and treated at MSKCC on sequential Institutional Review Board–approved trials for patients in the states of clinical metastases noncastrate and clinical metastases castrate disease were analyzed for eligibility under RECIST (10). Enrollment on these trials required progression of disease defined as any one of the following: a minimum of three increasing PSA levels obtained ≥2 weeks apart where the range of values is at least 25%, new or progressive (25% bidimensional increase) soft tissue masses on computerized tomography or magnetic resonance imaging, or new lesions on radionuclide bone scan. All imaging studies were reviewed in a blinded fashion by a single radiologist (L.H. Schwartz). The metastatic sites for each patient on the protocol were categorized as a ‘‘target’’ or ‘‘nontarget’’ lesion using the RECIST criteria. Sites considered were recurrent/persistent disease in the prostate or prostate bed, lymph nodes, viscera (e.g., liver and lung), and bone.
All of the manifestations were then related to the end points developed to assess objective tumor regression as defined by RECIST. Separately, the definitions for complete response, partial response, and stable and progressive disease were applied to the specific sites of prostate cancer spread. Overall response assessments defined by RECIST were also analyzed and related to the therapies currently approved for the systemic treatment of prostate cancer. A similar analysis was conducted for the manifestations of 31 patients with noncastrate metastatic disease treated on a study of rapid hormonal cycling and chemotherapy (11), and separately for patients in the states of localized disease and a rising PSA (noncastrate and castrate). An alternative set of phase II end points are proposed that are applicable to patients who occupy a given clinical state.
Results
Eligibility criteria
Target lesions
RECIST categorizes sites of tumor spread as either measurable (target lesions) or nonmeasurable (nontarget lesions), with the recommendation that up to 10 target lesions per patient be recorded and monitored. The RECIST criteria further recommend that only patients with target lesions be registered to clinical trials. We examined the distribution of disease in contemporary group of patients representing the different prostate cancer clinical states to determine what proportion had target lesions that would satisfy the eligibility criteria for enrollment on clinical trials under RECIST. Per RECIST, metastases to lymph nodes, liver, or lung may qualify as target lesions if they have a minimum size that is more than twice the slice thickness of the scanning instrument used (10).
Table 1 shows the proportion of patients with castrate metastatic disease with measurable visceral and/or nodal spread (target lesions), and osseous disease (nontarget lesions) from 124 men enrolled on three sequential Institutional Review Board–approved protocols (12–14). As noted, 54 (44%) had disease limited to bone, 12 (10%) disease limited to soft tissue, whereas 58 (47%) had disease in bone and soft tissue. Overall, 63 patients (51%) had lesions that could be measured in soft tissue and 7 (6%) had lesions that were considered too small to measure. In the 63 patients with measurable tumor, 54 patients (44%) had a total of 104 lesions that were ≥2 cm in size. The median number of lesions ≥2 cm per patient was 2 (range, 1–6). The sites of spread were lymph nodes in 76% (48% retroperitoneal, 36% pelvic, 7% mediastinal, 5% abdominal, 2% axillary, and 2% unspecified), liver in 13%, lung in 3%, recurrent in the prostate bed in 2%, and adrenal spread in 1% of cases. Nine (7%) additional patients had lesions in the range of 1.0 to 1.9 cm, which would qualify for enrollment using spiral computed tomography. The median size of all lesions was 2.5 cm (range, 1–8 cm); the median size is 3 cm (range, 2–8 cm) if only lesions ≥2 cm in size were considered. The distribution of lesions seen in the MSKCC series compared with those reported in recently completed multicenter trials is shown in Table 2 (15–17).
Table 1.
Distribution of disease in patients with progressive castrate metastatic disease enrolled on three sequential clinical trials at MSKCC
| Category | Overall |
|---|---|
| Total no. patients reviewed | 124 |
| TEC (1997–1999) | 41 |
| Hi-TEC (1999–2000) | 28 |
| Epothilone (2001–2003) | 55 |
| Median PSA at baseline (all studies) | 91.4 (range, 0.52–2282) |
| Patients | |
| No. patients with | |
| Bone only | 54 (44%) |
| Bone and soft tissue | 58 (47%) |
| Soft tissue only | 12 (10%) |
| No. patients with measurable soft tissue disease (≥1.0 cm) | 63 (51%) |
| No. patients with soft tissue disease too small to measure | 7 (6%) |
| No. patients with any measurable nodal disease | 48 |
| No. patients with any measurable visceral disease | 15 |
| No. patients with any measurable soft tissue lesions ≥2 cm | 54 |
| No. patients with measurable soft tissue lesions <2 cm | 9 |
| Lesions | |
| No. measurable lesions | 147 |
| No. lesions ≥2 cm | 104 (70.7%) |
| Lymph nodes | 84 (80.8%) |
| Mediastinum | 6 |
| Pelvic | 30 |
| Axilla | 2 |
| Retroperitoneal | 40 |
| Abdominal | 4 |
| Lymph node (unspecified) | 2 |
| Soft tissue | 20 (19.2%) |
| Liver/visceral (3 lung, 1adrenal) | 18 |
| Prostate/prostate bed | 2 |
| No. lesions <2 cm | 43 (29.2%) |
| Lymph nodes | 40 (93.0%) |
| Mediastinum | 1 |
| Pelvic | 16 |
| Retroperitoneal | 23 |
| Soft tissue | 3 (7.0%) |
| Liver/visceral (2 lung) | 3 |
| Median size of lesions ≥2 cm | 3 cm (range, 2–8) |
| Median no. lesions ≥2 cm per patient | 2 (range,1–6) |
Table 2.
Comparison of MSKCC distribution of disease with contemporary phase III studies (ranges are given to express the range of percentages across multiple arms of the studies cited)
| No. patients | No. patients with bone metastases | No. patients with nodal disease | No. patients with liver or visceral disease | |
|---|---|---|---|---|
| MSKCC data set | 124 | 112 (90%) | 48 (39%) | 15 (12%) |
| SWOG 99-16 (58) | 770 | 84–88% | 24–26% | 18–19% |
| TAX 327 trial (15) | 464 | 90–92% | Not stated* | 22–24% |
| Mitoxantrone/prednisone (17) | 120 | 79–86% | 18% | 6% |
Abbreviation: SWOG, Southwest Oncology Group.
Thirty-nine to forty percent had measurable disease (including visceral disease).
Table 3 shows a comparable analysis of 31 patients with clinical metastasis noncastrate disease. As shown, 13% had disease limited to bone, 16% to soft tissue, whereas 71% had both osseous and soft tissue disease. In this cohort, 12 (39%) patients had 15 target lesions of which 7 (47%) were ≥2 cm in size. Lymph node disease was again predominant. Five had soft tumor disease and no osseous spread.
