

Abstract
CMV tegument protein pp65 and CMV immediate early gene product IE1 are both considered immunodominant targets of cell-mediated immunity (CMI) and potentially capable of controlling CMV infection. To better assess their role in host defense, we have constructed a novel MVA transfer vector named pZWIIA and generated a recombinant MVA (rMVA) expressing both full-length pp65 and exon4 of IE1 (pp65-IE1-MVA) at high levels, followed by the genetic removal of the bacterial marker gene used to distinguish recombinant forms. Immunogenicity evaluation indicates that pp65-IE1-MVA not only can induce robust primary CMI to both antigens in HLA A2.1 Tg mice, but also can stimulate vigorous expansion of memory T lymphocyte responses to pp65 and IE1 in PBMC of CMV-positive donors. These properties make the MVA-based vaccine ideal for the dual role of priming and boosting CMV-specific T cell immunity as a means to control CMV disease in recipients of hematopoietic cell or solid organ transplantation (HCT or SOT). pZWIIA alone or in combination with other MVA transfer vectors can be used to generate MVA based multiple-antigen vaccine which have application in vaccine development for a wide spectrum of infectious diseases and cancer.
Keywords: Vaccinia, Cytomegalovirus, Vaccine, pp65, IE1
1. Introduction
The attenuated poxvirus, Modified Vaccinia Ankara (MVA), was historically developed as a safer alternative to licensed vaccinia derivatives as a prophylactic smallpox vaccine [1]. Its favorable safety profile was established during the 1970s when it was administered to 120,000 high-risk individuals, including children and the elderly, with minimal morbidity [1]. Although MVA can infect and replicate DNA efficiently; it is avirulent and unable to produce infectious progeny in all mammals including humans [2]. Since MVA has a well-established safety record and versatility for the production of heterologous proteins, it has been developed more recently as a viral vector for vaccine development for both cancer and infectious diseases. Depending upon the antigen, it is capable of stimulating potent CMI or humoral immunity, or both. Its potential value for the clinic stems from its capacity to cause protective immunity for both microbial pathogens and tumors [3,4]. An increasing number of MVA vector-based vaccines have entered into clinical trials [5–12]. Recent studies demonstrated that administration of MVA carrying HIV-1 Gag p24/p17 to HIV-1 infected individuals was safe and feasible [13]. However, no such MVA-based cytomegalovirus (CMV) vaccine has yet to reach the clinic.
Human CMV continues to be a major risk factor in patients undergoing allogeneic HCT and SOT despite advancement of antiviral therapy. HCT and SOT patients require CMV-specific CMI to control viral replication as a prerequisite to remain disease free [14–16]. Recent studies using a whole genome proteomic approach have confirmed that pp65 and IE1 are immunodominant, and recognized more frequently in healthy volunteers than most other less well immunologically characterized antigens [17]. Similar to healthy adults, CMV tegument protein pp65 and CMV IE1 are frequently recognized by human CD4+ and CD8+ T cells in HCT and SOT recipients and CMI against them may be linked to protection against viremia. [18–25].
It would be advantageous to incorporate more than one foreign gene into MVA to expand the number of antigens targeted by one vector. Since MVA has an exceedingly large capacity for foreign DNA, potentially multiple disease targets could be incorporated into one vaccine, thereby reducing manufacturing costs and dosing to patients. An MVA transfer vector targeting deletion III of MVA named pLW51 derived at NIAID could address some of these requirements [26]. It has two vaccinia promoters (vaccinia synthetic and modified H5 promoters) to express two foreign antigens and the bacterial marker gene is removable by ongoing recombination. We found limitations in this vector design, including excessive unwanted recombinants which remove the foreign gene insert and not the marker gene. Reliance on the facile color marker became impractical, so immunostaining had to be performed at each round of selection to eliminate those unwanted rMVA. To overcome the practical limitations of the pLW51 design and broaden the sites available for gene insertion, we designed and constructed a novel transfer vector based on the pLW51 design that targets deletion II of MVA and promotes removal of the bacterial marker gene. Two promoter cassettes facilitate two genes to be expressed simultaneously at similar levels. In this report, we successfully generated marker gene-free rMVA with simultaneous high level expression of CMV pp65 and IE1 using pZWIIA. We found robust stimulation of primary immunity both in vivo in mouse transgenic models and in vitro recall CMI in human PBMCs.
2. Methods
2.1 Cells, virus and animals
Primary chicken embryo fibroblast (CEF) cells prepared from specific pathogen-free chicken eggs were purchased from Charles River SPAFAS (North Franklin, Conn.). MVA virus stock was kindly provided by Drs. Linda Wyatt and Bernard Moss at the National Institutes of Allergy and Infections Diseases (NIAID), the National Institute of Health (NIH). HHD II Tg mice [27] were bred and maintained under specific pathogen-free conditions in the AALAC-approved animal care facility at City of Hope.
2.2 Construction of MVA transfer vector pp65-IE1-pZWIIA
The homologous recombination plasmid called pZWIIA was specifically redesigned from pLW22 and pLW51. IE1 exon4(IE1)-pLW22 and pp65-pSC11 were described previously [26]. IE1-exon 4 was used in favor of full-length IE1 as described in our recent report [25,26]. Briefly, a DNA region including a vaccinia synthetic promoter region and the gus gene flanked by two direct repeats (DR) from pLW51 was amplified by primers (5′ CGGGGTACCGACTATTGTTCTATATTATATATGGTTGTTGA 3′ and 5′ TTGGCGCGCCAGGCCTTCGCTAGCGGCCGCTATTTATTATTTATAGCATAG 3′) to yield a 2.5kbp PCR fragment as shown in Figure 1(i). It was then digested with Asc I and partially digested with Kpn I, gel purifed and ligated to pLW22 plasmid between the Kpn I and the Asc I sites to replace the β-galactosidase marker gene with the gus gene flanked by DR to yield pZWIIA. The 1.7 Kb pp65 and 1.2 Kb IE1 genes were PCR amplified from existing plasmids, and cloned into pZWIIA to yield pp65-IE1-pZWIIA shown in Figure 1(ii). pp65-IE1-pZWIIB and pp65-IE1-pZWIIC were also constructed in a similar manner, except both promoters were arranged in the same direction in pZWIIB and in back to back orientation in pZWIIC as shown in Figure 1(iii, iv). pZWIIA, pZWIIB and pZWIIC were all verified by restriction enzyme digestion and DNA sequencing analysis.
