

Abstract
The folding of Escherichia coli dihydrofolate reductase was examined at pH 7.8 and 15°C by using stopped-flow fluorescence and absorbance spectroscopies. The formation of a highly fluorescent intermediate occurs with relaxation times ranging between 142 and 343 msec, whereas stopped-flow absorbance spectroscopy using methotrexate binding assays shows a distinct lag phase during these time frames for the native state. The lag in absorbance kinetics and the lack of fast-track folding events indicate that the formation of this ensemble of intermediates is an obligatory step in the folding reaction.
The question of whether experimentally detectable intermediates are important in kinetic mechanisms has been a point of controversy. This question has been a matter of discussion not only among biochemists. For example, mechanistic organic chemists were involved in a fierce debate as to whether specific intermediates (i) exist and (ii) are important for particular chemical reactions (1–3). It was not until detailed kinetic studies were performed that this issue was resolved. Recently, those interested in the protein-folding problem have been engaged in a similar debate (4). Theoretical developments have proposed the possibility of two kinds of intermediates, on-route and off-route, during the folding event (5). The on-route idea is attractive, because it allows for a way to reduce the time needed for a full conformational search by forming a population of intermediates (6). Others have shown that populated partially folded forms may impede the formation of the native state (7, 8). One limitation in analysis of protein folding reactions is that many intermediates are formed within the dead time of the available instrumentation. Thus, detailed kinetic studies do not reveal the role of these “burst-phase” intermediates.
Dihydrofolate reductase (DHFR) from Escherichia coli is a particularly amenable system with which to address the role of intermediates in the formation of native protein. The folding reaction has been studied in detail (9–11). Parallel routes in refolding proceed through the population of two classes of intermediate species. The first detectable event is the formation of a marginally stable “burst-phase” intermediate, with a high degree of secondary structure and an exposed hydrophobic core (12, 13). After this event, the next detectable phase occurs in a time frame of hundreds of milliseconds and characterizes an ensemble of metastable species, designated as the highly fluorescent intermediate, IHF, which contain specific tertiary contacts (10). The formation of this intermediate can be monitored easily by stopped-flow fluorescence and CD spectroscopies. Native protein is formed in parallel reactions, with relaxation times ranging from 1–100 sec. Methotrexate, a potent inhibitor of DHFR, binds to the native state but not to IHF and shows a maximum in absorbance difference at 380 nm when bound. Thus, we can monitor the formation of the inhibitor binding pocket under conditions where protein signals do not contribute. Using stopped-flow absorbance and fluorescence spectroscopies, we demonstrate the absence of a fast track to native protein that would bypass intermediate formation. In addition, a distinct lag phase is detected, during which the highly fluorescent species accumulate. Both these findings confirm the role of an on-route species essential to the formation of native protein (14). In contrast to the behavior of small two-state folding proteins, we suggest that the topological constraints of folding a protein of greater than ≈100 amino acids may require the formation of an on-pathway intermediate(s) (15). The stability and nature of these on-pathway species would depend not only on the native topology but also on additional energetic factors.
Materials and Methods
Purification of DHFR.
Purification of DHFR was performed according to Jennings et al. (10). DHFR was dialyzed extensively against 10 mM phosphate buffer (10 mM potassium phosphate/0.2 mM K2 EDTA/1 mM β-mercaptoethanol), pH 7.80. All experiments were carried out under these conditions. The concentration of DHFR was measured by absorbance at 280 nm by using a molar extinction coefficient of 3.11 × 104 M−1 cm−1 (9). A 1 mM stock solution of methotrexate was prepared in 10 mM phosphate buffer, and the concentration was determined by measuring absorbance at 302 nm in the presence of 0.1 N NaOH, by using the molar extinction coefficient of 2.21 × 104 M−1cm−1 (9).
Stopped-Flow Fluorescence.
The stopped-flow fluorescence data were collected on the Applied Photophysics (Surrey, U.K.) SX-17MV unit with a path length of 0.2 cm and the entrance and exit slits set to 5 nm. The excitation wavelength was set to 293 nm and the data collected with the 320-nm cut-off filter in place. The protein was unfolded in 6 M urea and allowed to refold to a final concentration of 0.54, 0.75, 1.5, and 2 M urea and a final protein concentration of 11 μM at 15°C. The refolding process was monitored in the split-time base mode and collected for 50–500 sec depending on the final denaturant concentration.
Stopped-Flow Absorbance Studies.