Table 3.
Distribution of disease in patients with non-castrate metastatic prostate cancer
| Category | Overall |
|---|---|
| Patients reviewed | 31 |
| Median age (range) | 61 (47–79) |
| Median pretreatment PSA (range) | 19.85 (0.63–74.87) |
| Number of patients with: | |
| Bone only | 4 (13%) |
| Bone and soft tissue | 22 (71%) |
| Soft tissue only | 5 (16%) |
| Patients | |
| No. patients with measurable lesions | 12 (39%) |
| Nodes | 10 |
| Visceral | 2 |
| No. patients with lesions ≥2 cm | 5 (16%) |
| No. patients with lesions <2 cm | 7 (23%) |
| Lesions | |
| No. measurable lesions | 15 |
| No. lesions ≥2 cm | 7 (47%) |
| Lymph nodes | 5 (71%) |
| Pelvic | 4 |
| Lymph node (unspecified) | 1 |
| Soft tissue | 2 (29%) |
| Liver/other viscera | 1 |
| Prostate/prostate bed | 1 |
| No. lesions <2 cm | 8 (53%) |
| Lymph nodes | 7 (88%) |
| Pelvic | 5 |
| Retroperitoneal | 2 |
| Soft tissue | 1 (13%) |
| Prostate/bed | 1 |
| Median size of lesions ≥2 cm | 2.4 cm (range, 2–3.3) |
| Median size of lesions <2 cm | 1.6 cm (range, 0.9–1.8) |
Bone
Bone lesions are ‘‘truly nonmeasurable’’ and are recorded as nontarget lesions. Tumors that have eroded through bone into the epidural space or base of skull may in some cases be measurable. However, for most phase II trials, neurologic compromise is an automatic exclusion because these patients require immediate relief of symptoms using radiation therapy or surgery. As shown in Table 1, 54 (44%) of patients in the castrate data set and four patients (13%) in the noncastrate data set had disease limited to bone. These patients, by RECIST criteria, would be ineligible for trial entry.
Localized or locally recurrent disease
Disease in this location occurs in the newly diagnosed patient with clinically localized or metastatic disease, and those who have recurred after surgery or radiation therapy with or without metastases. In the MSKCC data set of patients with castrate metastatic disease, only one patient had a target lesion recorded in the prostate bed.
Tumor markers
The median PSA values for patients with castrate metastatic disease was 91 ng/mL (range, 0.5–2,282), for noncastrate metastatic disease was 19.8 ng/mL (range, 0.6–75), and in a series of patients with an increasing PSA was 3.2 ng/mL (range, 0.08–275; ref. 11). By RECIST criteria, marker values are recorded as ‘‘normal’’ or ‘‘abnormal’’ and increasing values are not required for entry (10). However, what constitutes an abnormal value varies by state, as reflected in the data above. For example, in a patient with localized disease who has been treated with external beam radiation therapy, a PSA of 1.5 ng/mL may signify the nonmalignant prostate tissue that also produces PSA after the malignant component has been eradicated, and not persistent or recurrent tumor. In contrast, any PSA above 0.2 or 0.4 ng/mL in a patient who has undergone radical prostatectomy indicates recurrent disease.
Most advanced-disease therapeutic trials require increasing values on entry (18, 19). Where trials may differ is in the absolute PSA value and/or the defined change over time (e.g., doubling time) required for entry. These aspects of PSA are not specified in RECIST, and increasing values are not required for trial entry.
Symptoms
The presence or absence of symptoms is not considered an eligibility criterion under RECIST criteria.
Eligibility criteria by Response Evaluation Criteria in Solid Tumors—a state-by-state view
Table 4 summarizes the disease manifestations across the disease states in relation with eligibility under RECIST. As shown, all patients with localized disease and an increasing PSA are excluded because there are no target lesions in these states. Even in patients with detectable metastatic lesions on imaging, whether castrate or noncastrate, the majority of patients do not have target lesions that would qualify for enrollment under RECIST.
Table 4.
Manifestations of prostate cancer by site and clinical state in relation to measurable disease (defined by RECIST)
| Clinical state
|
||||
|---|---|---|---|---|
| Disease manifestation | Localized disease | Increasing PSA | Clinical metastases, noncastrate | Clinical metastases, castrate |
| Primary tumor or local recurrence following radiation or primary hormones | Yes but nontarget | Possible, but nontarget | Nontarget, if present (0% at MSKCC) | Nontarget, if present (0.8% at MSKCC) |
| Prostate bed, following prostatectomy | No (by definition) | Possible, but nontarget | Target, but infrequent (0% at MSKCC) | Target, but infrequent (0.8% at MSKCC) |
| Abnormal PSA | Yes | Yes, depending on definition of normal (MSKCC median 3.15; range, 0.08–274.54) | Yes (19.9; range, 0.6–75) | Yes, depending on definition of normal (91.4 MSKCC median; range, 0.53–2282) |
| Lymph nodes | No (by definition) | No (by definition) | Target (32% at MSKCC) | Target (39% at MSKCC) |
| Viscera | No (by definition) | No (by definition) | Target, but infrequent (3% at MSKCC) | Target, but infrequent (12% at MSKCC) |
| Bone | No (by definition) | No (by definition) | Yes, but nontarget (84% at MSKCC) | Yes, but nontarget |
| Symptoms | Yes (not considered by RECIST) | No (by definition) | Yes (not considered by RECIST) | Yes (not considered by RECIST) |
| Eligible for trial under RECIST? | No (no target lesions) | No (no target lesions) | Yes,12 (39%) | Yes, 63 (51%) |
Outcome measures (response)
Table 5 lists sites of disease and clinical state and relates them to the definitions of complete response, partial response, and stable and progressive disease. These are considered below.
Table 5.
RECIST response categories using tumor regression applied to the individual manifestations of prostate cancer
| Complete response | Partial response | Stable disease | Progressive disease | |
|---|---|---|---|---|
| Primary tumor | No disease on pathology | No definition | No definition | No definition |
| Local recurrence | N/A | No definition | No definition | No definition |
| Measurable tumor | ||||
| Nodes, lungs, viscera | Disappearance of target lesions | 30% decrease in change of sum of longest diameters of target lesions | Neither progression nor partial response criteria met | 20% increase in change sum of longest diameters of target lesions |
| Abnormal PSA | Normalization | Abnormal levels | Abnormal levels | Not specified |
| Bone | Resolution | Persistence | Persistence | New lesions |
| Symptoms | Not considered | Not considered | Not considered | Not considered |
Abbreviation: N/A, not applicable.