Figure 1. Schematic map of MVA transfer vectors.
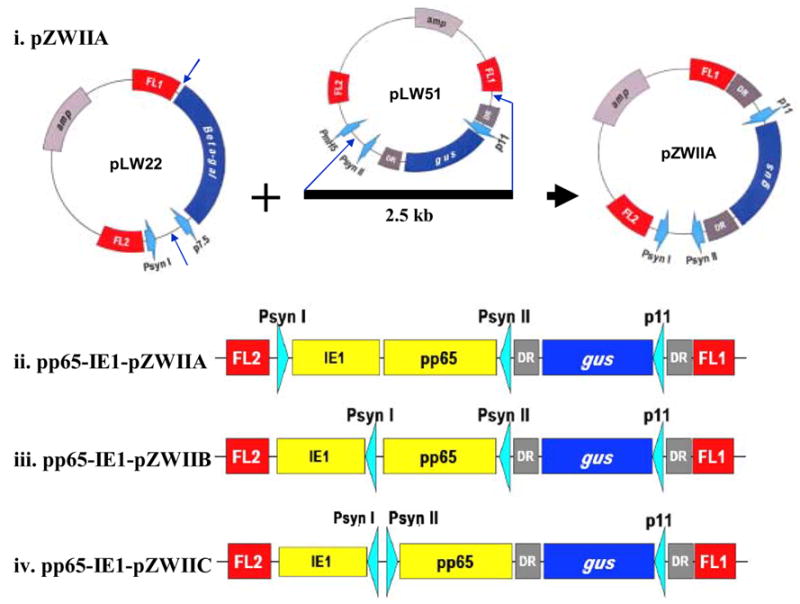
i) pZWIIA plasmid construction. A 2.5 Kb DNA segment including the synthetic promoter II (Psyn II) together with gus gene flanking by two direct repeats from pLW51 was PCR amplified and cloned into pLW22 to replace β-galatosidase gene to yield pZWIIA. ii) pp65-IE1-pZWIIA plasmid construction. The 1.7 Kb pp65 and 1.2 Kb IE1 genes were PCR amplified from existing plasmids, and cloned into pZWIIA to yield pp65-IE1-pZWIIA. Two vaccinia synthetic promoters (Psyn 1 and Psyn II) are arranged in head to head direction in the pZWIIA vector design. iii) Schematic representation of linear pZWIIB. pZWIIB has two synthetic promoters in the same direction. (iv) Schematic representation of linear pZWIIC plasmid. pZWIIC has two synthetic promoters in back to back orientation.
2.3 Generation and characterization of marker gene-free pp65-IE1-MVA
wt MVA was propagated on CEF cells. pp65-IE1-MVA was generated on CEF cells via homologous recombination by transfection/infection method as described previously using pZWIIA [26,28]. pp65-IE1-MVA was isolated by six consecutive rounds of plaque purification on CEF cells in the presence of β-glucuronidase substrate (X-glcA).
gus gene-free pp65-IE1-MVA virus was isolated using a newly described method of limiting dilution. Briefly, pp65-IE1-MVA isolates were distributed into individual wells of two 96-well plates (average 0.1 or 1 virus particle per well) to infect CEF cells, and followed by adding X-glcA substrate. After 3 to 5 days, wells that were not blue and visually appeared to be infected were collected. Two sets of PCR were used to identify pp65-IE1-MVA that lost the gus marker gene. The first PCR amplifies a 700bp DNA fragment using two primers that target gus gene and the flanking region of deletion II as shown in left panel of Figure 4A (primer a: 5′ GCA TAA TTA CGA ATA TCT GCA T 3′ and primer c: 5′ TGC ATT TAA GGC GGA TGTC 3′ as shown in Figure 4A) to detect the loss of the gus gene. The second PCR used primers that target pp65 and flanking region of deletion II as shown in left panel of Figure 4B (primer b: 5′ GCTCTTTCCACTGGTTCTGC 3′ and primer c) to amplify from pp65 to flanking region to generate a 1Kbp PCR product, indicative that the gus gene is deleted.
Figure 4.

A) Schematic map of pp65 and IE1 expression cassette in pp65-IE1-MVA and PCR primers to amplify the gus gene is shown on left panel. Forward primer a targets the gus gene. Reverse primer c targets the FL1 region of MVA genome. Right panel is PCR amplification of the gus gene using primers a and c. Expected PCR product is 0.7kb. Lanes #4, #8, #9, #16, #17 and #22 are total DNA extracted from CEF cells infected with collected samples. Total 22 samples were tested. Only PCR results of #4, #8, #9, #16, #17 and #22 out of 22 samples were shown in gel. B) Schematic map of pp65 and IE1 expression cassette in marker gene-free pp65-IE1-MVA is shown on left panel. Forward primer b targets the pp65 gene. Reverse primer c targets the FL1 region of the MVA genome. Right panel is PCR amplification of the pp65 gene in marker gene-free pp65-IE1-MVA. The expected size of PCR product is 1Kb if gus gene is deleted. C) Western blot detection of pp65 and IE1 expression level in pp65-IE1-MVA of passage 1 to 4 infected CEF cells. Lane 1, 2, 3 and 4 were cell lysate of passage 1 to 4 of pp65-IE1-MVA infected CEF cells. Top panel is pp65 expression level and bottom panel is IE1 expression levels.