The stopped-flow studies were carried out on the Applied Photophysics SX-17MV stopped-flow unit with the slits set to 5 mm for the entrance and 1 mm for the exit, and the absorbance monitored at 380 nm with a 1-cm pathlength cuvette. The data were collected at 15°C in the split-time base mode, with the times set to 1 sec and 50 sec for the 0.54-M refolding jump. All other refolding jumps were collected in single-time base mode. The final conditions were 11 μM DHFR and 12 μM methotrexate (MTX), giving a binding stoichiometry of 1.1:1.
Dead-Time Binding of MTX.
The binding of MTX to native DHFR was monitored by stopped-flow absorbance at 380 nm; the maximal signal change on binding of MTX to DHFR occurs at this wavelength. The reaction was initiated by a 1:1 mixing of the protein (22 μM in 0.54 M urea) and MTX (24 μM in 0.54 M urea). The final conditions were 11 μM DHFR and 12 μM MTX in 0.54 M urea, giving a binding stoichiometry of 1.1:1
Results and Discussion
Kinetic Methods.
There are several problems in discerning the role of intermediates in protein folding reactions. Intermediates formed in the dead time of an instrument may be on-pathway but cannot be distinguished from those formed in an off-pathway event (16–20). Characterization of intermediates formed in the millisecond (or longer) time frame is often complicated by the fact that folding reactions are generally followed by optical signals that may have contributions from all species (U,I,N), making it difficult to extract accurately the time-dependent population of a particular species (21), whereas in cases where rapid quench techniques are used, only a limited number of points can be collected, and the time course has inherently more error (22, 23). To best test the role of an intermediate during folding, the following conditions are optimal:
(i) The N and I species should have signals distinct from one another.
(ii) The rate of formation of I and N are similar to one another.
(iii) The time-dependent population of N is monitored continuously.
(iv) The intermediate is relatively stable.
and
(v) The rate of formation of I and N are measurable with conventional mixing techniques.
Given that these conditions are met, one can differentiate between on- and off-pathway kinetic events.
To illustrate the kinetic distinctions between these two cases, we performed simulations based on two simple mechanisms:
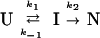 |
Scheme 1
and
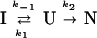 |
Scheme 2
In Scheme 1, the formation of I is necessary for the production of N, and I is an on-pathway obligatory intermediate. In Scheme 2, I is an off-pathway trap. In both cases, the starting point for the simulation is U, based on the kinetic criterion that the rate of formation of I is measurable. A representative plot of the expected fractional population of various species (U,I,N) as a function of time for the two simple folding schemes is shown in Fig. 1. Fig. 1 a and b demonstrate the expected effect for the population of intermediate, I, on the rate of formation of N for (i) an on- or (ii) an off-pathway reaction. In these simulations, the respective rate constants were kept constant. In the on-pathway scheme, a distinct lag in the formation of N is apparent in the first ≈120 msec of the reaction (24). In the off-pathway reaction scheme, it is important to note that, whereas the population of N increases faster in the early part of the progress curve, the overall increase in the population of N is slowed relative to that observed in Fig. 1a by the unfavorable preequilibrium in an off-pathway event. Importantly, no lag in the production of N is observed in the off-pathway simulation. Note, however, that it is possible to distinguish between these two mechanisms only when both kinetic steps are resolved. To illustrate this point, we simulated the same two mechanisms but made the rate of formation of I very fast relative to that of N. Clearly, any lag is obscured under these conditions. Although the intrinsic rate constant for the formation of N remains the same in simulations a–d (Fig. 1 a–d), the apparent rate constant for the formation of N is slowed dramatically in Fig. 1d. Again, this reflects the unfavorable preequilibrium of U with an off-pathway I. In Fig. 1 b and d, it is clear that the off-pathway event results in a trap such that the overall apparent rate of formation of N is slowed, suggesting a higher barrier for formation of N despite the fact that the intrinsic rate constant is not altered.
Figure 1.

Kinetic simulations of on- (Scheme 1) and off- (Scheme 2) pathway simple folding models. The starting point for all simulations is 100% U, where the U to I transition is designated as k1, whereas the I to U transition is k−1 and the ratio of k1/k−1 is kept constant. (a) Plot of the kinetic simulations for a simple on-pathway folding mechanism by using kinsim with k1 = 3.33 s−1, k−1 = 0.833 s−1 and k2 = 1 s−1 with the population of native species shown in red. (b) Simulation for an off-pathway folding model with k1 = 3.33 s−1, k−1 = 0.833 s−1 and k2 = 1 s−1. (c) Plot of the simulation depicted in a, but with k1 = 333 s−1, k−1 = 83.3 s−1 and k2 = 1 s−1. (d) Depicts the off-pathway folding mechanism in b, with k1 = 333 s−1, k−1 = 83.3 s−1 and k2 = 1 s−1.