Target lesions
The majority of soft tissue target lesions in patients with clinical metastases enrolled on trials are lymph nodes. A partial response classification by RECIST requires a ≥30% decrease, and progression of disease a ≥20% increase in the sum of the largest diameters of the individual target lesions. Using the target lesions shown in Table 1, this change represents a decrease from 3 to 2.1 cm, or increase from 3 to 3.6 cm in the greatest diameter. Lesions of this size are subject to greater measurement error and subsequent misclassification than larger tumors even when they are read by a single radiologist (20). In addition, lymph nodes do not disappear even if the tumor is eradicated and a response may represent a shrinkage of minimally enlarged node to normal size, e.g., 2.2 to 1.8 cm (21).
A separate consideration is whether changes in the size of a lymph node actually parallel changes in the tumor in bone. Given the insensitivity of standard radiographic techniques to assess disease in bone, radiographic changes in lymph nodes may not be paralleled by bone scintigraphy (22). Similarly, whether distinct trials should be designed for the 15% of patients with disease limited to soft tissue or disease limited to osseous sites is unknown.
Nontarget lesions
The defined response categories are not applicable to patients with disease limited to bone, which are nontarget lesions. Further, there are no accepted standard criteria to describe a favorable outcome for treated osseous metastases by bone scan.
Similarly, the definitions of progression are also not standardized, although most investigators accept the appearance of one or more new lesions and/or unequivocal progression of existing nontarget lesions. Consequently, changes in bone scan are best characterized as ‘‘improved,’’ ‘‘stable,’’ or ‘‘progressing.’’ The issue of ‘‘pseudoprogression,’’ the appearance of new lesions or an increase in intensity of existing lesions with successful therapy, is not addressed. In these cases, a patient who is actually benefiting from therapy may be recorded as having progression of disease (23).
Prostate or prostate bed (newly diagnosed or recurrent)
Although the size of the prostate can be estimated using ultrasound or magnetic resonance imaging, in most cases only a proportion of the gland is involved by tumor and no dominant lesion is visible. By ultrasound, there are no reproducible findings that can distinguish tumor- from non-tumor-bearing regions reliably, although discrete lesions can be seen in some instances. Magnetic resonance imaging with spectroscopy has the potential to improve detection and monitoring (24), but is still considered investigational at this time. Recurrent disease in the prostatic bed is rarely palpable on physical examination or visible on an imaging study. Whereas magnetic resonance imaging may show abnormalities that are not detected using other modalities, it is not used routinely and is rarely ‘‘measurable.’’ (25) ProstaScint scanning is a Food and Drug Administration – approved imaging modality that uses a radiolabeled antibody to an internal epitope of prostate-specific membrane antigen to detect sites of disease (26, 27). However, the images obtained do not qualify as target lesions under RECIST and cannot be used on a serial basis to assess changes in tumor growth. In clinical practice, the presence or absence of disease in the prostatic bed following radical surgery is ‘‘predicted’’ on the basis of factors such as the pathologic stage, margin status, the time from primary treatment to the documentation of recurrence, PSA doubling time, and current PSA level (28–31). Even the performance of a biopsy to confirm or exclude disease in the prostate bed is not reliable enough to be recommended on a routine basis, and even if it were positive, would not qualify as a target lesion under RECIST. Consequently, the prostate is a ‘‘nonmeasurable,’’ nontarget site whether the patient is newly diagnosed or has persistent or recurrent disease in the gland.
As the tumor within the prostate itself cannot be accurately measured, the terms complete response, partial response, progressive disease, or stable disease are not applicable to patients with localized disease in which the sole site of disease is the newly diagnosed primary tumor itself, or a tumor that has persisted in the prostate after radiation therapy or prostate bed after radical prostatectomy. The exception is the rare case (3% in the current series) in which it has met the criteria for a measurable target lesion at entry.
Tumor markers
Tumor markers alone are not used to assess outcome by RECIST, and patients with an increasing PSA as the sole manifestation of disease are not eligible for trials. Thus, for patients enrolled on trials on the basis of progressing target lesions and an increasing PSA to be classified in a complete response category requires a disappearance of the target lesion and normalization of the PSA. However, what is considered normal will vary depending on whether the patient has received treatment to the prostate, and if so, of what type. Take, for example, three patients with no osseous disease who have with multiple target lesions that have regressed completely. The first had progressed after radical prostatectomy, the second after radiation therapy, and the third never received treatment to the primary tumor. For the first patient, a normal PSA is one that is undetectable; for the second, <0.5 or 1.0 ng/mL; for the third, 2.5 or perhaps as high as 4.0 ng/mL.
Symptoms
Symptoms that are present are recorded and monitored, but are not used to assess response. Worsening of preexisting symptoms or the development of new ones can, at the investigator’s discretion, be used to show disease progression once a patient is on a study, although it is often difficult to distinguish progressive disease from drug-related adverse events in this patient group.
Response criteria by Response Evaluation Criteria in Solid Tumors—a state-by-state view of best overall response
RECIST require the assignment of a best overall response of either complete response, partial response, or stable or progressive disease on the basis of defined changes in target lesions and some nontarget lesions. As shown in Table 5, the difficulties of categorizing changes in the individual sites of disease into a complete response, partial response, or stable disease category renders the best overall response classifications of limited value. Bone disease is particularly problematic because most techniques to assess changes in bone metastases are too insensitive to detect change, be it favorable or unfavorable, on a serial basis. In cases where plain radiographs are abnormal, resolution of the osteoblastic change rarely occurs and improvements in a radionuclide bone scan may not occur for many months even if all disease in bone were eliminated by the treatment. The criteria do require confirming a ‘‘favorable’’ change in an imaging study within 4 weeks of its original documentation, but this interval is too short to show a meaningful change on a bone scan.
A second difficulty is the failure to incorporate contemporary uses of serial changes in the PSA to assess treatment effects. Associations with posttreatment PSA changes and survival have been reported for patients with an increasing PSA after local therapy (32, 33), as well as for those castrate metastatic disease in retrospective analyses (34–38). Equally important, RECIST does not address the significance of increasing values as an indication of disease progression. As a result, a patient with an increasing PSA could be considered as responding favorably if his scans were without evidence of progressive disease.
Discussion
The difficulties associated with drug development in prostate cancer have long been recognized. To address this, consensus groups of experts have established guidelines to conduct clinical trials addressing questions for patients in the states of clinical metastases castrate disease (18), and more recently for the state of an increasing PSA (19). As illustrated by these efforts, and a contemporary review of trials for patients with detectable metastases on imaging, fewer than half had disease manifestations that would be recognized as target lesions under RECIST. Even when documented, lesions were generally few in number and small in size, and >80% of the sites were in lymph nodes. The most common manifestations of metastatic prostate cancer, osseous metastases with an increasing PSA, are not adequately addressed. For these and for other prostate cancer clinical states, eligibility and outcome measures designed to assess tumor regression simply do not apply.
