

Abstract
Objective
Because ouabain activates several pathways that are critical to cardioprotective mechanisms such as ischemic preconditioning, we tested if this digitalis compound could protect the heart against ischemia-reperfusion injury through activation of the Na+,K+-ATPase/c-Src receptor complex.
Methods and Results
In Langendorff-perfused rat hearts, a short (4 min) administration of ouabain 10 μM followed by an 8-minute washout before 30 minutes of global ischemia and reperfusion improved cardiac function, decreased lactate dehydrogenase release and reduced infarct size by 40%. Western blot analysis revealed that ouabain activated the cardioprotective phospholipase Cγ1/protein kinase Cε (PLC-γ1/PKCε) pathway. Pre-treatment of the hearts with the Src kinase family inhibitor 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolol[3,4-d]pyrimidine (PP2) blocked not only ouabain-induced activation of PLC-γ1/PKCε pathway, but also cardiac protection. This protection was also blocked by a PKCε translocation inhibitor peptide (PKCε TIP).
Conclusion
Short exposure to a low concentration of ouabain protects the heart against ischemia/reperfusion injury. This effect of ouabain on the heart is most likely due to the activation of the Na+,K+-ATPase/c-Src receptor complex and subsequent stimulation of key mediators of preconditioning, namely PLC-γ1 and PKCε.
Introduction
Ouabain and other cardiac glycosides all share the property of being potent and highly specific inhibitors of the purified Na+,K+-ATPase, the ubiquitous transmembrane enzyme that transports Na+ and K+ across the plasma membrane by hydrolysis of ATP (1–3). In that context, it had been accepted for years that the physiological and pharmacological actions of these compounds were due to the inhibition of the ATP-driven transport of Na+ and K+ ion across the cell membrane. However, in the past few years, it has become apparent that binding of ouabain and other cardiac glycosides to the Na+,K+-ATPase also triggers a series of changes in protein-protein interactions, resulting in the activation of protein kinases within the Na+,K+-ATPase signalplex (4). This has been shown to take place within the caveolar structures of various tissues and different types of cells where the Na+,K+-ATPase and c-Src interact and form a receptor complex (5–8). Binding of ouabain to this receptor complex activates the Na+,K+-ATPase-associated Src, resulting in the activation of multiple signaling cascades. For instance, the ouabain-activated Na+,K+-ATPase/Src complex is able to recruit and activate PLC-γ1 in LLC-PK1 cells (9). Significantly, activation of this particular pathway has been demonstrated in the cultured neonatal cardiac myocytes (10) and is apparently associated with ouabain-induced inotropy in isolated heart preparations (11).
There is strong evidence that induction of myocardial protection against ischemia/reperfusion injury by a treatment given before the onset of ischemia, known as cardiac preconditioning, is a receptor-mediated process that involves the activation of Src, ERKs, and PKC isozymes (12). Specifically, activation of PLC/PKCε pathway has been demonstrated during ischemic preconditioning as well as in many G protein-coupled receptor-mediated preconditioning (13–16). Interestingly, there is evidence that direct activation of PKCε via genetic manipulation is sufficient to protect the heart from ischemia/reperfusion injury (15). Ouabain at inotropic doses (50–100 μM) activates Src and increases overall PKC activity in the isolated heart (10–11). However, it is not known whether lower doses, with little or no effect on contractility, would be capable of activating Src and PKC in this model. Accordingly, as a first step towards an understanding of the physiological and pharmacological impact of the newly appreciated signaling function of the Na+,K+-ATPase, we tested whether a low dose of ouabain (10 μM) can trigger cardiac preconditioning by activation of the Src/PLC-γ1/PKCε pathway in the Langendorff-perfused rat heart.
Methods
Isolated Perfused Rat Heart Model
Experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institute of Health (NIH Publication No. 85–23, revised 1996). Twelve weeks old Male Sprague Dawley rats (350–400 g) were anesthetized with 50 mg pentobarbital sodium injected intraperitoneally. Hearts were rapidly removed and plunged into ice cold (4°C) Krebs-Henseleit solution. Within 40 sec, hearts were perfused in Langendorff mode with an oxygenated Krebs-Henseleit solution containing (in mmol/L) NaCl (118.0), KCl (4.0), CaCl2 (1.8), KH2PO4 (1.3), MgSO4 (1.2), Ethylene glycol-bis (2-aminoethylether)-N, N, N′, N′-tetraacetic acid (0.3), NaHCO3 (25), D-glucose (11). The gas used for oxygenation was a mixture of O2 (95%) and CO2 (5%), resulting in a pO2 of 618 mm Hg, a pCO2 of 30 mm Hg and a pH of 7.5. The coronary flow was kept constant at a rate initially set to obtain a coronary perfusion pressure of about 100 mm Hg (15 ml min−1, 17). A drainage cannula was inserted into the apex of the left ventricular cavity to vent the Thebesian flow. Isovolumic left ventricular developed pressure (LVDP, the difference between systolic and diastolic left ventricle pressures) was measured by inserting a water-filled latex balloon into the left ventricle connected to a P23XL Becton Dickinson pressure transducer, a CP122 AC/DC strain gage amplifier and a Grass Telefactor recording system. The end diastolic pressure (EDP) was adjusted initially at 10 mm Hg. Hearts were paced at 4.5 Hz and the latex-balloon was kept inflated throughout the experiment, including during ischemia. The temperatures of the buffer and isolated heart were maintained at 37°C with a temperature-controlled, circulating water bath. After 10 to 15 minutes of equilibration, the experiment began, and the left ventricular developed pressure (LVDP) and coronary perfusion pressure were monitored throughout the experiment.
Choice of Ouabain Dose
The dose of 10 μM was chosen based on pharmacological and physiological considerations. We wanted to test a model where the observed effect of ouabain on contractility would be minimal, to keep the interpretation of potential protection as straightforward as possible. According to previous studies (18), the dose of 10 μM met this requirement.
Experimental Protocol
Six groups of hearts were studied. The ischemia-reperfusion group (IR) was perfused for 20 min, subjected to 30-min zero-flow ischemia, and then reperfused for 120 min. In the ouabain ischemia-reperfusion group (OIR), ouabain 10 μM was added for 4 min, 12 min before the onset of ischemia. The PP2+OIR and PP2+IR groups were perfused for 20 min with 2 μM of the c-Src family kinase inhibitor 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolol[3,4-d]pyrimidine (PP2, Calbiochem, San Diego, CA) in the presence or absence of ouabain, respectively. The TIP+OIR and TIP+IR groups were perfused for 20 min with 5 μM of the protein kinase C epsilon (PKCε) translocation inhibitor peptide EAVSLKPT (TIP, Calbiochem, San Diego, CA) in the presence or absence of ouabain, respectively. After ischemia, all hearts were reperfused with standard Krebs-Henseleit solution. As indicated in Figure 1, left ventricles for analysis of PLCγ1 and PKCε phosphorylation/activities were frozen at time 12 min (n=3/group). LDH release in coronary effluent and contractile function were monitored until time 80 min. At time 170 min, hearts were processed for infarct size measurements (n=5–7/group).
Figure 1. Experimental protocols.