The expression level of pp65 and IE1 from MVA-infected cells was determined by western blot using an enhanced chemiluminescence (ECL) kit (Amersham Pharmacia Biotech, Buckinghamshire, United Kingdom). Western blot membranes were incubated first with purified mAb 28–103 [29] against pp65 or mAb p63-27 [30] against IE1, and then followed with a peroxidase-labeled goat anti–mouse Ab according to manufacturer’s instructions. pp65-IE1-MVA virus for mouse immunization was purified once by 36% sucrose density ultracentrifugation and resuspended in PBS containing 5% lactose[31].
2.4 Mouse immunization procedures and Chromium release assay (CRA)
Eight (8 to 10 weeks old) HHD II (Tg for HLA A2.1) mice divided into four groups, were immunized with 5 x 107 pfu of purified pp65-IE1-MVA in 100 μL by intraperitoneal (i.p.) route. Three weeks after immunization, spleens were removed and splenocyte mixtures from two individual mice were stimulated in vitro once for 1 week with lipopolysaccharide (LPS) blasts as antigen presenting cells (APCs) [26]. Separate cultures were stimulated with APC loaded with either pp65495-503 or IE1316-324 CTL epitopes [32]. Cytotoxic activity of stimulated splenocytes was measured by 4-hour CRA as described previously [33].
2.5 Human PBMC Studies
We obtained informed consent from two HLA A*0201 healthy volunteers (UPN011 and UPN031). The CD8+ T cell population was separated from 60 ml peripheral blood by magnetic bead separation method as described previously [34]. Autologous Epstein-Barr virus-transformed lymphoblastoid cell lines (EBVLCLs) infected with pp65-IE1-MVA at MOI=5 for 2 hours were used as APCs for IVS. After 10 days incubation, the stimulated T cell population was harvested. ICS was performed on the aliquots of IVS cells using pp65495-503 or IE1316-324 epitopes or irrelevant peptide as stimulators. Cells were stained with fluorescein isothiocyanate (FITC)-conjugated CD8 antibody and APC-conjugated anti-IFN-γ (BD Biosciences, San Jose, CA) after permeabilization and treatment with Brefeldin A as described [32]. Both CD8+ and IFN-γ + cells were represented as a percentage of the CD8+ T cell population. Other PBMC aliquots were incubated in a 4-hour CRA using autologous EBVLCL as targets loaded with pp65495-503 or IE1316-324, as described previously [32].
3. Results
3.1 MVA transfer vector construction
We designed and constructed a novel MVA transfer vector called pZWIIA based on previous transfer vectors developed in the Moss laboratory (pLW51 and pLW22) as shown in Figure 1 (i) [28,35]. The pLW51 transfer vector targets Del III of the MVA genome by incorporating Flanking region 1 (FL1) between 148533 to 149313 and Flanking region 2 (FL2) from 149345 to 149866 of MVA [U94848 [gi:2772662]. The direct repeat (DR) sequence from pLW51 used to construct pZWIIA is derived from 149058 to 149298 of the MVA genome. pZWIIA has four essential components: 1) Sequence flanking Del II of MVA corresponding to genome position 20718 to 21284 for FL1 and 19972 to 20666 for FL2, originally incorporated into pLW22 [36]. 2) An expression cassette, which includes two vaccinia synthetic promoters (Psyn I from pLW22 and Psyn II from pLW51) arranged head to head to independently promote foreign insert gene expression. 3) gus marker gene obtained from pLW51 directed by the P11 vaccinia promoter [2]. 4) DR sequence derived from pLW51 to facilitate gus gene removal from rMVA via intragenomic homologous recombination. The 1.7 Kb pp65 and 1.2 Kb IE1 genes were PCR amplified from existing plasmids, and cloned into pZWIIA to yield pp65-IE1-pZWIIA as shown in Figure 1(ii). Similar transfer vectors were also constructed, which have two synthetic promoters arranged back to back or in the same orientation as shown in Figure 1 (iii and iv).
3.2 Generation and preliminary characterization of pp65-IE1-MVA
Marker gene-free pp65-IE1-MVA was made successfully using pp65-IE1-pZWIIA as shown in Figure 2. pp65-IE1-MVA was generated by transfecting pp65-IE1-pZWIIA plasmid DNA into CEF cells infected with wt MVA, followed by six consecutive rounds of plaque purification in the presence of X-glcA [37]. Western blot was performed on pp65-IE1-MVA-infected CEF cells to determine both pp65 and IE1 expression levels. As shown in Figure 3, both pp65 and IE1 protein were expressed at equivalently high levels. We also found that after six rounds of purification, >95% of plaques simultaneously express high levels of pp65 and IE1 (data not shown). Two other MVA transfer vectors shown in Figure 1 (iii and iv) were also used to generate rMVA. We found that rMVA generated using pZWIIB expressed high levels of pp65, but low levels of IE1, whereas rMVA generated using pZWIIC had low expression levels of both pp65 and IE1 (data not shown). These results maybe attributed to both promotors interfering with each other if they are assembled in the same direction or back to back. Only pp65-IE1-MVA generated by pZWIIA as shown in Figure 1(i) was further investigated.
Figure 2. Generation of marker gene-free pp65-IE1-MVA.

pZWIIA was transfected into wt MVA infected CEF cells to generate pp65-IE1-MVA via homologous recombination. The gus gene was removed by intragenomic homologous recombination between the two DRs to yield marker gene-free pp65-IE1-MVA.