Prolongation of life is ultimately the primary objective of therapy directed toward prostate cancers representing any clinical state. It can be achieved by eliminating the disease surgically, with radiation or by medical means, or, in theory by slowing the disease to the point where the patient dies of other, nonprostate cancer–related causes. There are, however, other treatment objectives of importance to patients in addition to prolongation of life, illustrated by the therapies approved for use for patients with castration resistant disease. Among them are mitoxantrone and prednisone, strontium-89 (Metastron), and samarium-153 (Quadramet), all of which were approved to palliate pain (39–43). Zometa was approved on the basis of reducing the risk of skeletal complications (40), whereas mitoxantrone and prednisone were shown to reduce the constitutional symptoms of an increasing disease burden (39). Noteworthy is that none of these therapies were approved on the basis of trials using an end point of tumor regression, and none were approved on the basis of an effect on PSA. In fact, most had no effect on PSA, and the clinical benefits were shown without prolongation of life. Only docetaxel, recently approved by the Food and Drug Administration for the treatment of patients with castration-resistant disease, was shown to prolong life in a randomized prospective clinical trial (15).
These examples show the importance of tailoring drug development to what is routinely done in clinical practice: focusing the eligibility and outcomes on the clinical reason that a therapy is being offered and what it is expected to achieve. The outcome measures must account for the putative action of the drug, whether it is cytotoxic or cytostatic, potentially curative or palliative, and whether it affects the cancer cell directly or the interaction between the tumor and host. The short-term measures may be effects on PSA, on symptoms, or, depending on the context, more traditional measures of disease regression. Assuming that these milestones are achieved, the more critical issue becomes the durability of these effects with a focus on clinically meaningful measures of disease progression.
By separating the short-term indices of a favorable outcome that justify continuing treatment and the longer-term measures of the duration of benefit to show when a drug is no longer working, trials are not limited to studying drugs solely on the basis of their effects on PSA, tumor regression, or other parameters of uncertain clinical significance. Disease progression is indicated by any clinically significant parameter that reflects a disease manifestation that is worsening, including an objective in the size of a target lesion, the failure to palliate or to control of symptoms, the development of new symptoms for patients at high risk, the increase in the size of or development of new lesions on a bone scan, or an unfavorable change in PSA. The crucial issue is that the definition of disease progression be tailored specifically to the reason that an intervention is being offered and the clinical state or clinical scenario of the patient group in which it is being evaluated, as shown in Table 6.
Table 6.
State-specific eligibility and outcome measures
| Disease manifestation | Eligibility/outcomes | Intermediate endpoint | Progression |
|---|---|---|---|
| Localized disease. Therapeutic aim: cure, no biochemical evidence of relapse | |||
| Demographics | Prior history (BPH, use of finasteride) | ||
| PSA | Pretreatment risk assessment | NMA after prostatectomy <0.5–1.0 after radiation | Detectable PSA after prostatectomy |
| Pathology | Gleason score, number of cores, percentage of cores, Tstage | Radical prostatectomy: all cancer removed with negative surgical margins
Radiation therapy: negative biopsy ≥2 y after treatment |
Persistent or recurrent cancer |
| Measurable | N/A | Not applicable | Detectable disease |
| Bone | N/A | Not applicable | Detectable disease |
| Quality of life | Pain, obstructive symptoms, potency | Improved on validated scale | Worse on validated scale |
| Increasing PSA (systemic). Therapeutic aim: prevent metastatic disease | |||
| Demographics | Prior treatment, probability of relapse on pretreatment risk assessment | ||
| PSA | Rate of increase and other kinetic properties | Predefined alteration of PSA kinetics (dependent on mechanism of intervention) | No change in PSA kinetics |
| Pathology | Initial tumor characteristics | Negative biopsy | Biopsy proven metastatic disease |
| Measurable | N/A | Not applicable | Detectable |
| Bone | N/A | Not applicable | Detectable |
| Quality of life | Baseline assessment of urinary function, potency, bowel function | No change from baseline on validated scale | Worse than baseline on validated scale secondary to disease. |
| Clinical metastases: noncastrate. Therapeutic aim: increase and prolong response to androgen deprivation, reduce and prevent symptoms, prolong life | |||
| Demographics | Prior treatment | ||
| PSA | Absolute value or kinetic properties | Predefined alteration of value (% decline, % change in doubling time) | |
| Pathology | Gleason score, molecular determinants | Negative biopsy | Positive biopsy |
| Measurable | Identification of target lesions | RECIST | RECIST |
| Bone | Extent of disease assessment | Resolution, no new lesions (excluding flare) | New (three or more) lesions |
| Quality of life | Pain, performance status, other validated scales | Improvement on validated scale | Worsening on validated scale |
| Clinical metastases: castrate. Therapeutic aim: reduce and/or prevent symptoms, delay progression, prolong life | |||
| Demographics | Prior treatment | ||
| PSA | Absolute value or kinetic properties | Predefined alteration of value (e.g., ≥50% decline)
No increase vs any increase at12 wk |
Increasing values |
| Pathology | Gleason score, molecular determinants | Negative biopsy | Positive biopsy |
| Measurable | Identification of target lesions | RECIST | RECIST |
| Bone | Extent of disease assessment | Resolution, no new lesions (excluding flare) | New (three or more) lesions |
| Quality of life | Pain, performance status, other validated scales | Improvement on validated scale | Worsening on validated scale |
Abbreviations: NMA, no measurable amount; BPH, benign prostatic hypertrophy.
To apply this approach clinically, all manifestations and potential sites of disease must be monitored at fixed time intervals. Failure at any site would be indicative of disease progression. The overall effects of an intervention are then assessed by examining either the proportion of patients who have not progressed at a fixed time point, the proportion who have not progressed to the next state, or an overall reduction in the hazard ratio of progression. The principle can be applied to single-arm phase II trials and phase II randomized trials. The overall risk/benefit for the intervention could then be estimated by relating the adverse events noted on the study to the duration of benefit that was observed. These will be considered on a state-by-state basis.