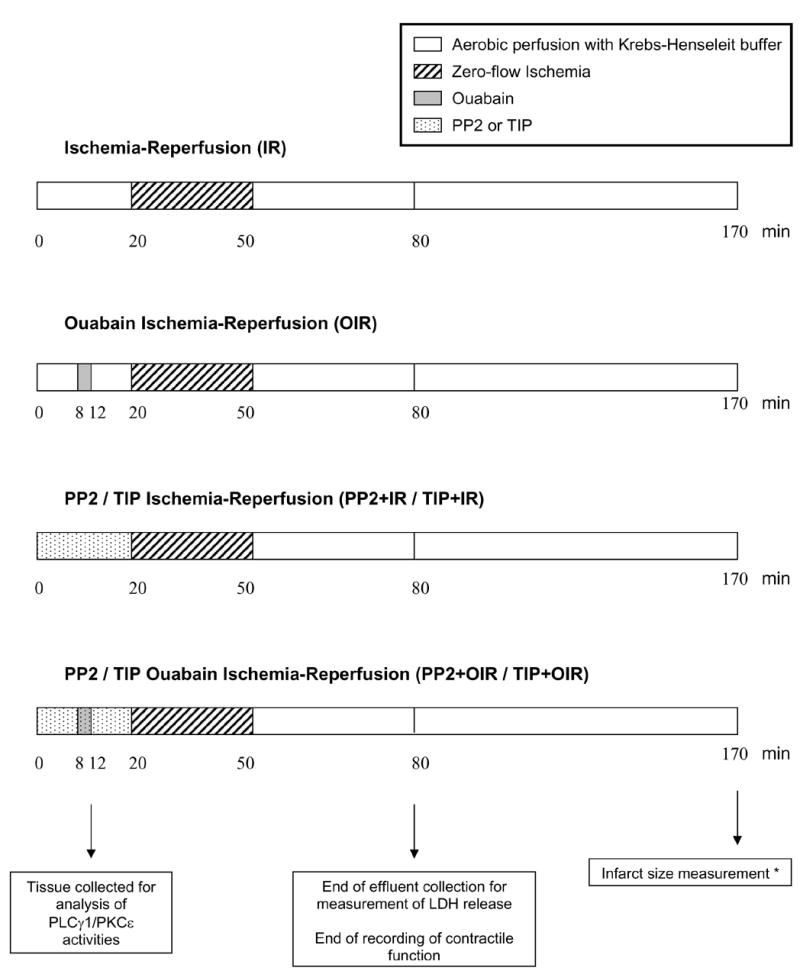
Timing of interventions is shown in relation to ischemia. Ouabain was perfused at 10 μM. The c-Src kinase inhibitor PP2 (4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolol[3,4-d]pyrimidine, 2 μM) and the (PKCε) translocation inhibitor peptide (TIP, 5 μM) were given before, during and after ouabain. After ischemia, all hearts were reperfused with standard Krebs-Henseleit solution. * Except for TIP groups.
Lactate dehydrogenase (LDH) activity measurement
Coronary effluent was collected for 30 sec at time 20 min (just before the onset of ischemia), and then at 0, 1, 2, 3, 4, 5, 10, 15, 20, and 30 min of reperfusion. LDH activity was determined colorimetrically using a standard assay (TOX 7 kit, Sigma, Saint-Louis, MO, USA), according to the manufacturer recommendation.
Tissue preparation, SDS-PAGE, immunoprecipitation and immunoblotting
For each experimental group, 3 hearts were quick-frozen in liquid nitrogen after 12 min of perfusion according to their respective protocols (Figure 1). One hundred mg of powdered left ventricle was placed into a ice-cold buffer containing 30 mM histidine, 250 mM sucrose, 1 mM EDTA, 1mM phenylmethylsulfonyl fluoride, 1 mM sodium orthovanadate, 1 mM NaF, 10 nM okadaic acid, 10 μg/ml aprotinin, 10 μg/ml leupeptin and then homogenized in a 30ml homogenizer by repeating 5 times a series of 8 up-and-down strokes separated by 30 sec intervals. For immunoprecipitation experiments, homogenates were diluted 1 to 3 in an ice-cold solution containing 1% Nonidet P40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA, 1mM phenylmethylsulfonyl fluoride, 1 mM sodium orthovanadate, 1 mM NaF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 50 mM Tris-HCl (pH 7.4) (11). Lysates were centrifuged at 16,000 X g for 15 min, and supernatants (2mg) were immunoprecipitated using an anti-PKCε rabbit polyclonal or an anti-PLC-γ1 monoclonal- antibody (both from Santa Cruz, CA). The immunoprecipitates were dissolved in 2X Laemmli sample buffer, boiled for 5 min, separated on 10% SDS-PAGE, transferred to a nitrocellulose membrane and probed with anti-phospho-PKCs (pan, Cell Signaling Technology, Beverly, MA) or anti-phospho-PLC-γ1 antibody (Santa Cruz). To assay PKCε translocation from cytosolic to particulate fraction, the same homogenates were centrifuged at 100,000 X g for 1h at 4°C. The supernatant designated as the cytosolic fraction was removed and saved. The pellet was homogenized in the solution containing 1% Triton and the particulate fraction was prepared as we previously described (11).
Histomorphometry
After 120 min of reperfusion, hearts were frozen (−20°C, 2h), sliced into 3 mm thick transverse sections and incubated in triphenyltetrazolium chloride solution (TTC, 1% in phosphate buffer, pH 7.4) at 37°C for 20 min, then further incubated for 20 min in formalin. TTC stains the viable tissue in a bright-red color, which allows the discrimination between viable (red) and nonviable (pale yellow) tissue. Slices were electronically scanned and subsequently analyzed using Image J (version 1.32j) software (National Institutes of Health, USA http://rsb.info.nih.gov/ij/). The amount of necrosis was then estimated with a macro modified from reference (19) to detect poor uptake of TTC. Infarct size was expressed as a percentage of the risk zone (equivalent to total cardiac muscle mass), as reported previously (20).
Statistical Analysis
Differences between untreated and pharmacologically treated groups were analyzed by one-way ANOVA followed by Tukey’s multiple comparison post hoc test. When 2 groups were compared, unpaired bilateral student’s t test was used. P<0.05 was considered as statistically significant.
Results
Cardiac Function
We first determined the effect of ouabain exposure on the recovery of cardiac function. Preliminary experiments on continuously perfused Langendorff rat hearts showed that after a 10–15 min equilibration period, cardiac performance was stable with a decrease < 10% over the next 80-min perfusion period (data not shown). Baseline parameters were identical in all groups (Figures 2 and 6). Four min infusion of 10 μM ouabain caused a small and transient increase in LVDP (<10%, time = 8min, Figure 2), followed by a subsequent recovery to baseline value during washout (time = 12 to 20 min, Figure 2). This is in accordance with the fact that ouabain dissociates from the Na+,K+-ATPase α1 isoform rapidly when the drug is removed from the perfusate. As expected, ischemia resulted in cardiac arrest in all groups. Although cardiac performance resumed within seconds of reperfusion, it was significantly compromised in all groups as shown by the significant decrease in LVDP (time = 55 min, upper panel of Figure 2) and the increase in EDP (time = 55 min, lower panel of Figure 2). However, a significant protection by ouabain pre-treatment was clearly detected at 10 min of reperfusion and after (time = 60 to 80 min), and the recovery in the ouabain-treated group was faster and greater. At the end of the 30 min reperfusion, the LVDP recovery was 21.3 ± 3.0% of the pre-ischemic value in control hearts and 75.7 ± 9.8% in ouabain-treated hearts (P<0.001, Figure 2). To determine if the ouabain effects on cardiac performance were mediated by the activation of Src, PP2 was administered before, during and after ouabain perfusion. This treatment significantly reduced ouabain-induced improved recovery of LVDP and EDP (Figure 2).