Figure 3. Western blot detection of pp65 and IE1 exon4 protein from pp65-IE1-MVA infected CEF cells.

Lane 1: cell lysate of rMVA expressing pp65 as (+) control; Lane 4: cell lysate of rMVA expressing IE1 as (+) control. Lane 2 and 5: cell lysate of mock-infected CEF as (−) control; Lane 3 and 6: cell lysate of pp65-IE1-MVA infected CEF cells. Lanes 1, 2 and 3 were blotted with mAb 28-103 against pp65. Lane 4, 5 and 6 were blotted with mAb p63-27 against IE1. Non-specific cellular protein was also detected in Lane 4, 5 and 6 in addition to IE1.
3.3 Removal of gus gene
As the gus gene is flanked by repetitive sequences, we hypothesized that it is stochastically deleted from pp65-IE1-MVA viral genome by intragenomic homologous recombination during viral replication. We tested whether pp65-IE1-MVA deleting the gus marker gene can be selected out by simple limiting dilution. To test our assumption, pp65-IE1-MVA virus stock was titrated, diluted and distributed in 96-well plates with CEF cells in the presence of X-glcA substrate as described in Materials and Methods. We collected and extracted DNA samples from 22 wells which appeared to be infected, but had no color.
To identify pp65-IE1-MVA that lost the gus gene, two sets of PCR were designed. One PCR targeted the gus (700 bp) gene (primer a) and the FL1 of deletion II (primer c), as shown in Figure 4A (left panel), to demonstrate the loss of gus gene. The expected PCR product would be approximately 700bp if the gus gene were present. As shown in the right panel of Figure 4A, all six candidates appeared to have lost the gus gene. This PCR alone did not unequivocally prove the existence of marker gene-free pp65-IE1-MVA, a second PCR, targeting the inserted pp65 gene (primer b) and the FL1 of deletion II (primer c), as shown in Figure 4B (left panel), was carried out to ensure that pp65-IE1-MVA isolates were marker gene free. If pp65-IE1-MVA were marker gene-free, then the expected size of the PCR product will be 1.0 kb. If the gus marker gene is present, then the expected size of PCR product would be 2.9 kb. As shown in Figure 4B (right panel), all six samples have only a 1.0 kb PCR product band, indicating that these samples are all marker gene-free rMVA. In addition, the expected PCR product was the same size in each instance, suggesting that the gus gene was deleted by a similar mechanism for all six samples. Correct insertion of the pp65 and IE1 expression cassettes into deletion II region of MVA was verified by PCR and DNA sequencing (data not shown). We have passaged the marker-gene-free version of pp65-IE1-MVA 4 times on CEF cells. We observed that pp65-IE1-MVA grows equivalently well as wt MVA, and maintains similar pp65 and IE1 protein expression levels at each successive passage as shown in Figure 4C.
3.4 In vivo immunogenicity of pp65-IE1-MVA
The ability of pp65-IE1-MVA to elicit pp65- and IE1-specific cellular immune responses was evaluated in HHD II mice. HHD II mice have minimal endogenous murine MHC Class I presentation, to potentially interfere with the chimeric HHD transgene that restricts antigen presentation based on human HLA A2 peptide binding motifs [26,27,38]. HHD II mice were immunized via i.p. route with 50M pfu of pp65-IE1-MVA. 3 weeks after immunization, the splenocytes were harvested and CTL responses were determined one week after IVS. As shown in Figure 5, we found substantial cytotoxicity levels against human T2 cell targets loaded with either the HLA A2-restricted pp65495-503 or IE1316-324 peptides at all E/T ratios, after a single immunization. Minimal cytolytic background activity was found when target cells were loaded with an irrelevant p53 HLA A2-restricted epitope. Our results show that a single administration of pp65-IE1-MVA to HHD II mice simultaneously elicits both pp65- and IE1-specific cellular immune responses.
Figure 5. In vivo immunogenicity analysis of pp65-IE1-MVA in HHD II Tg mice.

pp65495-503 (⋄ diamond) and IE1316-324 (□ rectangle) A2 CTL peptide loaded T2 cells were used as targets. Non-related p53149-157 A2 CTL peptide loaded T2 cells (♦ solid diamond ■ solid rectangle) were negative control of targets. Means and SE were calculated at each effector-target (E/T) ratio for all 4 groups of mice.
3.5 In vitro expansion of CMV-specific T cells from human PBMC using pp65-IE1-MVA
An additional application of pp65-IE1-MVA is IVS of human PBMC for expansion of T cells usable in adoptive transfer. CMV-specific T cells can potentially be administered to severely immunosuppressed patients who might not respond to a direct vaccination. Prior examples using less robust approaches exist in the literature for CMV [14,39]. The ability of pp65-IE1-MVA to expand CMV-specific memory T cells from PBMC was evaluated in two healthy CMV positive HLA A*0201 donors (UPN011 and UPN031). Autologous EBVLCLs infected with pp65-IE1-MVA were used as APCs to amplify 1 x 106 CD4, CD56 and CD16 magnetically depleted PBMCs from two donors (UPN011 and UPN031) for 10 days. More than 1 x 107 cells from UPN011 and more than 1.8 x 107 cells from UPN031 were recovered after a 10-day expansion (estimated 10-fold amplification for UPN011 and 18-fold for UPN031). The frequency of IFN-γ+ CD8+ T cells were determined by ICS, and the functional cytotoxic activity of CMV-specific T cells expanded in vitro was measured by CRA. Table 1 shows that the expansion of both pp65 and IE1 specific T cells and the functional activity of expanded CMV-specific T cells against pp65495-503 and IE1316-324 loaded targets was substantial for both donors. These data demonstrate the feasibility of the expansion of pp65-specific and IE1-specific T cells simultaneously by pp65-IE1-MVA.