DHFR.
One well-characterized folding system that follows the above kinetic criteria for testing the role of intermediates in folding is DHFR. A plot of the relative fluorescence intensity as a function of time for a refolding jump to 0.54 M urea, pH 7.80, is shown in Fig. 2. The first 10 sec of the reaction are shown for clarity. The fluorescence signal rises to a maximal value with a relaxation time of 142 msec followed by a decrease in intensity in a series of reactions that characterize the formation of the native protein. This nonmonotonic fluorescence behavior is indicative of the transient population of a highly fluorescent intermediate species before formation of the native protein.
Figure 2.

Plot of the change in the relative fluorescence intensity as a function of time for a refolding jump from 6 M to 0.54 M urea at pH 7.80 and 15°C, measured on the Applied Photophysics SX.17MV stopped-flow unit. Tryptophan fluorescence was followed by using an excitation wavelength of 293 nm and by monitoring the emission for wavelengths >320 nm.
To detect the formation of native protein, we took advantage of the fact that the inhibitor MTX binds tightly to native protein and gives rise to an optical absorbance difference signal at 380 nm, a wavelength beyond any optical protein signals. In addition, MTX binds with a nanomolar binding constant in the dead time of stopped-flow experiments (10). A series of refolding experiments to final denaturant concentrations of 0.54, 0.76, 1.5, and 2 M urea were performed in the absence and presence of a 1.1-fold excess of MTX. Representative plots of the absorbance changes at 380 nm as a function of time for a refolding reaction to 0.54 M and 2 M, as monitored by MTX binding, are shown in Fig. 3, with the first 2 sec shown (Inset) for clarity. In all cases, there is no signal change in the dead time of the instrument. The optical signal increases with time from the initial zero point with relaxation rates expected for the rate of formation of native protein, in agreement with CD and fluorescence spectroscopy (10, 13). There is no fast-track folding to the native state for DHFR, in contrast to what has been observed for lysozyme (8). An additional interesting feature of the observed reaction is the apparent lag in the formation of N observed in the first few hundred milliseconds (see Fig. 3 Insets) over the denaturant range of 0.54–2 M urea. A plot of the first 1 sec of the folding reaction to 0.54 M urea, as detected by fluorescence, and the absorbance change on MTX binding are shown in Fig. 4 a and b, respectively. The lag phase in absorbance data, which monitors native-state formation, corresponds to the build up of intermediate detected by the change in fluorescence intensity. Control experiments with the native protein in 0.54 M urea and 1 sec after the initiation of refolding to 0.54 M urea indicate that the protein binds the inhibitor in the dead time of the experiment. Thus the observed lag is not a result of slow inhibitor binding but an inherent feature of the folding reaction. The length of the lag varies from 250 to 400 msec as a function of final denaturant, as is expected from the denaturant dependence of the formation of IHF and the native protein.
Figure 3.

(a) Plot of the difference in absorbance on methotrexate binding as a function of time for a refolding jump to 0.54 M urea. (b) Plot of the absorbance difference for a refolding jump to 2 M urea. Spectra were acquired on the Applied Photophysics SX.17MV stopped-flow unit. The change in signal was observed at 380 nm. The solid lines correspond to the simulation of the proposed four-channel folding model. Insets show the first 2 sec of the reaction. The data were collected at pH 7.80 and 15°C.
Figure 4.

(a) Plot of the change in relative fluorescence intensity as a function of time for a refolding jump from 6 M to 0.54 M urea at pH 7.80 and 15°C, measured on the Applied Photophysics SX.17MV stopped-flow unit. Tryptophan fluorescence was monitored by an excitation wavelength of 293 nm and emission observed for wavelengths >320 nm. (b) Plot of the difference in absorbance on methotrexate binding as a function of time for a refolding jump from 6 M to 0.54 M urea, pH 7.80 and 15°C.
The Formation of an On-Pathway Intermediate.