Figure 2. Effect of Ouabain and PP2 on Myocardial Function.
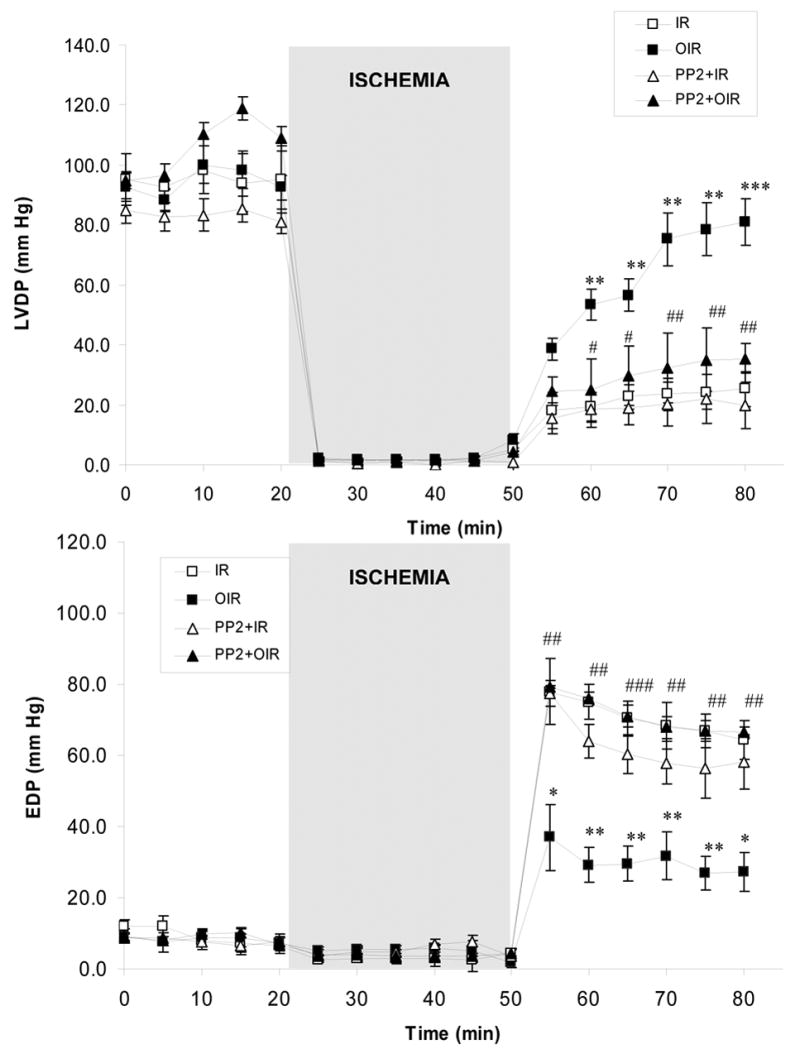
Left ventricular developed pressure (LVDP, upper panel) and end diastolic pressure (EDP, lower panel) were measured in Langendorff-perfused hearts submitted to no treatment (IR, □), ouabain (OIR, ▪), PP2 (PP2+IR, ▵) or PP2+ouabain (PP2+OIR, ▴). Values are means ± SEM of 5–7 independent experiments for each group. ** P<0.01 and *** P<0.001 vs. IR. ## P<0.01 and ### and P<0.001 vs. OIR.
Figure 6. Effect of PKCε inhibition on ouabain-induced preconditioning.
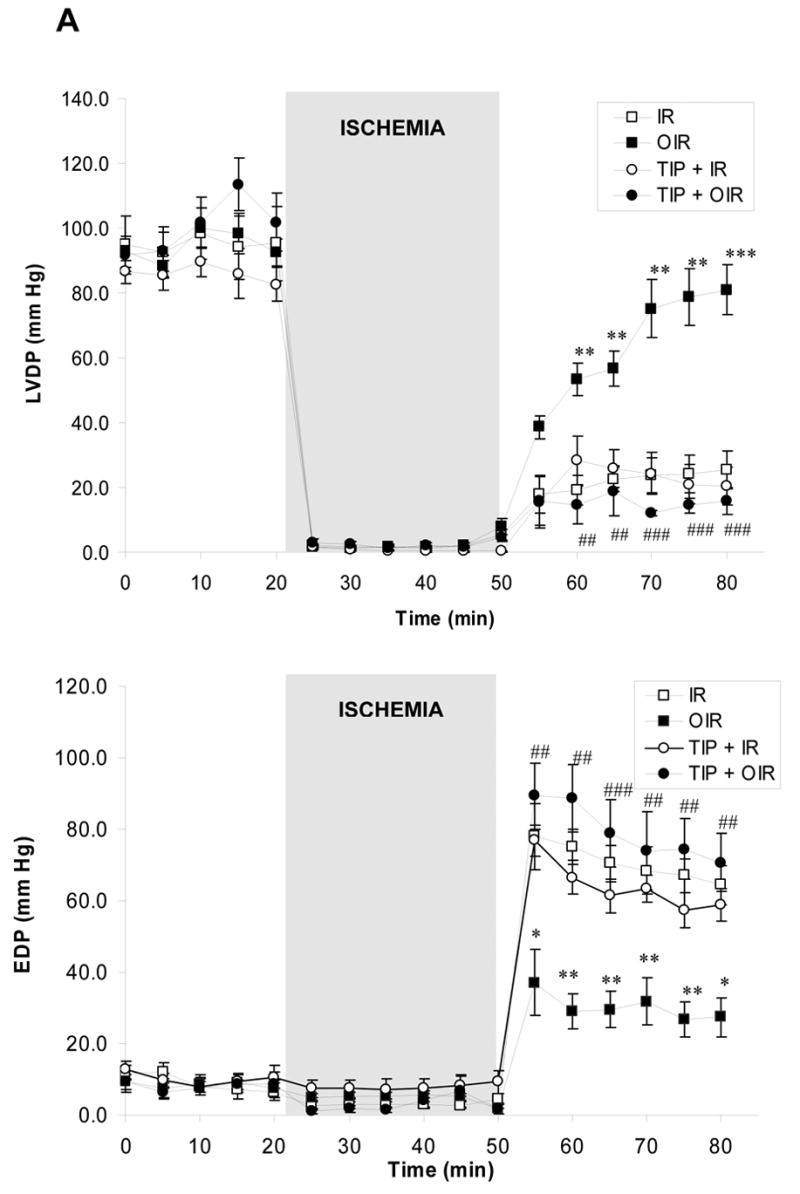
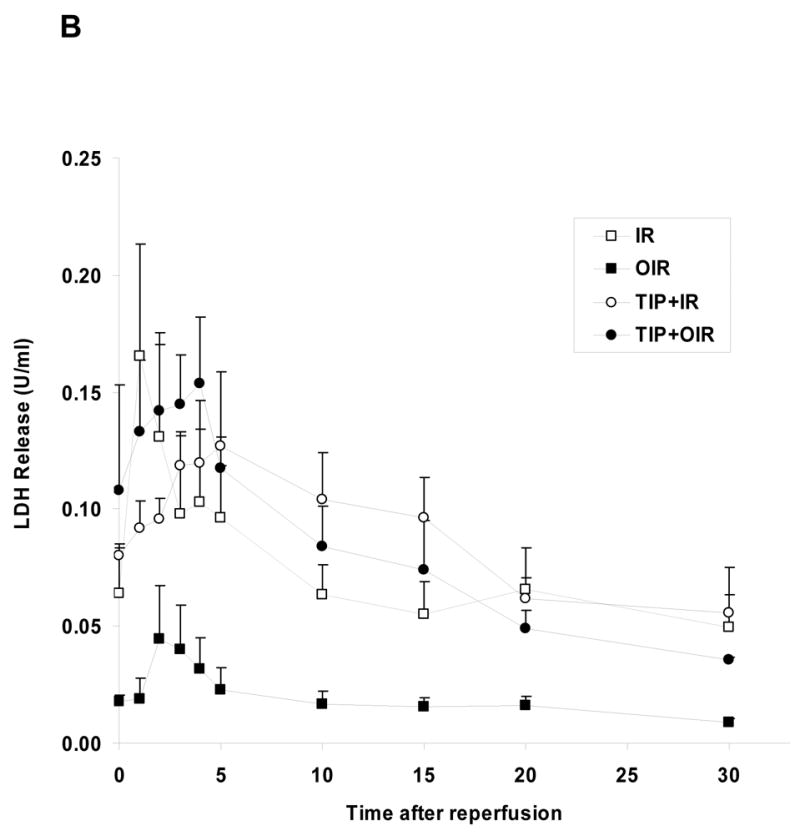
A. Left ventricular developed pressure (LVDP, upper panel) and end diastolic pressure (EDP, lower panel) were measured in Langendorff-perfused hearts submitted to no treatment (IR, □), ouabain (OIR, ▪), TIP (○) or TIP+ouabain (TIP+OIR, ●). * P<0.05, ** P<0.01, and *** P<0.001 vs. IR. ## P<0.05, ## P<0.01, and ### P<0.001 vs. OIR. B.LDH release over the 30 minutes of reperfusion was IR: 32.3 ± 8.4 U, OIR: 8.0 ± 2.6 U (P<0.05 vs. IR), TIP+IR: 39.0 ± 7.7 U and TIP+OIR: 34.7 ± 6.4 U (P<0.05 vs. OIR). Values are means ± SEM of 5–7 independent experiments for each group.
Enzyme release (LDH)
Coronary effluent was collected as detailed in the Methods section. The total LDH release over the 30 minutes of reperfusion (calculated from areas under the curves presented in Figure 3) was 32.3 ± 8.4 U in the untreated group. Ouabain treatment reduced LDH release to 8.0 ± 2.6 U (P<0.05). PP2 had no significant effect by itself (24.0 ± 8.6 U) but completely blocked the protective effect of ouabain on LDH release (15.1 ± 1.8 U, P<0.05 vs. OIR).