Localized disease
The objectives of treatment for a patient with localized disease are to eliminate the cancer completely and to prevent its recurrence in the primary site and systemically. The probability of eliminating the cancer can be estimated using risk models or nomograms (44, 45). For the patient who has undergone a prostatectomy, the first indication of ‘‘success’’ is whether or not an undetectable PSA is achieved postoperatively. If it is, recurrence or progression is signified when the PSA becomes detectable, e.g., >0.2 or 0.4 ng mL, and the abnormality is confirmed and continues to increase. For radiation therapy–treated patients, a different definition of PSA failure is used (46). The longer-term objective is to prevent an increasing PSA (or PSA recurrence), which antedates radiographic and clinical progression in virtually all cases. Neither definition of biochemical relapse addresses whether the failure is local or systemic, and if the latter, whether the patient is likely to develop metastasis or die of disease; a more relevant intermediate end point might be the proportion of patients who fail with a rapid PSA-DT, which has been shown to associated with a higher risk of prostate cancer metastases and death (33). This requires prospective validation.
Rising prostate-specific antigen: noncastrate
For patients with an increasing PSA after surgery or radiation, the first question is whether the increase in PSA represents recurrent/persistent disease in the primary site, systemic disease, or both. Locally persistent/recurrent disease in the prostate or prostate bed with no metastasis may still be curable or controllable with additional therapy to the prostate or prostate bed. As is the case for the newly diagnosed patient with localized disease, the first indication of success of a salvage prostatectomy or salvage radiation therapy is whether the PSA becomes undetectable.
For those in whom the increasing PSA represents systemic disease, the primary clinical objective is to prevent a transition to a state of clinical metastases, a point in the disease where the risk of a prostate cancer–specific death is significantly increased (47). The short-term objective may be achieved by eliminating the disease, slowing tumor growth, or inhibiting the metastatic process without directly affecting tumor growth. Here again, the mechanism of the agent must be considered. Some agents, such as those that promote differentiation, may induce short-term PSA increases that do not signify treatment failures. Others, such as immunologic agents or antiangiogenic drugs, may alter the rate of increase but not induce a PSA decline. The effect on PSA may also be delayed. As it is unlikely that patients with increasing PSA values will remain on therapy or be observed until an end point of radiographic progression is reached, it has been suggested that only agents with activity in more advanced clinical states should be tested here (19). Ultimately, the clinically significant end point is metastasis-free survival or overall survival, which are a better gauge of whether the drug is changing the natural history of the disease and not simply modulating PSA levels independent of an effect on tumor growth (19).
Rising prostate-specific antigen: castrate
Based on maturing data showing the prognostic significance of PSA doubling times and metastatic progression and prostate cancer–specific mortality (31, 32, 47), an increasing number of patients are receiving androgen deprivation before overt metastases are documented on an imaging study. It follows that patients are treated with androgen deprivation when an increasing PSA is the sole manifestation of disease progress in a similar manner—with an increasing PSA despite castrate levels of testosterone. The result is an entirely new disease state—increasing PSA: castrate—whose natural history is not well described. The overall treatment objectives are similar to those described for patients with increasing PSA: noncastrate, recognizing that the anticipated survival times following the development of metastases in the setting of castrate levels of testosterone carries a more ominous prognosis (48).
Clinical metastases: noncastrate
Elimination of all disease is rarely possible for patients with clinically detectable metastases. Instead, shorter-term therapeutic objectives are focused on strategies to increase the degree and the duration of response to androgen deprivation, whereas longer-term objectives include preventing the development of resistance, delaying the progression of established lesions or the development of new ones, and preventing new symptoms of disease (49). Given the high response proportions observed with androgen deprivation alone, these trials tend to be large and carry considerable risk of not demonstrating a significant difference in outcome for the experimental treatment. Such has been the case in trials studying the question of whether combined androgen blockade in superior to testosterone lowering monotherapies (50).
Noteworthy, however, is that PSA-based diagnosis, treatment, and monitoring has resulted in a decrease in the proportion of men that are being diagnosed with detectable metastases. The majority of men with detectable metastases do not have disease in the primary site and lower overall tumor burdens than in the past. In these cases as well, the probability of achieving an undetectable PSA varies as a function of disease extent (11). It remains an open question whether a proportion of these patients may be curable as well.
Clinical metastases: castrate
Patients who have progressed on hormones face a high likelihood of experiencing cancer-related morbidity and dying of their cancers. The short-term objective of treatment in these cases includes the control of disease-related symptoms. The longer-term objective is to prevent their occurrence or recurrence and to prolong life. With the recent approval of docetaxel for castrate metastatic disease on the basis of a survival benefit (15, 38), drug development efforts are now focused in two contexts: patients who have not received taxane-based chemotherapy (first-line therapy) and those who have (second-line therapy). Regardless of whether the drug is being explored as first-line or second-line therapy, an initial treatment effect could be assessed by using the proportion of patients who achieve a predetermined change in PSA, the proportion who show a significant reduction in soft tissue disease by standard criteria, or those who do not progress by bone scintigraphy (3, 51, 52). For those with symptoms, the proportion of patients who enjoy a palliative benefit is of interest, but equally important is the durability of these benefits. Accordingly, many trials consider a change in oncolytic therapy, such as the administration of radiation therapy to new symptoms of disease or a change in chemotherapy, as an indication that the treatment under study was no longer effective. This approach addresses the clinical reality that therapy is rarely stopped if the PSA is declining or stable even if a 50% benchmark is not reached. It is also and end point that can be validated prospectively in randomized comparisons of a first-line and second-line cytotoxic approach.
As a screening method for active treatments, many trials report the proportion of patients who achieve a defined posttherapy change (e.g., the proportion showing a 50% or 80% decline from baseline) as suggested by the Prostate Specific Antigen Working Group (18), while monitoring all sites of disease to ensure concordance (6). Whether a posttherapy decline in PSA can serve as a ‘‘surrogate’’ for clinical benefit, however, remains controversial (53–55). Recent work by our group has shown an association between a ‘‘stable’’ bone scan at 6 months after the start of therapy and survival (56). This observation, coupled with our observation of the prognostic significance of a ‘‘no increase’’ versus ‘‘increase’’ in PSA at 12 weeks (54), has led us to propose a composite end point that requires that the levels of PSA do not increase and that the bone scan does not progress for a minimum of 6 months as a potential intermediate end point for more definitive trials. The 6-month landmark represents the median time to progression observed in the docetaxel/estramustine arm of the Southwest Oncology Group study 99-16 (38), in which a survival benefit was shown relative to the control treatment. It also represents the median time to a change in chemotherapy for patients enrolled on first-line chemotherapy trials were subsequently enrolled on a second-line chemotherapy trial (Beekman), representing a change in disease status that was significant enough to require a change in systemic chemotherapy.