The proposed kinetic folding mechanism of DHFR involves a series of parallel reactions to a set of native conformers (N1–N4). Each parallel path proceeds through the formation of a collapsed burst phase intermediate (IBP) followed by the formation of the collection of highly fluorescent intermediates (IHF), which are then processed to the corresponding native conformer (10). Because interconversions between channels are slow relative to folding (25), the mechanism for each channel can be represented by a four-state model:
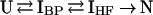 |
In this model, U, IBP, IHF, and N correspond to the unfolded protein, the burst-phase intermediate, the HF intermediate, and the native protein, respectively. Simulations of the expected MTX-binding kinetics based on the proposed folding mechanism were performed with kinsim and are compared with the experimental data in Fig. 3 (26). In these simulations, the rate of formation of IBP and IHF is held constant at the experimentally determined rate in all the channels, whereas the rate of formation of the native conformer is fixed at the experimentally determined rate for each channel. The amount of material flowing through each channel was fixed at the previously determined value (9). Because the fourth channel comprises only 6% of the material refolding and is too slow to be detected by stopped-flow techniques, it was not included in the simulations. The good agreement between the simulated and experimental data is readily apparent in Fig. 3. Not only the full time course but also the duration of the lag phase in the observed data is reproduced in the simulation. All attempts to simulate the data with IHF being off-pathway failed. Because IBP forms in the dead time of the instrument, it cannot be determined if it is an on- or off-pathway intermediate, but the formation of IHF is monitored easily and will simulate only to an on-pathway model. We conclude that, (i) there is no fast track to N, and (ii) formation of IHF is an essential step in the folding of DHFR.
Structural Description of the Intermediates.
Stopped-flow CD and mutagenic studies have shown that two tryptophans, Trp-47 and Trp-74, adopt an edge/face orientation in IHF as well as in the native protein. These findings suggest that the adenine-binding domain that encompasses these residues is folded, because the residues are 27 positions removed from each other in the primary sequence (Fig. 5) (13). Thus, the experimental data indicate that the nucleotide-binding domain is folded early in the mechanism. Detailed studies of the folding mechanism of staphylococcal nuclease by Roder and colleagues demonstrated that a global fit of the denaturant dependence of the folding reaction could give information on the change in solvent accessible surface area of transient intermediates during folding (21). We used a similar approach to analyze the denaturant dependence of the folding of DHFR. The fraction of buried surface was determined to be 0.20 and 0.65 for IBP and IHF, respectively. The change in solvent accessible surface area seen in the formation of IBP likely represents a collection of highly heterogeneous states with a high degree of secondary structure and some protection from hydrogen–deuterium exchange, in agreement with previous studies (27). In contrast, IHF is more highly folded, with at least 2/3 of the molecule in a compact state on formation of the IHF ensemble. Interestingly, theoretical calculations based on purely topological considerations agree with this conclusion (15). Although it is certain that IHF is composed of an ensemble of states, the macroscopic features are similar, suggesting that the region affords enough structure and plasticity to accommodate folding of mutant proteins while containing specific elements of secondary and tertiary structure (27). Our studies using a specific and continuous assay to probe the native structure of the wild-type protein provide clear evidence that the IHF intermediate is essential in the folding of DHFR over a range of denaturant concentrations. It would be interesting to determine whether drastically altering the primary sequence by circular permutation alters the observed obligatory nature of the folding route.
Figure 5.

Ribbon diagram of E. coli dihydrofolate reductase by using the x-ray coordinates from the Bolin et al. structure (28). The molecule is oriented to show the two subdomains with the interconnecting loops. The two tryptophans forming the exciton coupling (47 and 74) are shown in purple and blue, respectively, whereas the substrate-binding pocket is shown with the bound methotrexate in red. The program molscript was used to prepare this diagram (29).
Acknowledgments
We thank J. Adams and J. Onuchic for critical reading of the manuscript, C. R. Matthews and P. Taylor for the use of their stopped-flow instruments, and L. A. Gross for helpful discussions. This work was supported in part by the Hellman Fellowship (P.A.J.), the Sloan Fellowship (P.A.J.), National Institutes of Health Grants GM 54038 (P.A.J.), CA 09523 (M.R.), and GM 08326 (D.K.H.).