Figure 3. Effect of Ouabain and PP2 on LDH release.
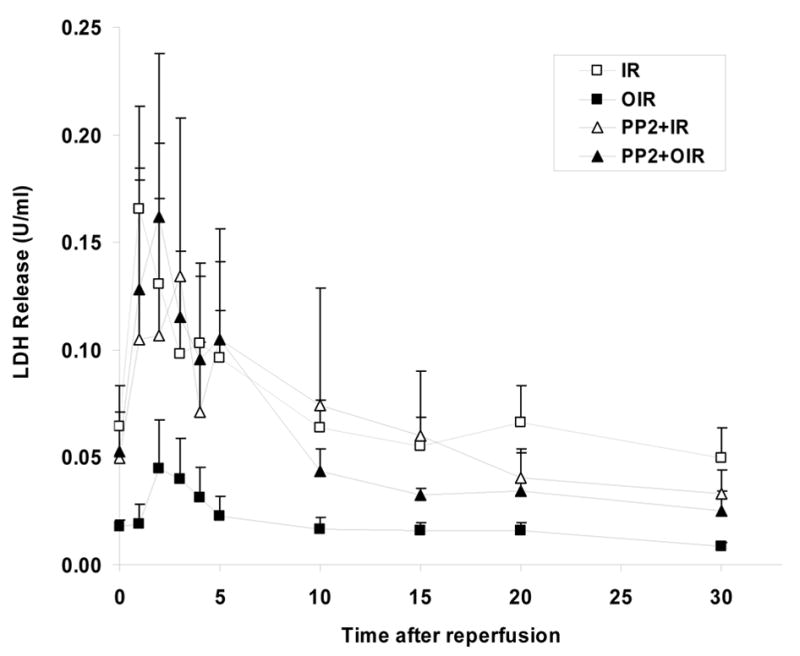
LDH release over the 30 minutes of reperfusion was IR: ischemia/reperfusion (□), 32.3 ± 8.4 U, OIR: ouabain + ischemia/reperfusion (▪): 8.0 ± 2.6 U (P<0.05 vs. IR), PP2+IR: PP2 + ischemia/reperfusion (▵), 24.0 ± 8.6 U and PP2+OIR: PP2 + ouabain + ischemia/reperfusion (▴) 15.1 ± 1.8 U (P<0.05 vs. OIR).
Tissue Necrosis
TTC staining in 5–7 samples demonstrated that 30 minutes of global ischemia followed by 2h of reperfusion in the untreated heart brought about the infarction of 43.5 ± 1.3 % of the area at risk. As shown in Figure 4, ouabain induced a protective effect. In those hearts, infarct size (26.1 ± 2.3 % of the area at risk) was reduced by 40% with respect to that of untreated hearts (P<0.001). PP2 treatment did not modify the infarct size by itself but blocked ouabain effect.
Figure 4. Effect of Ouabain and PP2 on Infarct Size.
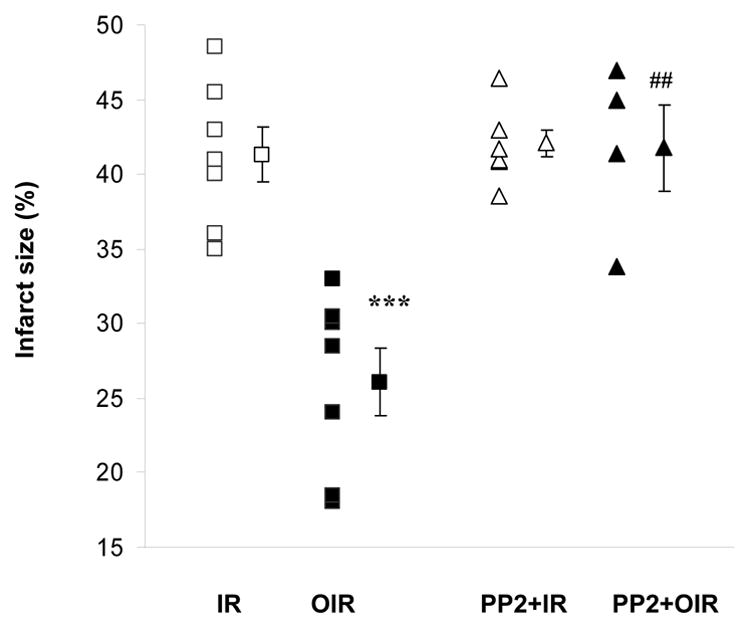
Infarct size expressed as a percentage of the risk zone was measured after 120 min of reperfusion. IR: ischemia/reperfusion (□); OIR: ouabain + ischemia/reperfusion (▪); PP2+IR: PP2 + ischemia/reperfusion (▵); PP2+OIR: PP2 + ouabain + ischemia/reperfusion (▴). Shown are individual experimental data and means ± SEM of 5–7 independent experiments for each group. *** P<0.001 vs IR, ##P<0.001 vs. OIR.