Composite end points, however, are not without pitfalls. An example is provided by a recent comparative trial of the endothelin receptor-A antagonist atrasentan, in which a protocol definition of progression mandated removal of patients from the study. The definition, the development of two or more lesions on a bone scan, did not address the lag that may occur between the elevations in PSA required for entry on study and changes in radionuclide bone scan. As a result, over half of the patients came off trial at 3 months in spite of stable or improving symptoms and PSA values. In fact, the continued analysis of the trial has shown that all the other end points, save the bone scan, favored the atrasentan arm (57).
Critical to the use of composite end points is tailoring each element to the class of drug being tested. For example, an increasing PSA may not signify a treatment failure for certain biological agents, such as those that induce differentiation or those that slow progression without cytotoxic effects. By the same token, patients should not be taken off study for a worsened bone scan 8 to 12 weeks following initiation of cytotoxic therapy in the context of a declining PSA or improving symptoms, as the scintigraphic findings may also represent flare phenomena. Similarly, a pain flare following treatment with certain hormonal agonist/antagonists or bone-seeking radiopharmaceuticals may indicate response rather than treatment failure. Ultimately, each element of the composite—be it PSA, scintigraphy, radiography, biological assay, or symptomatology—must be independently defined using the putative mechanism of the drug and the preclinical data as part of the trial design process.
Summary
The purpose of the phase II trial is to determine whether a treatment produces a sufficient effect in a sufficient proportion of patients for a sufficient period of time to justify further development. How activity is defined should be determined by the treatment objective for the patients to whom it is being offered. The manifestations of prostate cancer do not lend themselves to the ‘‘packaging’’ assessments of treatment effects into the response categories proposed by RECIST. More important than defining ‘‘response’’ is to define objective, meaningful, measures of disease progression at which point it can be stated more precisely that the therapy under evaluation is no longer effective. This methodology can be applied across the clinical spectrum of the disease, is independent of the specific intervention used, and circumvents the controversies of assessing response using changes in PSA and bone, the most common manifestations of the disease.
Footnotes
Grant support: Memorial Sloan-Kettering Cancer Center Specialized Programs of Research Excellence in Prostate Cancer, PepsiCo, and the Prostate Cancer Foundation, NIH grant CA102544, and CA05826.
References
- 1.Johnson JR, Williams G, Pazdur R. End points and United States Food and Drug Administration approval of oncology drugs. J Clin Oncol. 2003;21:1404–11. doi: 10.1200/JCO.2003.08.072. [DOI] [PubMed] [Google Scholar]
- 2.Yagoda A, Watson RC, Natale RB, et al. A critical analysis of response criteria in patients with prostatic cancer treated with cis-diamminedichloride platinum II. Cancer. 1979;44:1553–62. doi: 10.1002/1097-0142(197911)44:5<1553::aid-cncr2820440502>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 3.Scher HI, Mazumdar M, Kelly WK. Clinical trials in relapsed prostate cancer: defining the target. J Natl Cancer Inst. 1996;88:1623–34. doi: 10.1093/jnci/88.22.1623. [DOI] [PubMed] [Google Scholar]
- 4.Fazzari M, Heller G, Scher HI. The phase II/III transition: towards the proof of efficacy in cancer clinical trials. Control ClinTrials. 2000;21:360–8. doi: 10.1016/s0197-2456(00)00056-8. [DOI] [PubMed] [Google Scholar]
- 5.Scher HI, Yagoda A. Clinical trials in prostatic cancer: methodology and controversies. In: Bruce AW, Trachtenberg J, editors. Adenocarcinoma of the prostate. NewYork: Springer-Verlag; 1987. pp. 197–220. [Google Scholar]
- 6.Figg WD, Ammerman K, Patronas N, et al. Lack of correlation between prostate-specific antigen and the presence of measurable soft tissue metastases in hormone-refractory prostate cancer. Cancer Invest. 1996;14:513–7. doi: 10.3109/07357909609076896. [DOI] [PubMed] [Google Scholar]
- 7.Kang Y, Siegel PM, Shu W, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537–49. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- 8.Shah RB, Mehra R, Chinnaiyan AM, et al. Androgen-independent prostate cancer is a heterogeneous group of diseases: lessons from a rapid autopsy program. Cancer Res. 2004;64:9209–16. doi: 10.1158/0008-5472.CAN-04-2442. [DOI] [PubMed] [Google Scholar]
- 9.Scher HI, Heller G. Clinical states in prostate cancer: towards a dynamic model of disease progression. Urology. 2000;55:323–7. doi: 10.1016/s0090-4295(99)00471-9. [DOI] [PubMed] [Google Scholar]
- 10.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. JNatl Cancer Inst. 2000;92:205–16. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 11.Beekman K, Morris M, Slovin S, et al. Androgen deprivation for minimal metastatic disease: the threshold for achieving an undetectable PSA. Urology. 2005;65:947–52. doi: 10.1016/j.urology.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 12.Kelly WK, Curley T, Slovin S, et al. Paclitaxel, estramustinephosphate, and carboplatinin patients with advanced prostate cancer. J Clin Oncol. 2001;19:44–53. doi: 10.1200/JCO.2001.19.1.44. [DOI] [PubMed] [Google Scholar]
- 13.Solit DB, Morris M, Slovin S, et al. Clinical experience with intravenous estramustine phosphate, paclitaxel, and carboplatin in patients with castrate, metastatic prostate adenocarcinoma. Cancer. 2003;98:1842–8. doi: 10.1002/cncr.11754. [DOI] [PubMed] [Google Scholar]