Abbreviations
- DHFR
dihydrofolate reductase
- MTX
methotrexate
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.100547697.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.100547697
References
- 1.Blackburn G M, Jencks W P. J Am Chem Soc. 1968;90:2638–2645. [Google Scholar]
- 2.Robinson D R. J Am Chem Soc. 1970;92:3138–3146. [Google Scholar]
- 3.Cunningham B A, Schmir G L. J Am Chem Soc. 1966;88:551–558. [Google Scholar]
- 4.Baldwin R L. J Biomol NMR. 1995;5:103–109. doi: 10.1007/BF00208801. [DOI] [PubMed] [Google Scholar]
- 5.Socci N D, Onuchic J N, Wolynes P G. Proteins Struct Funct Genet. 1998;32:136–158. [PubMed] [Google Scholar]
- 6.Xu Y, Mayne L, Englander S W. Nat Struct Biol. 1998;5:774–778. doi: 10.1038/1810. [DOI] [PubMed] [Google Scholar]
- 7.Sosnick T R, Hiller M R, Englander S W. Nat Struct Biol. 1994;1:149–156. doi: 10.1038/nsb0394-149. [DOI] [PubMed] [Google Scholar]
- 8.Kiefhaber T. Proc Natl Acad Sci USA. 1995;92:9029–9033. doi: 10.1073/pnas.92.20.9029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Touchette N A, Perry K M, Matthews C R. Biochemistry. 1986;25:5445–5452. doi: 10.1021/bi00367a015. [DOI] [PubMed] [Google Scholar]
- 10.Jennings P A, Finn B E, Jones B E, Matthews C R. Biochemistry. 1993;32:3783–3789. doi: 10.1021/bi00065a034. [DOI] [PubMed] [Google Scholar]
- 11.Gegg C V, Bowers K E, Matthews C R. Protein Sci. 1997;6:1885–1892. doi: 10.1002/pro.5560060909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones B E, Beechem J M, Matthews C R. Biochemistry. 1995;34:1867–1877. doi: 10.1021/bi00006a007. [DOI] [PubMed] [Google Scholar]
- 13.Kuwajima K, Garvey E P, Finn B E, Matthews C R, Sugai S. Biochemistry. 1991;30:7693–7703. doi: 10.1021/bi00245a005. [DOI] [PubMed] [Google Scholar]
- 14.Creighton T E. Biochem J. 1990;270:1–16. doi: 10.1042/bj2700001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clementi C, Jennings P A, Onuchic J N. Proc Natl Acad Sci USA. 2000;97:5871–5876. doi: 10.1073/pnas.100547897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Capaldi A P, Ferguson S J, Radford S E. J Mol Biol. 1999;286:1621–1632. doi: 10.1006/jmbi.1998.2588. [DOI] [PubMed] [Google Scholar]
- 17.Raschke T M, Kho J, Marqusee S. Nat Struct Biol. 1999;6:825–831. doi: 10.1038/12277. [DOI] [PubMed] [Google Scholar]
- 18.Tsui V, Garcia C, Cavagnero S, Suizdak G, Dyson H J, Wright P E. Protein Sci. 1999;8:45–49. doi: 10.1110/ps.8.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jennings P A, Dyson H J, Wright P E. In: Statistical Mechanics, Protein Structure, and Protein Substrate Interactions. Doniach S, editor. New York: Plenum; 1994. pp. 7–18. [Google Scholar]
- 20.Bai Y. Proc Natl Acad Sci USA. 1999;96:477–480. doi: 10.1073/pnas.96.2.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walkenhorst W F, Green S M, Roder H. Biochemistry. 1997;36:5795–5805. doi: 10.1021/bi9700476. [DOI] [PubMed] [Google Scholar]
- 22.Heidary D K, Gross L A, Roy M, Jennings P A. Nat Struct Biol. 1997;4:725–731. doi: 10.1038/nsb0997-725. [DOI] [PubMed] [Google Scholar]
- 23.Schmid F X. In: Protein Folding. Creighton T, editor. New York: Freeman; 1992. pp. 213–221. [Google Scholar]
- 24.Connors K A. Chemical Kinetics: The Study of Reaction Rates in Solution. New York: VCH; 1990. pp. 60–77. [Google Scholar]
- 25.Jones B E, Jennings P A, Pierre R A, Matthews C R. Biochemistry. 1994;33:15250–15258. doi: 10.1021/bi00255a005. [DOI] [PubMed] [Google Scholar]
- 26.Barshop B A, Wrenn R F, Frieden C. Anal Biochem. 1983;130:134–145. doi: 10.1016/0003-2697(83)90660-7. [DOI] [PubMed] [Google Scholar]
- 27.Jones B E, Matthews C R. Protein Sci. 1995;4:167–177. doi: 10.1002/pro.5560040204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bolin J T, Filman D J, Matthews D A, Hamlin R C, Kraut J. J Biol Chem. 1982;257:13650–13662. [PubMed] [Google Scholar]
- 29.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]