Role of PKCε activation in ouabain-induced protection
We have shown that sustained treatment with ouabain at inotropic doses (50–100 μM, 10–25 min without washout) activates c-Src and PKCs in the isolated heart (11) and more specifically c-Src/PKCε in cultured cardiac myocytes (10). Recently, we also reported that ouabain stimulated Src/PLC-γ pathway in cultured LLC-PK1 cells (9). These previous observations led to the hypothesis that preconditioning treatment (low dose and transient) applied in the present study may stimulate the Src/PLC-γ/PKCε pathway in the isolated heart, resulting in cardiac protection. To test our working hypothesis, we first measured the effect of ouabain on PKCε translocation from the cytosolic to the particulate fraction as described in Methods. As shown in Figure 5, ouabain induced a significant increase in PKCε translocation. PP2 reversed ouabain effect (Figure 5). Concomitantly, both PLC-γ1 and PKCε phosphorylation were activated by Arabian. These effects were reversed in the presence of PP2 (not shown). To directly address the role of PKCε activation in ouabain-induced protection, we used the PKCε specific translocation inhibitor peptide TIP as previously described (21). TIP did not have a significant effect by itself but totally blocked ouabain-induced PKCε translocation (Figure 5). Likewise, TIP did not significantly modify the amount of LDH released over the 30 min of reperfusion (39.0 ± 7.7 U for TIP + IR, P>0.05 vs. 32.3 ± 8.4 U for IR, Figure 6B), but totally prevented the ouabain-induced decrease (34.7 ± 6.4 U for TIP + OIR, P<0.05 vs. OIR, Figure 6B). Finally, in terms of recovery of cardiac function, no effect of TIP itself was observed. However, TIP treatment prevented ouabain-induced preconditioning as shown by a limited recovery of LVDP and EDP, similar to the untreated group (Figure 6A). Although infarct size was not measured, these data strongly suggest that ouabain-induced activation the Src/PLC-γ/PKCε pathway in the isolated heart preparation is critical to ouabain-induced preconditioning.
Figure 5. Effect of Ouabain and PP2 and TIP on PKCε activation.
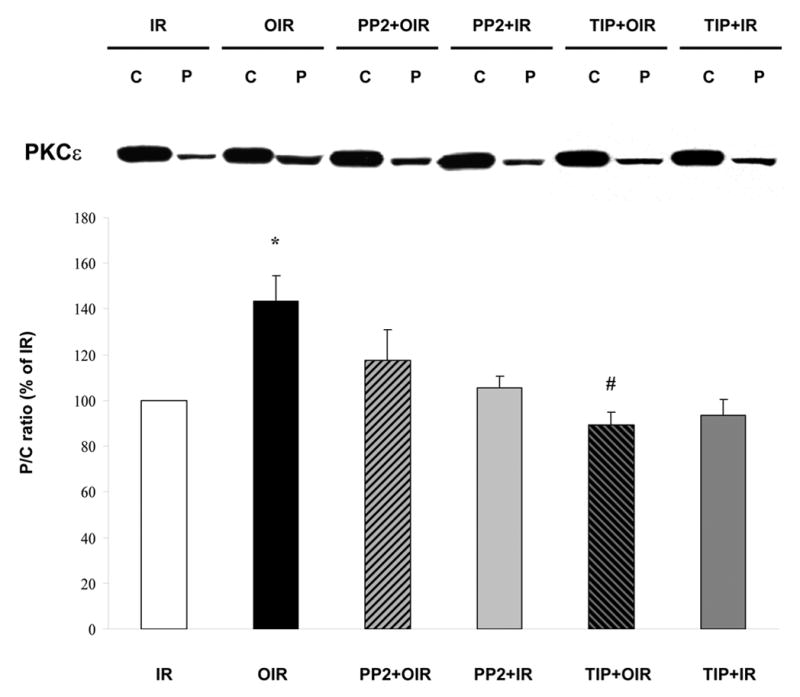
Hearts were perfused for 12 min according to the protocols described in figure 1. Cytosolic (C) and particulate (P) fractions obtained from tissue lysates were then assayed and compared for their contents in PKCε. IR: ischemia/reperfusion; OIR: ouabain + ischemia/reperfusion; PP2+IR: PP2 + ischemia/reperfusion; PP2+OIR: PP2 + ouabain + ischemia/reperfusion. TIP: PKCε-specific translocation inhibitor peptide. TIP+IR: TIP + ischemia/reperfusion; TIP+OIR: TIP + ouabain + ischemia/reperfusion. Upper panel: representative western blot. Lower panel: means ± SEM of 3 separate experiments. *P<0.01 vs IR. # P<0.01 vs OIR.
Discussion
Using the rat Langendorff-perfused isolated heart model, the present study shows that ouabain induces myocardial protection against ischemia/reperfusion injury. This is documented by an improved recovery of cardiac function, a reduction in cardiac enzyme release and a reduction of tissue necrosis. The protection occurred at the low dose of 10 μM, required c-Src activation and involved the activation of the cardioprotective PLC-γ1/PKCε. This suggests the signaling function of the Na/K-ATPase/c-Src receptor complex as the cellular mechanism of this effect.
Ouabain Preconditioning vs. Ischemic Preconditioning (IP)
IP was the first described and remains the best-characterized form of preconditioning. It is defined as a protective mechanism whereby hearts exposed to brief, sublethal ischemic insults are more resistant against subsequent prolonged ischemia, reducing myocellular death and postischemic cardiac dysfunction (22). Being the first attempt to demonstrate a preconditioning effect of the digitalis ouabain, the present study was designed to allow comparison with previous studies on ischemic injury and/or IP. Accordingly, the protocol described in Figure 1 was established in a widely used model of rat isolated heart, exposed to 30 minutes of ischemia and 30 minutes of reperfusion. The recovery of cardiac mechanical function usually reported ranges from 4% to 40% (23–27) of the initial value in the absence of preconditioning. We observed a recovery of 21.3 ± 3% of the LVDP, suggesting that this protocol allowed us to characterize a protection against a fairly standard and previously reported extend of injury. In comparable protocols, the reported recovery of mechanical function when the heart is preconditioned ischemically prior to the sustained ischemia typically reaches 60 to 90% (25–27). Therefore, the recovery of 75.7 ± 9.8 % afforded by ouabain 10 μM is quite super-imposable to the reported effect of IP. Like the protection afforded by IP, the effects of ouabain on EDP and myocellular death were significant. The latter was revealed by the significant decrease of LDH release over the course of the reperfusion and the reduction by about 40% of the infarct size compared with the untreated ischemic group.