- 14.Galsky MD, Small EJ, Oh WK, et al. Multi-institutional randomized phase II trial of the epothilone B analog ixabepilone (BMS-247550) withor without estramustine phosphate in patients with progressive castrate metastatic prostate cancer. J Clin Oncol. 2005;23:1439–46. doi: 10.1200/JCO.2005.09.042. [DOI] [PubMed] [Google Scholar]
- 15.Tannock IF, de Wit R, Berry WR, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. NEnglJMed. 2004;351:1502–12. doi: 10.1056/NEJMoa040720. [DOI] [PubMed] [Google Scholar]
- 16.Eisenberger MA, De Wit R, Berry W, et al. A multi-center phase III comparison of docetaxel (D) + prednisone (P) in patients with hormone-refractory prostate cancer (HRPC) Proc Am Soc Clin Onc. 2004;23:2. [Google Scholar]
- 17.Berry W, Dakhil S, Modiano M, Gregurich M, Asmar L. Phase III study of mitoxantrone/low-dose prednisone versus low-dose prednisone alone in patients with asymptomatic hormone-refractory carcinoma of the prostate. J Urol. 2002;168:2439–43. doi: 10.1016/S0022-5347(05)64163-8. [DOI] [PubMed] [Google Scholar]
- 18.Bubley GJ, Carducci M, Dahut W, et al. Eligibility and response guidelines for phase II clinical trials in androgen-independent prostate cancer: recommendations from the PSA Working Group. J Clin Oncol. 1999;17:3461–7. doi: 10.1200/JCO.1999.17.11.3461. [DOI] [PubMed] [Google Scholar]
- 19.Scher HI, Eisenberger M, D’Amico AV, et al. Eligibility and outcomes reporting guidelines for clinical trials for patients in the state of a rising PSA: Recommendations from the Prostate-Specific Antigen Working Group. J Clin Onc. 2004;22:537–56. doi: 10.1200/JCO.2004.07.099. [DOI] [PubMed] [Google Scholar]
- 20.Erasmus JJ, Gladish GW, Broemeling L, et al. Inter-observer and intraobserver variability in measurement of non-small-cell carcinoma lung lesions: implications for assessment of tumor response. J Clin Oncol. 2003;21:2574–82. doi: 10.1200/JCO.2003.01.144. [DOI] [PubMed] [Google Scholar]
- 21.Cheson BD, Horning SJ, Coiffier B, et al. Reportofan international workshop to standardize response criteria fornon-Hodgkin’slymphomas. NCI Sponsored International Working Group. J Clin Oncol. 1999;17:1244. doi: 10.1200/JCO.1999.17.4.1244. [DOI] [PubMed] [Google Scholar]
- 22.Scher HI, Yagoda A, Ahmed T, Watson RC. Methyl-glyoxal-bis(guanylhydrazone) in hormone-resistant adenocarcinoma of the prostate. J Clin Oncol. 1985;3:224–8. doi: 10.1200/JCO.1985.3.2.224. [DOI] [PubMed] [Google Scholar]
- 23.Smith PH, Bono A, Calais da Silva F, et al. Some limitations of the radioisotope bone scan in patients with metastatic prostatic cancer. Cancer. 1990;66:1009–16. doi: 10.1002/cncr.1990.66.s5.1009. [DOI] [PubMed] [Google Scholar]
- 24.Dhingsa R, Qayyum A, Coakley FV, et al. Prostate cancer localization with endorectal MR imaging and MR spectroscopic imaging: effect of clinical data on reader accuracy. Radiology. 2004;230:215–20. doi: 10.1148/radiol.2301021562. [DOI] [PubMed] [Google Scholar]
- 25.Sella T, Schwartz LH, Swindle PW, et al. Suspected local recurrence after radical prostatectomy: endorectal coil MR imaging. Radiology. 2004;231:379–85. doi: 10.1148/radiol.2312030011. [DOI] [PubMed] [Google Scholar]
- 26.Rosenthal SA, Haseman MK, Polascik TJ. Utility of capromab pendetide (ProstaScint) imaging in the management of prostate cancer. Tech Urol. 2001;7:27–37. [PubMed] [Google Scholar]
- 27.Kahn D, Williams RD, Manyak MJ, et al. 111-Indium Capromab pendetide in the evaluation of patients with residual or recurrent prostate cancer after radical prostatectomy. The ProstaScint Study Group. J Urol. 1998;159:2041–6. doi: 10.1016/S0022-5347(01)63239-7. discussion 2046–7. [DOI] [PubMed] [Google Scholar]
- 28.Partin AW, Pound CR, Clemens JQ, Epstein JI, Walsh PC. Serum PSA following anatomical radical prostatectomy: The Johns Hopkins experience after ten years. Urol Clin North Am. 1993;20:713–25. [PubMed] [Google Scholar]
- 29.Pound CR, Partin AW, Epstein JI, Walsh PC. Prostate-specific antigen after anatomic radical retropubic prostatectomy. Patterns of recurrence and cancer control. Cancer. 1997;79:528–37. doi: 10.1016/s0094-0143(05)70386-4. [DOI] [PubMed] [Google Scholar]
- 30.Han M, Partin AW, Zahurak M, Piantadosi S, Epstein JE, Walsh PC. Biochemical (prostate specific antigen) recurrence probability following radical prostatectomy for clinically localized prostate cancer. J Urol. 2003;169:517–23. doi: 10.1097/01.ju.0000045749.90353.c7. [DOI] [PubMed] [Google Scholar]
- 31.D’Amico AV, Cote K, Loffredo M, Renshaw AA, Schultz D. Determinants of prostate cancer-specific survivalafter radiation therapy for patients with clinicallylocalized prostate cancer. J ClinOncol. 2002;20:4567–73. doi: 10.1200/JCO.2002.03.061. [DOI] [PubMed] [Google Scholar]
- 32.D’Amico AV, Moul JW, Carroll PR, Sun L, Lubeck D, Chen M-H. Cancer specific mortality following surgery or radiation for patients with clinically localized prostate cancer managed during the PSA era. J Natl Cancer Inst. 2003;95:1376–83. [Google Scholar]
- 33.D’Amico AV, Moul JW, Carroll PR, et al. Intermediate end point for prostate cancer-specific mortality following salvage hormonal therapy for prostate-specific antigen failure. J Natl Cancer Inst. 2004;96:509–15. doi: 10.1093/jnci/djh086. [DOI] [PubMed] [Google Scholar]
- 34.Kelly WK, Scher HI, Mazumdar M, Vlamis V, Schwartz M, Fossa SD. Prostate specific antigen as a measure of disease outcome in hormone-refractory prostatic cancer. J Clin Oncol. 1993;11:607–15. doi: 10.1200/JCO.1993.11.4.607. [DOI] [PubMed] [Google Scholar]
- 35.Sridhara R, Eisenberger MA, Sinibaldi VJ, Reyno LM, Egorin MJ. Evaluation of prostate-specific antigen as a surrogate marker for response of hormone-refractory prostate cancer to suramin therapy. J Clin Oncol. 1995;13:2944–53. doi: 10.1200/JCO.1995.13.12.2944. [DOI] [PubMed] [Google Scholar]