Table 1. Frequency of IFN-γ+ and CD8+ T cells and functional activity of CMV-specific T cells (pre-IVS and post-IVS).
PBMCs of two healthy donors (UPN011, UPN031) were co-cultured with EBVLCL infected with pp65-IE1-MVA as APCs. PBMCs before IVS (pre-IVS) and after IVS (post-IVS) were used in ICS and CRA. For each subject, the percentages of CD8+ T cells producing IFN-γ upon 18-hour stimulation with HLA A2 peptide (pp65495-503, IE1316-324 and p53264-272) are shown. For flow analysis, a primary gate was set on lymphocytes by forward versus side scatter. A secondary plot was made of IFN-γ versus CD8. The remaining PBMC cells (pre-IVS and post-IVS) were used as effectors in CRA assays with autologous EBVLCL loaded with HLA A2 pp65495-503, IE1316-324 and p53264-272 peptides as targets. 4-hour CRAs were performed as described in Material and Methods. The cytotoxicity percentages at an effector to target ratio of (100:1) are shown.
| Subject | % CD8+IFN-γ+ | % cytotoxicity | ||||
|---|---|---|---|---|---|---|
| pp65 | IE1 | p53 | pp65 | IE1 | p53 | |
| UPN011 (pre-IVS) | 0.4 | 0.1 | 0.1 | 26.0 | 9.0 | 5.0 |
| UPN011 (post-IVS) | 32.6 | 15.2 | 4.0 | 94.0 | 50.0 | 30.0 |
| UPN031 (pre-IVS) | 3.2 | 1.8 | 0.1 | 9.7 | 3.6 | 0.0 |
| UPN031 (post-IVS) | 12.3 | 8.4 | 0.5 | 82.0 | 38.0 | 5.8 |
4. Discussion
In this study, pp65-IE1-MVA was generated successfully on CEF cells using pp65-IE1-pZWIIA with existing rMVA methodology. It was plaque purified by six-consecutive rounds via color screening. Marker gene-free pp65-IE1-MVA was easily isolated from 96-well plates by a limiting dilution method described in Results. These results are consistent with the hypothesis that marker gene-free pp65-IE1-MVA was derived spontaneously during virus expansion, and that intragenomic homologous deletion recombination of the gus gene between the two DR is efficient. There is the possibility of other intergenic recombination events, because the DR sequence within pZWIIA is derived from the Del III region of the MVA genome. Theoretically, intergenic recombination events could happen between Del II and Del III. However Del II is very far (130 Kb) from Del III, so the likelihood is small, and a virus resulting from such an event will have a large deletion making it unlikely it will both replicate and survive. We do not have any evidence for such unexpected deletions. We also observed that co-expression of both pp65 and IE1 remained stable for at least 4 passages. The expression stability of the two antigen genes, as well as the genetic stability of the insertion region, in long-term passage needs to be further investigated. Similar to the pLW51 transfer plasmid, the complete virus derivation process using pZWIIA can be adapted and performed on primary CEF cells under GLP conditions without the use of additional cell lines or selection in the presence of drug.
MVA was derived from vaccinia virus ankara through more than 570 serial passages in chicken embryos [40]. Analysis of the MVA genome revealed six major deletions [41]. Deletions II and III were developed and widely used for insertion of foreign genes to create recombinant MVA (rMVA) via homologous recombination [36,42]. To isolate rMVA from parental wt MVA, several screening methods were developed, including color development by metabolism of substrates by bacterial marker genes such as β-galactosidase or β-glucuronidase [43]. Selection regimes include neomycin resistance in the presence of the antibiotic G418, or E. coli guanine phosphoribosyltransferase (gpt) in the presence of mycophenolic acid [44], or host range gene selection using the properties of the K1L locus [45]. These approaches select for rMVA with stable integration of the selection marker gene, which is not optimal for clinical investigation, mainly because of the potential of the marker gene product to be immunogenic or allergenic[46–48]. Consequently, these derivation processes cannot be easily adapted to cGMP, because they require growth either in the presence of selection drug or in an unqualified rabbit kidney cell line (RK-13) for manipulation of the K1L locus.
There are few MVA transfer vectors freely available to the biomedical community that can be used to create marker gene-free rMVA for clinical purposes under cGMP conditions. One approach combined screening with β-glactosidase and gpt selection, targeting both the hemagglutinin (ha) and the thymidine kinase (tk) loci of MVA [49]. In some cases, limitations on the efficiency of the vector to produce the correct recombinant have increased the effort and time involved in deriving rMVA [28]. For example, when using the pLW51 transfer vector, immunostaining to detect foreign antigen gene expression has to be performed at each screening round due to rMVA expressing only the bacterial marker gene being inappropriately generated. In the original design by the Moss laboratory for the transfer vector pLW51, two identical copies of a 240 base pair sequence (DR) flanked the color marker gene (Figure 1). The choice of the sequence to be used as a DR was influenced by FDA concerns to avoid the introduction of additional foreign gene sequences into the virus. Therefore, since the Del III region flanking sequence (FL) was already used to promote recombination between the virus and shuttle vectors designed previously (pMC03) [37], it was thought that this experience and degree of characterization would be helpful in the predictability of the products of recombination. In fact, the sequences work well to generate recombinants, but some unavoidable consequences develop, including the generation of unintended recombinants. The reason is that the DR sequence of pLW51 is identical to a 240 bp segment of FL1 contained in pLW51 (148533-149313 of MVA genome), facilitating the generation of recombinants in which the foreign gene cassette is unintentionally removed (Figure 2).