Initiation of the ouabain Preconditioning Effect: Pumping vs. Signaling
Applying a new concept (signaling through Na+,K+-ATPase) to an old drug (ouabain) led us to this unexpected yet highly physiologically relevant finding. Indeed, the rationale for this study was clearly based on striking similarities observed between cardiac preconditioning- and ouabain-signaling cascades, both newly characterized and still under intensive investigation. However, based on the classical role of the cardiac enzyme and previous reports on digitalis/Na+,K+-ATPase in ischemia/reperfusion injury, exposure to a Na+,K+-ATPase inhibitor, even at a very low concentration, may not have been expected to induce myocardial protection. Indeed, since the pioneer work of Beller et al (28), alteration of myocardial Na+,K+-ATPase activity and remodeling have been repeatedly reported as one key adverse consequence of ischemic injury (26; 29–30). Although the exact mechanism is still under investigation (30–32), this alteration is critical in the intracellular Na+ accumulation and Ca2+ overload during ischemia and reperfusion, both of which are closely related to the outcome of myocardial damage. In apparent contradiction with the paradigm that we chose to investigate are the facts that 1) IP preserves myocardial Na+,K+-ATPase activity after ischemia/reperfusion injury (26; 33) and 2) high concentrations of ouabain given after IP blocked the protective effect of IP (34). Three important differences in the present study may explain the apparently contradictory outcome. First, we used a low concentration of ouabain (20 times lower than the above-mentioned study, ref 34). Second, the exposure was transient (4 min) and third, ouabain was given and washed out (consistent with the rapid dissociation rate of ouabain when bound to the rodent α1 isoform of Na+,K+-ATPase) before the onset of ischemia.
By the same token, it seems important to emphasize that the mechanism investigated here is different from the one involved in the well-known positive inotropic action of digitalis. The inotropic effect has long been known to involve an inhibition of the sarcolemmal Na+,K+-ATPase leading to a significant increase in intracellular Na+ and Ca++, and in the force of contraction (35). Although the more recently described signaling cascade is essential to the ouabain’s effect on Ca++ in rat cardiac myocytes (36), the signaling cascade itself can be initiated without changes in intracellular Ca++ concentration and relies on a pathway linked to ROS production (37). In ouabain-insensitive species like rat (about 1,000 time less sensitive than human), ouabain at the dose of 10 μM induces a limited inhibition of the cardiac enzyme activity, of about 20% (38). This resulted in less than 10% changes in LVDP (inotropic effect) in the present study, in agreement with previous reports (18). In other words, the ouabain exposure that was given here (low dose and transient) was targeting the initiation of the signaling cascade rather than the ion pumping activity of the enzyme at the time of reperfusion. Therefore, this preconditioning effect of ouabain is not in contradiction with a preservation of Na+,K+-ATPase activity at the time of reperfusion.
Taken together, these findings suggest that while ouabain-induced inotropy requires continued occupation of the receptor, its effect on preconditioning is solely dependent on the activation of the signaling cascade. In other words, once activated, this signaling cascade will lead to the activation of PLC-γ1/PKCε in the absence of continued receptor occupancy. Admittedly, important issues such as the role played by downstream intracellular messengers (ROS and Ca2+) or differences in the mechanisms triggered by continuous vs transient Na+,K+-ATPase receptor occupancy need to be further addressed in future studies.
Downstream effectors of the ouabain Preconditioning Effect
We have recently demonstrated that Na+,K+-ATPase and c-Src interact and form a receptor complex (8). Binding of ouabain to this receptor complex activates Src, leading to the recruitment and tyrosine phosphorylation of PLC-γ1 in pig kidney cells (9). Here we demonstrated that ouabain could also activate the Src/PLC-γ1 cascade in the isolated heart preparation. In addition, we observed that ouabain increased phosphorylation of PKCε in a Src-dependent manner. Because phosphorylation is required for PKCε translocation, it is conceivable that the concerted effects of ouabain on PKCε phosphorylation and on DAG generation via the activated PLC-γ1 shall lead to a full activation of PKCε in the isolated heart. This conclusion is consistent with the PKCε translocation data (Figures 5 and 6) and with our prior observation that inotropic doses of ouabain were sufficient to stimulate both c-Src and PKCs in the isolated hearts (11). Others have shown that the formation of c-Src/PKCε signaling module is an important step in IP (39). Activation of PKCε facilitates the formation of this complex leading to c-Src activation, which places PKCε upstream from c-Src at least in rabbit species (40). Evidence of a c-Src-dependant activation of PKCε, more in line with the present report, has also been reported (41). The current vision seems therefore to include the involvement of a c-Src-dependent and a c-Src-independent mechanism of PKCε activation, probably taking place in different subcellular compartments (membrane vs cytoplasmic) in IP. The present data suggest that the ouabain-induced cardioprotective effect is mediated primarily by the formation of a Na+,K+-ATPase/c-Src/PLC-γ1 tertiary complex preceding PKCε activation. In addition, it should be mentioned that opening of mitochondrial KATP channel and increases in ROS production may also be important ouabain-induced cardioprotective mediators linked to the PLC-γ1/PKCε pathway (42–43). Clearly, these issues need to be addressed in future studies.
Continuing research is providing an increasing number of agents capable of preconditioning the heart. Unfortunately, their translation into clinical therapy involves obvious practical challenges that increase the likelihood of significant unanticipated risks (44–45). Our data suggesting an effect of digitalis on resistance to ischemia/reperfusion injury raises the possibility that this well-studied drug could be used as a preconditioning agent. Clinicians have accumulated a large body of experience and expertise in the use of digitalis under numerous physiological conditions and dosing regimens (46–48). Nevertheless, the administration of digitalis for preconditioning in human patients is complicated by such obvious concerns as the differences in sensitivity to the drug exhibited by rodents and primates, as well as the distinction between the acute exposures used in this work and the typically chronic exposures in patients. Clearly, further study is needed to determine if an extrapolation to humans is warranted, but the present findings raise the possibility of including this traditional drug in a regimen for reducing ischemia/reperfusion injury.