- 36.Smith DC, Dunn RL, Strawderman MS, Pienta KJ. Change in serum prostate-specific antigen as a marker of response to cytotoxic therapy for hormone-refractory prostate cancer. J Clin Oncol. 1998;16:1835–43. doi: 10.1200/JCO.1998.16.5.1835. [DOI] [PubMed] [Google Scholar]
- 37.Small EJ, Halabi S, Ratain MJ, et al. Randomized study of three different doses of suramin administered with a fixed dosing schedule inpatients with advanced prostate cancer: results of intergroup 0159, cancer and leukemia group B 9480. J Clin Oncol. 2002;20:3369–75. doi: 10.1200/JCO.2002.10.022. [DOI] [PubMed] [Google Scholar]
- 38.Petrylak DP, Tangen CM, Hussain MH, et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. NEnglJMed. 2004;351:1513–20. doi: 10.1056/NEJMoa041318. [DOI] [PubMed] [Google Scholar]
- 39.Tannock IF, Osoba D, Stockler MR, et al. Chemotherapy with mitoxantrone plus prednisone or prednisone alone for symptomatic hormone-resistant prostate cancer: a Canadian randomized trial with palliative end points. J ClinOncol. 1996;14:1756–64. doi: 10.1200/JCO.1996.14.6.1756. [DOI] [PubMed] [Google Scholar]
- 40.Saad F, Gleason DM, Murray R, et al. A randomized, placebo-controlled trial of zoledronic acid in patients with hormone-refractory metastatic prostate carcinoma. J Natl Cancer Inst. 2002;94:1458–68. doi: 10.1093/jnci/94.19.1458. [DOI] [PubMed] [Google Scholar]
- 41.Serafini AN, Houston SJ, Resche I, et al. Palliation of pain associated with metastatic bone cancer using samarium-153 lexidronam: a double-blind placebo-controlled clinical trial. J Clin Oncol. 1998;16:1574–81. doi: 10.1200/JCO.1998.16.4.1574. [DOI] [PubMed] [Google Scholar]
- 42.Bolger JJ, Dearnaley DP, Kirk D, et al. Strontium-89 (Metastron) versus external beam radiotherapy in patients with painful bone metastases secondary to prostatic cancer: preliminary report of a multicenter trial. Semin Oncol. 1993;20:32–3. [PubMed] [Google Scholar]
- 43.Quilty PM, Kirk D, Russell JM, et al. Strontium-89 versus external beam radiotherapy for palliation of prostate cancer bony metastases (final results of the UK Metastron trial) Br J Radiol. 1992;65:1. [Google Scholar]
- 44.Kattan MW, Eastham JA, Stapleton AMF, Wheeler TM, Scardino PT. A preoperative nomogram for disease recurrence following radical prostatectomy for prostate cancer. J Natl Cancer Inst. 1998;90:766–71. doi: 10.1093/jnci/90.10.766. [DOI] [PubMed] [Google Scholar]
- 45.Ross PL, Scardino PT, Kattan MW. A catalog of prostate cancer nomograms. J Urol. 2001;165:1562–8. [PubMed] [Google Scholar]
- 46.Cox JD, Gallagher MJ, Hammond EH, Kaplan RS, Schellhammer PF. Consensus statements on radiation therapy of prostate cancer: guidelines for prostate re-biopsy after radiation and for radiation therapy with rising prostate-specific antigen levels after radical prostatectomy. American Society for Therapeutic Radiology and Oncology Consensus Panel. J Clin Oncol. 1999;17:1155. doi: 10.1200/JCO.1999.17.4.1155. [DOI] [PubMed] [Google Scholar]
- 47.Pound CR, Partin AW, Eisenberger MA, Chan DW, Pearson JD, Walsh PC. Natural history of progression to metastases an death from prostate cancer in men with PSA recurrence following radical prostatectomy. JAMA. 1999;281:1591–7. doi: 10.1001/jama.281.17.1591. [DOI] [PubMed] [Google Scholar]
- 48.Bianco FJ, Dotan ZA, Kattan MW, et al. Natural history of biochemically recurrent castrate-resistant disease in men treated with maximal androgen blockage for a rising PSA after radical prostatectomy. J Urol. 2005;173:309. [Google Scholar]
- 49.Scher HI. Prostate carcinoma: Defining therapeutic objectives and improving overall outcomes. Cancer. 2003;97:758–71. doi: 10.1002/cncr.11151. [DOI] [PubMed] [Google Scholar]
- 50.Schmitt B, Wilt TJ, Schellhammer PF, et al. Combined androgen blockade with nonsteroidal anti-androgens for advanced prostate cancer: a systematic review. Urology. 2001;57:727–32. doi: 10.1016/s0090-4295(00)01086-4. [DOI] [PubMed] [Google Scholar]
- 51.Dawson NA. Eligibility and response criteria in hormone refractory prostate cancer (HRPC). A need for consensus. Proc Am Soc Clin Oncol. 1997;16:317a. [Google Scholar]
- 52.Newling DW. Second line treatment of metastatic prostate cancer. Urol Res. 1997;25:73–8. doi: 10.1007/BF00941992. [DOI] [PubMed] [Google Scholar]
- 53.Verbel DA, Heller G, Kelly WK, Scher HI. Quantifying the amount of variation in survival explained by PSA. Clin Cancer Res. 2002;8:2576–9. [PubMed] [Google Scholar]
- 54.Scher HI, Kelly WK, Zhang Z-F, et al. Post-therapy serum prostate specific antigen level and survival in patients with androgen-independent prostate cancer. J Natl Cancer Inst. 1999;91:244–51. doi: 10.1093/jnci/91.3.244. [DOI] [PubMed] [Google Scholar]
- 55.Crawford ED, Pauler DK, Tangen CM, et al. Three-month change in PSA as surrogate endpoint for mortality in advanced hormone-refractory prostate cancer (HRPC): data from Southwest Oncology Group. Proc Am Soc Clin Oncol. 2004;23:382. [Google Scholar]
- 56.Galsky MD, Scher HI, Wilton A, Heller G, Kelly WK. The impact of post-treatment change in PSA, bone scans, and measurable disease on survival in patients with castrate metastatic prostate cancer (CMPC) Proc Am Soc Clin Oncol. 2005 Submitted. [Google Scholar]
- 57.Beekman KW, Fleming M, Scher HI, et al. Outcomes with second-line chemotherapy in castrate metastatic prostate cancer. Proc Multidisciplinary Prostate Cancer Symposium. 2005;180 abstract 291. [Google Scholar]
- 58.Carducci MA, Padley RJ, Breul J, et al. Effect of endothelin–a receptor blockade with atrasentan on tumor progression in men with hormone-refractory prostate cancer: a randomized, phase II, placebo-controlled trial. J Clin Oncol. 2003;21:679–89. doi: 10.1200/JCO.2003.04.176. [DOI] [PubMed] [Google Scholar]
- 59.Petrylak DP, Tangen C, Hussain M, et al. SWOG 99-16: Randomized phase III trial of docetaxel (D)/estramustine (E) versus mitoxantrone(M)/prednisone(p) in men with androgen-independent prostate cancer (AIPCA) Proc Am Soc Clin Oncol. 2004;23:2. [Google Scholar]