The DR sequences of pZWIIA are identical to those contained in pLW51 (Figure 1). However, both FL sequences in pZWIIA are derived from flanking sequence of Del II of MVA genome that originated in plasmid vector pLW22 (Figure 1), and share no homology with the DRs, which are derived from Del III [31]. The possibility to generate recombinant MVA carrying only the color marker gene (gus) via DR sequence recombination with FL1 or FL2 flanking Del II is substantially reduced. This leads to dramatically increased recovery of recombinants having both required elements for screening; the foreign gene cassette and the color marker gene. The requirement to immunostain as the only means to detect rMVA during screening is eliminated, and can be substituted by simply following the presence of the marker gene.
MVA holds great promise as a viral vector that can incorporate foreign antigens, elicit strong humoral and T cell responses toward these foreign antigens, and be delivered in a non-toxic manner to humans. For example, preliminary results from malaria vaccine clinical trials suggest that MVA carrying the potent immunogen ME-TRAP of p.falciparum exerts protection against the malaria pathogen. It has been proposed that a superior malaria vaccine will be multistage and multi-component requiring a high capacity vector such as MVA [50]. Using dendritic cells infected with poxvirus encoding CEA and co-stimulatory molecules B7-1, ICAM-1 and LFA-3 activates potent CEA-specific immune responses [51]. Recent clinical studies indicated that MVA expressing HIV-1 Gag coupled to other CD8+ T-cell epitopes could induce multifunctional HIV-1-specific T cells in healthy subjects or in individuals with chronic HIV-1 infection [13,52].
The safety of MVA has been established after testing it in immuno-compromised primate models and clinically in HIV patients, without findings of persistence or the development of adverse reactions [53–55]. Although MVA has not been tested in humans post-transplant, there are no properties of the virus that suggest it would be problematic or generate pathology. Consequently, we envision MVA could be used safely as a vaccine in either recipients or donors, and either before or after the transplant procedure. In addition, as we showed in prior publications, MVA acts as a powerful stimulator of lymphocyte proliferation in vitro, which makes it ideal to generate antigen-specific T cells for adoptive transfer into transplant recipients [26,32,56]. The favored approach depends upon the application, and the sophistication and experience of the transplant center. Since MVA can incorporate multiple genes in different regions of the virus, it is feasible to include tumor antigens which would give it a dual role as a vaccine against the malignancy and infectious complications post-transplant. However, a patient needs to be immuno-competent to respond to the vaccine, so complications early post- transplant might be best addressed using the property of in vitro stimulation of anti-viral effectors.
The usage of full-length antigens expressed from MVA is a powerful immunotherapeutic tool applicable regardless of an individual’s HLA type. We have published that a dual CMV antigen pp65/pp150 expressing MVA was strongly recognized in vitro by PBMC from CMV positive healthy subjects with HLA A*1101, HLA A*6801, HLA A*0301, and HLA B*0702 haplotypes [32]. The magnitude of T cell expansion was shown to be far greater than other published expansion techniques, especially in the 1–2 week comparison period evaluated in the study. In addition, there is increased safety of using highly attenuated MVA compared to other techniques which utilize live viruses. Procedures which depend on CMV lysate for T cell expansion are unlikely to be applicable to the clinic, since the reproducibility of the antigenic mixture is in question, as well as its composition that includes disrupted CMV virions. Other techniques that rely on purification of CMV-specific T cells using HLA tetramers, or through the combination of stimulating peptides and bi-specific capture antibodies are not designed to amplify T cells [14,21]. Consequently, they require more incubation time and larger volumes of blood, because they capture existing T cells, but do not stimulate T cell proliferation that is the hallmark of MVA. Further, synthetic peptides cannot substitute for full-length antigen to stimulate the full repertoire of CTL in bulk culture. Finally, the incorporation of the CMV lysate into the technique to elicit CD4 T-helper response is likely to encounter obstacles for clinical use. Riddell and colleagues showed that infusions of donor derived CD8+ T-cell clones effectively reconstitute CMV-specific CTL responses, after HSCT [39]. However, the approach was logistically complex and incapable of inducing CD4+ T-helper cells, to support CTL persistence. The technique is also plagued by the requirement of using live CMV infected fibroblasts to stimulate CMV-specific CTL. Thus the proposed approach using CMV-MVA appears qualitatively more robust than previous ones, since it is able to elicit clinically relevant numbers of T-cells, which are specific for the full-length immunodominant pp65 and IE proteins using a recombinant vector considered safe for vaccination of immunosuppressed individuals.
There are a number of replication-defective viral vectors available for vaccine development. These include adeno-associated virus (AAV)[57], replication-defective adenovirus (AdV) [58–60], Vesicular stomatitis viruses (VSV) [61] and Venezuelan equine encephalitis (VEE) [62,63]. However they all have the common problem of limited capacity to incorporate foreign genes. In contrast, MVA has the potential for large foreign gene capacity because the genome suffered six major deletions totaling nearly 30Kb [2], during its passage in CEF [41]. There is ample space to insert multiple genes from CMV involved in multiple stages of the virus life cycle as an approach to disable virus transmission. A small problem to overcome is the difficulty of MVA to express glycoproteins at high density, because the attenuation of the virus weakens it sufficiently to prevent it from propagating in even permissive lines. To overcome that deficiency, we took advantage of a strategy first employed by Plotkin and collaborators to delete the transmembrane portion of cell surface glycoproteins [64]. The example we used of CMV-gB, could only be made into a recombinant MVA when the TM region was deleted [28]. It is likely that other surface glycoproteins, including gM, gN [65,66], gL, gO and gH [67] are involved in virus neutralization, so it is incumbent to find structures that will allow MVA virus formation. If those practical issues can be overcome, then an MVA composed of a combination of strong humoral immunity inducing antigens, along with the antigens we have discussed in this report for stimulating cellular immunity might be useful in the setting of congenital infection. Tests of the concept pre-clinically in relevant animal models are essential, such as the guinea pig [68] and rhesus macaque [69]. Recombinant MVA generated via pZWIIA and similar vectors can generate vaccines with broad enough coverage for inclusion as a candidate vaccine for prevention of congenital CMV infection.