Acknowledgments
The authors gratefully acknowledge Dr Benedict R. Lucchesi, Department of Pharmacology, University of Michigan, for his support with the calibration of the isolated heart perfusion experiments and Dr Joseph I. Shapiro, Department of Internal Medicine, Medical University of Ohio, for his help with the assessment of the infarct area. Jennifer Seminerio was supported by an Institutional Medical Student Summer Research Award. This research was supported by grants HL67963, HL36573 and HL67842 from the National Heart, Lung, and Blood Institute and by a grant from Inserm.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Skou JC. Nobel Lecture. The identification of the sodium pump. Biosci Rep. 1998;18:155–69. doi: 10.1023/a:1020196612909. [DOI] [PubMed] [Google Scholar]
- 2.Lingrel JB, Kuntzweiler T. Na+,K+-ATPase. J Biol Chem. 2002;263:19659–62. [PubMed] [Google Scholar]
- 3.Kaplan JH. Biochemistry of Na+,K+-ATPase. Annu Rev Biochem. 2002;71:511–35. doi: 10.1146/annurev.biochem.71.102201.141218. [DOI] [PubMed] [Google Scholar]
- 4.Xie Z, Cai T. Na+,K+-ATPase-mediated signal transduction: from protein interaction to cellular function. Mol Interv. 2003;3:157–68. doi: 10.1124/mi.3.3.157. [DOI] [PubMed] [Google Scholar]
- 5.Liu L, Mohammadi K, Aynafshar B, Wang H, Li D, Liu J, et al. Role of caveolae in signal-transducing function of cardiac Na+,K+-ATPase. Am J Physiol Cell Physiol. 2003;284:C1550–60. doi: 10.1152/ajpcell.00555.2002. [DOI] [PubMed] [Google Scholar]
- 6.Wang H, Haas M, Liang M, Cai T, Tian J, Li S, et al. Ouabain assembles signaling cascades through the caveolar Na+,K+-ATPase. J Biol Chem. 2004;279:17250–9. doi: 10.1074/jbc.M313239200. [DOI] [PubMed] [Google Scholar]
- 7.Liu L, Abramowitz J, Askari A, Allen JC. Role of caveolae in ouabain-induced proliferation of cultured vascular smooth muscle cells of the synthetic phenotype. Am J Physiol Heart Circ Physiol. 2004;287:H2173–82. doi: 10.1152/ajpheart.00352.2004. [DOI] [PubMed] [Google Scholar]
- 8.Tian J, Cai T, Yuan Z, Wang H, Liu L, Haas M, et al. Binding of Src to Na+,K+-ATPase Forms a Functional Signaling Complex. Mol Biol Cell. 2006;17:317–26. doi: 10.1091/mbc.E05-08-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuan Z, Cai T, Tian J, Ivanov AV, Giovannucci DR, Xie Z. Na+,K+-ATPase Tethers Phospholipase C and IP3 Receptor into a Calcium-regulatory Complex. Mol Biol Cell. 2005;16:4034–45. doi: 10.1091/mbc.E05-04-0295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mohammadi K, Komeniati P, Xie Z, Askari A. Role of protein kinase C in the signal pathways that link Na+,K+-ATPase to ERK 1/2. J Biol Chem. 2001;276:42050–6. doi: 10.1074/jbc.M107892200. [DOI] [PubMed] [Google Scholar]
- 11.Mohammadi K, Liu L, Tian J, Kometiani P, Xie Z, Askari A. Positive inotropic effect of ouabain on isolated heart is accompanied by activation of signal pathways that link Na+,K+-ATPase to ERK1/2. J Cardiovasc Pharmacol. 2003;41:609–14. doi: 10.1097/00005344-200304000-00014. [DOI] [PubMed] [Google Scholar]
- 12.Armstrong SC. Protein kinase activation and myocardial ischemia/reperfusion injury. Cardiovasc Res. 2004;61:427–36. doi: 10.1016/j.cardiores.2003.09.031. [DOI] [PubMed] [Google Scholar]
- 13.Munakata M, Stamm C, Friehs I, Zurakowski D, Cowan DB, Cao-Danh H, et al. Protective effects of protein kinase C during myocardial ischemia require activation of phosphatidil-inositol specific phospholipase C. Ann Thorac Surg. 2002;73:12236–45. doi: 10.1016/s0003-4975(01)03594-9. [DOI] [PubMed] [Google Scholar]
- 14.Baines CP, Zhang J, Wang GW, Zheng YT, Xiu JX, Cardwell EM, et al. Mitochondrial PKCε and MAPK form signaling modules in the murine heart. Enhanced mitochondrial PKCγ-MAPK interactions and differential MAPK activation in PKCε-induced cardioprotection. Circ Res. 2002;90:390–7. doi: 10.1161/01.res.0000012702.90501.8d. [DOI] [PubMed] [Google Scholar]
- 15.Inagaki K, Hahn HS, Dorn IIGW, Mochly-Rosen D. Additive protection of the ischemic heart ex vivo by combined treatment with δ-protein kinase C inhibitor and ε-protein kinase C activator. Circulation. 2003;108:869–75. doi: 10.1161/01.CIR.0000081943.93653.73. [DOI] [PubMed] [Google Scholar]
- 16.McCarthy J, McLeod CJ, Minners J, Essop MF, Ping P, Sack MN. PKCepsilon activation augments cardiac mitochondrial respiratory post-anoxic reserve-a putative mechanism in PKCepsilon cardioprotection. J Mol Cell Cardiol. 2005;38:697–700. doi: 10.1016/j.yjmcc.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 17.Sutherland FJ, Shattock MJ, Baker KE, Hearse DJ. Mouse isolated perfused heart: characteristics and cautions. Clin Exp Pharmacol Physiol. 2003;30:867–78. doi: 10.1046/j.1440-1681.2003.03925.x. [DOI] [PubMed] [Google Scholar]
- 18.Favereau X, Dunica S, Chevalier B, Mouas C, Swynghedeauw B. The antagonistic effects of ouabain and calcium channel blockers on the contractility of the isolated rat heart. J Pharmacol. 1986;17:651–6. [PubMed] [Google Scholar]
- 19.Kennedy DJ, Vetteth S, Periyasamy SM, Kanj M, Fedorova L, Khouri S, et al. Central role for the cardiotonic steroid marinobufagenin in the pathogenesis of experimental uremic cardiomyopathy. Hypertension. 2006;47:488–95. doi: 10.1161/01.HYP.0000202594.82271.92. [DOI] [PubMed] [Google Scholar]
- 20.Ytrehus K, Liu Y, Tsuchida A, Miura T, Liu GS, Yang XM, et al. Rat and rabbit heart infarction: effects of anesthesia, perfusate, risk zone, and method of infarct sizing. Am J Physiol. 1994;267:H2383–H2390. doi: 10.1152/ajpheart.1994.267.6.H2383. [DOI] [PubMed] [Google Scholar]
- 21.Bae S, Zhang L. Gender differences in cardioprotection against ischemia/reperfusion injury in adult rat hearts: focus on Akt and protein kinase C signaling. JPET. 2005;315:1125–35. doi: 10.1124/jpet.105.090803. [DOI] [PubMed] [Google Scholar]
- 22.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–36. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 23.Dos Santos P, Kowaltowski AJ, Laclau MN, Seetharaman S, Paucek P, Boudina S, et al. Mechanisms by which opening the mitochondrial ATP- sensitive K(+) channel protects the ischemic heart. Am J Physiol Heart Circ Physiol. 2002;283:H284–95. doi: 10.1152/ajpheart.00034.2002. [DOI] [PubMed] [Google Scholar]
- 24.Penna C, Alloatti G, Cappello S, Gattullo D, Berta G, Mognetti B, et al. Platelet-activating factor induces cardioprotection in isolated rat heart akin to ischemic preconditioning: role of phosphoinositide 3-kinase and protein kinase C activation. Am J Physiol Heart Circ Physiol. 2005;288:H2512–20. doi: 10.1152/ajpheart.00599.2004. [DOI] [PubMed] [Google Scholar]
- 25.Xu ZL, Endoh H, Ishihata A, Takahashi E, Doi K. Effect of ischemic preconditioning on myocardial oxygen consumption during ischemia. J Mol Cell Cardiol. 1998;30:2165–74. doi: 10.1006/jmcc.1998.0771. [DOI] [PubMed] [Google Scholar]
- 26.Elmoselhi AB, Lukas A, Ostadal P, Dhalla NS. Preconditioning attenuates ischemia-reperfusion-induced remodeling of Na+,K+-ATPase in hearts. Am J Physiol Heart Circ Physiol. 2003;285:H1055–63. doi: 10.1152/ajpheart.00865.2002. [DOI] [PubMed] [Google Scholar]
- 27.Harrison GJ, Willis RJ, Headrick JP. Extracellular adenosine levels and cellular energy metabolism in ischemically preconditioned rat heart. Cardiovasc Res. 1998;40:74–87. doi: 10.1016/s0008-6363(98)00123-0. [DOI] [PubMed] [Google Scholar]
- 28.Beller GA, Conroy J, Smith TW. Ischemia-induced alterations in myocardial Na+,K+-ATPase and cardiac glycoside binding. J Clin Invest. 1976;57:341–50. doi: 10.1172/JCI108285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim MS, Akera T. O2 free radicals: cause of ischemia-reperfusion injury to cardiac Na+,K+-ATPase. Am J Physiol. 1987;252:H252–7. doi: 10.1152/ajpheart.1987.252.2.H252. [DOI] [PubMed] [Google Scholar]
- 30.Inserte J, Garcia-Dorado D, Hernando V, Soler-Soler J. Calpain-mediated impairment of Na+,K+-ATPase activity during early reperfusion contributes to cell death after myocardial ischemia. Circ Res. 2005;97:465–73. doi: 10.1161/01.RES.0000181170.87738.f3. [DOI] [PubMed] [Google Scholar]
- 31.Bersohn MM. Sodium pump inhibition in sarcolemma from ischemic hearts. J Mol Cell Cardiol. 1995;27:1483–9. doi: 10.1016/s0022-2828(95)90161-2. [DOI] [PubMed] [Google Scholar]
- 32.Fuller W, Eaton P, Bell JR, Shattock MJ. Ischemia-induced phosphorylation of phospholemman directly activates rat cardiac Na+,K+-ATPase. FASEB J. 2004;18:197–9. doi: 10.1096/fj.03-0213fje. [DOI] [PubMed] [Google Scholar]
- 33.Yorozuya T, Adachi N, Dote K, Nakanishi K, Takasaki Y, Arai T. Enhancement of Na+,K+-ATPase and Ca2+-ATPase activities in multi-cycle ischemic preconditioning in rabbit hearts. Eur J Cardiothorac Surg. 2004;26:981–7. doi: 10.1016/j.ejcts.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 34.Imahashi K, Nishimura T, Yoshioka J, Hideo Kusuoka. Role of intracellular Na+ kinetics in preconditioned rat heart. Circulation Res. 2001;88:1176–82. doi: 10.1161/hh1101.092139. [DOI] [PubMed] [Google Scholar]
- 35.Akera T, Brody TM. The role of Na+,K+-ATPase in the inotropic action of digitalis. Pharmacol Rev. 1997;29:187–220. [PubMed] [Google Scholar]
- 36.Tian J, Gong X, Xie Z. Signal-transducing function of Na+,K+-ATPase is essential for ouabain’s effect on [Ca2+]i in rat cardiac myocytes. Am J Physiol. 2001;281:H1899–H1907. doi: 10.1152/ajpheart.2001.281.5.H1899. [DOI] [PubMed] [Google Scholar]
- 37.Liu J, Tian J, Haas M, Shapiro JI, Askari A, Xie Z. Ouabain interaction with cardiac Na+,K+-ATPase initiates signal cascades independent of changes in intracellular Na+ and Ca2+ concentrations. J Biol Chem. 2000;275:27838–44. doi: 10.1074/jbc.M002950200. [DOI] [PubMed] [Google Scholar]
- 38.Charlemagne D, Maixent JM, Preteseille M, Lelievre LG. Ouabain binding sites and Na+,K+-ATPase activity in rat cardiac hypertrophy. J Biol Chem. 1986;261:185–9. [PubMed] [Google Scholar]
- 39.Song C, Vondriska TM, Wang GW, Klein JB, Cao X, Zhang J, et al. Molecular conformation dictates signaling module formation: example of PKCepsilon and Src tyrosine kinase. Am J Physiol Heart Circ Physiol. 2002;282:H1166–71. doi: 10.1152/ajpheart.00830.2001. [DOI] [PubMed] [Google Scholar]
- 40.Ping P, Zhang J, Zheng YT, Li RC, Dawn B, Tang XL, et al. Demonstration of selective protein kinase C-dependent activation of Src and Lck tyrosine kinases during ischemic preconditioning in conscious rabbits. Circ Res. 1999;85:542–50. doi: 10.1161/01.res.85.6.542. [DOI] [PubMed] [Google Scholar]
- 41.Hattori R, Otani H, Uchiyama T, Imamura H, Cui J, Maulik N, et al. Src tyrosine kinase is the trigger but not the mediator of ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2001;281:H1066–74. doi: 10.1152/ajpheart.2001.281.3.H1066. [DOI] [PubMed] [Google Scholar]
- 42.Tian J, Liu J, Garlid KD, Shapiro JI, Xie Z. Involvement of mitogen-activated protein kinases and reactive oxygen species in the inotropic action of ouabain on cardiac myocytes. A potential role for mitochondrial KATP channels. Mol Cell Biochem. 2003;242:181–7. [PubMed] [Google Scholar]
- 43.Garlid KD, Dos Santos P, Xie Z, Costa ADT, Paucek P. Mitochondrial potassium transport: the role of the mitochondrial ATP-sensitive K+ channel in cardiac function and cardioprotection. Biochim Biophys Acta. 2003;1606:1–21. doi: 10.1016/s0005-2728(03)00109-9. [DOI] [PubMed] [Google Scholar]
- 44.Bolli R, Becker L, Gross G, Mentzer R, Jr, Balshaw D, Lathrop DA. Myocardial protection at a crossroads: the need for translation into clinical therapy. Circ Res. 2004;95 :125–34. doi: 10.1161/01.RES.0000137171.97172.d7. [DOI] [PubMed] [Google Scholar]
- 45.Kloner RA, Rezkalla SH. Cardiac protection during acute myocardial infarction: where do we stand in 2004? J Am Coll Cardiol. 2004;44:276–86. doi: 10.1016/j.jacc.2004.03.068. [DOI] [PubMed] [Google Scholar]
- 46.Gheorghiade M, Adams KF, Jr, Colucci WS. Digoxin in the management of cardiovascular disorders. Circulation. 2004;109:2959–64. doi: 10.1161/01.CIR.0000132482.95686.87. [DOI] [PubMed] [Google Scholar]
- 47.Young JB. Whither Withering’s Legacy? Digoxin’s role in our contemporary pharmacopeia for heart failure. J Am Coll Cardiol. 2005;46:505–7. doi: 10.1016/j.jacc.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 48.Adams KF, Jr, Patterson JH, Gattis WA, O’Connor CM, Lee CR, Schwartz TA, et al. Relationship of serum digoxin concentration to mortality and morbidity in women in the digitalis investigation group trial: a retrospective analysis. J Am Coll Cardiol. 2005;46:497–504. doi: 10.1016/j.jacc.2005.02.091. [DOI] [PubMed] [Google Scholar]