Acknowledgments
We wish to thank Drs. Linda Wyatt and Bernard Moss of the Laboratory of Viral Diseases, NIAID in NIH for providing us not only with the pLW22, pLW51, and pSC11 plasmids and MVA, but also suggestions on construction of rMVA. The personnel at the Animal Resource Center of the City of Hope are acknowledged for their help in maintaining the mice. GCRC personnel, especially Maria Madrigal, Brenda Williams, and Ann Geva, are acknowledged for their assistance in recruitment of human subjects for evaluation. We thank Donna Packer and Julia Santos for assistance of manuscript preparation and managing the laboratory.
These studies have been partially supported by grants from the National Cancer Institute (NCI; R01-CA77544 and P01-CA30206, Project III and a subcontract from AI52065 to DJD. The City of Hope Comprehensive Cancer Center is supported by the NCI (CA33572). The Laboratory of Vaccine Research is partially supported by the Edwin and Bea Wolfe Charitable Foundation. Support for the City of Hope General Clinical Research Center (GCRC; satellite of University of Southern California GCRC) is from the NIH (MO1-RR0043-38).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mayr A. Historical review of smallpox the eradication of smallpox, the attenuated smallpox MVA vaccine. Berl Munch Tierarztl Wochenschr. 1999;112(9):322–328. [PubMed] [Google Scholar]
- 2.Sutter G, Moss B. Nonreplicating vaccinia vector efficiently expresses recombinant genes. Proc Natl Acad Sci U S A. 1992;89(22):10847–10851. doi: 10.1073/pnas.89.22.10847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sutter G, Wyatt LS, Foley PL, Bennink JR, Moss B. A recombinant vector derived from the host range-restricted and highly attenuated MVA strain of vaccinia virus stimulates protective immunity in mice to influenza virus. Vaccine. 1994;12(11):1032–1040. doi: 10.1016/0264-410x(94)90341-7. [DOI] [PubMed] [Google Scholar]
- 4.Taylor GS, Haigh TA, Gudgeon NH, et al. Dual stimulation of Epstein-Barr Virus (EBV)-specific CD4+- and CD8+-T-cell responses by a chimeric antigen construct: potential therapeutic vaccine for EBV-positive nasopharyngeal carcinoma. J Virol. 2004;78(2):768–778. doi: 10.1128/JVI.78.2.768-778.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harrer E, Bauerle M, Ferstl B, et al. Therapeutic vaccination of HIV-1-infected patients on HAART with a recombinant HIV-1 nef-expressing MVA: safety, immunogenicity and influence on viral load during treatment interruption. Antivir Ther. 2005;10(2):285–300. [PubMed] [Google Scholar]
- 6.Cebere I, Dorrell L, McShane H, et al. Phase I clinical trial safety of DNA- and modified virus Ankara-vectored human immunodeficiency virus type 1 (HIV-1) vaccines administered alone and in a prime-boost regime to healthy HIV-1-uninfected volunteers. Vaccine. 2006;24(4):417–425. doi: 10.1016/j.vaccine.2005.08.041. [DOI] [PubMed] [Google Scholar]
- 7.Corona Gutierrez CM, Tinoco A, Navarro T, et al. Therapeutic vaccination with MVA E2 can eliminate precancerous lesions (CIN 1, CIN 2, and CIN 3) associated with infection by oncogenic human papillomavirus. Hum Gene Ther. 2004;15(5):421–431. doi: 10.1089/10430340460745757. [DOI] [PubMed] [Google Scholar]
- 8.Bejon P, Peshu N, Gilbert SC, et al. Safety profile of the viral vectors of attenuated fowlpox strain FP9 and modified vaccinia virus Ankara recombinant for either of 2 preerythrocytic malaria antigens, ME-TRAP or the circumsporozoite protein, in children and adults in Kenya. Clin Infect Dis. 2006;42(8):1102–1110. doi: 10.1086/501459. [DOI] [PubMed] [Google Scholar]
- 9.Webster DP, Dunachie S, McConkey S, et al. Safety of recombinant fowlpox strain FP9 and modified vaccinia virus Ankara vaccines against liver-stage P. falciparum malaria in non-immune volunteers. Vaccine. 2006;24(15):3026–3034. doi: 10.1016/j.vaccine.2005.10.058. [DOI] [PubMed] [Google Scholar]
- 10.Dunachie SJ, Walther M, Vuola JM, et al. A clinical trial of prime-boost immunisation with the candidate malaria vaccines RTS, S/AS02A and MVA-CS. Vaccine. 2006;24(15):2850–2859. doi: 10.1016/j.vaccine.2005.12.041. [DOI] [PubMed] [Google Scholar]
- 11.Meyer RG, Britten CM, Siepmann U, et al. A phase I vaccination study with tyrosinase in patients with stage II melanoma using recombinant modified vaccinia virus Ankara (MVA-hTyr) Cancer Immunol Immunother. 2005;54(5):453–467. doi: 10.1007/s00262-004-0616-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Di Nicola M, Carlo-Stella C, Mortarini R, et al. Boosting T cell-mediated immunity to tyrosinase by vaccinia virus-transduced, CD34(+)-derived dendritic cell vaccination: a phase I trial in metastatic melanoma. Clin Cancer Res. 2004;10(16):5381–5390. doi: 10.1158/1078-0432.CCR-04-0602. [DOI] [PubMed] [Google Scholar]
- 13.Dorrell L, Yang H, Ondondo B, et al. Expansion and Diversification of Virus-Specific T Cells following Immunization of Human Immunodeficiency Virus Type 1 (HIV-1)-Infected Individuals with a Recombinant Modified Vaccinia Virus Ankara/HIV-1 Gag Vaccine. J Virol. 2006;80(10):4705–4716. doi: 10.1128/JVI.80.10.4705-4716.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rauser G, Einsele H, Sinzger C, et al. Rapid generation of combined CMV-specific CD4+ and CD8+ T-cell lines for adoptive transfer into recipients of allogeneic stem cell transplants. Blood. 2004;103(9):3565–3572. doi: 10.1182/blood-2003-09-3056. [DOI] [PubMed] [Google Scholar]
- 15.Einsele H, Roosnek E, Rufer N, et al. Infusion of cytomegalovirus (CMV)-specific T cells for the treatment of CMV infection not responding to antiviral chemotherapy. Blood. 2002;99(11):3916–3922. doi: 10.1182/blood.v99.11.3916. [DOI] [PubMed] [Google Scholar]
- 16.Walter EA, Greenberg PD, Gilbert MJ, et al. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N Engl J Med. 1995;333(16):1038–1044. doi: 10.1056/NEJM199510193331603. [DOI] [PubMed] [Google Scholar]
- 17.Sylwester AW, Mitchell BL, Edgar JB, et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J Exp Med. 2005;202(5):673–685. doi: 10.1084/jem.20050882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elkington R, Walker S, Crough T, et al. Ex vivo profiling of CD8+-T-cell responses to human cytomegalovirus reveals broad and multispecific reactivities in healthy virus carriers. J Virol. 2003;77(9):5226–5240. doi: 10.1128/JVI.77.9.5226-5240.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Riddell SR, Reusser P, Greenberg PD. Cytotoxic T cells specific for cytomegalovirus: a potential therapy for immunocompromised patients. Rev Infect Dis. 1991;13 (Suppl 11):S966–973. doi: 10.1093/clind/13.supplement_11.s966. [DOI] [PubMed] [Google Scholar]
- 20.Wills MR, Carmichael AJ, Mynard K, et al. The human cytotoxic T-lymphocyte (CTL) response to cytomegalovirus is dominated by structural protein pp65: frequency, specificity, and T-cell receptor usage of pp65-specific CTL. J Virol. 1996;70(11):7569–7579. doi: 10.1128/jvi.70.11.7569-7579.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cobbold M, Khan N, Pourgheysari B, et al. Adoptive transfer of cytomegalovirus-specific CTL to stem cell transplant patients after selection by HLA-peptide tetramers. J Exp Med. 2005;202(3):379–386. doi: 10.1084/jem.20040613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peggs KS, Verfuerth S, Pizzey A, et al. Adoptive cellular therapy for early cytomegalovirus infection after allogeneic stem-cell transplantation with virus-specific T-cell lines. Lancet. 2003;362(9393):1375–1377. doi: 10.1016/S0140-6736(03)14634-X. [DOI] [PubMed] [Google Scholar]
- 23.Cwynarski K, Ainsworth J, Cobbold M, et al. Direct visualization of cytomegalovirus-specific T-cell reconstitution after allogeneic stem cell transplantation. Blood. 2001;97(5):1232–1240. doi: 10.1182/blood.v97.5.1232. [DOI] [PubMed] [Google Scholar]
- 24.Bunde T, Kirchner A, Hoffmeister B, et al. Protection from cytomegalovirus after transplantation is correlated with immediate early 1-specific CD8 T cells. J Exp Med. 2005 doi: 10.1084/jem.20042384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gyulai Z, Endresz V, Burian K, et al. Cytotoxic T lymphocyte (CTL) responses to human cytomegalovirus pp65, IE1-Exon4, gB, pp150, and pp28 in healthy individuals: reevaluation of prevalence of IE1-specific CTLs. J Infect Dis. 2000;181(5):1537–1546. doi: 10.1086/315445. [DOI] [PubMed] [Google Scholar]
- 26.Wang Z, La Rosa C, Mekhoubad S, et al. Attenuated Poxviruses Generate Clinically Relevant Frequencies of CMV-Specific T cells. Blood. 2004;104(3):847–856. doi: 10.1182/blood-2003-10-3469. [DOI] [PubMed] [Google Scholar]
- 27.Pascolo S, Bervas N, Ure JM, Smith AG, Lemonnier FA, Perarnau B. HLA-A2.1-restricted education and cytolytic activity of CD8(+) T lymphocytes from beta2 microglobulin (beta2m) HLA-A2.1 monochain transgenic H-2Db beta2m double knockout mice. J Exp Med. 1997;185(12):2043–2051. doi: 10.1084/jem.185.12.2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Z, La Rosa C, Maas R, et al. Recombinant modified vaccinia virus Ankara expressing a soluble form of glycoprotein B causes durable immunity and neutralizing antibodies against multiple strains of human cytomegalovirus. J Virol. 2004;78(8):3965–3976. doi: 10.1128/JVI.78.8.3965-3976.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Britt WJ, Auger D. Identification of a 65 000 dalton virion envelope protein of human cytomegalovirus. Virus Res. 1985;4(1):31–36. doi: 10.1016/0168-1702(85)90018-8. [DOI] [PubMed] [Google Scholar]
- 30.Plachter B, Britt W, Vornhagen R, Stamminger T, Jahn G. Analysis of proteins encoded by IE regions 1 and 2 of human cytomegalovirus using monoclonal antibodies generated against recombinant antigens. Virology. 1993;193(2):642–652. doi: 10.1006/viro.1993.1172. [DOI] [PubMed] [Google Scholar]
- 31.Moss B, Earl PL. Current protocols in Molecular Biology. John Wiley & Sons, Inc; New York, N.Y: 1998. Expression of proteins in mammalian cells using vaccinia virus vectors; pp. 16.15.11–16.21.19. [Google Scholar]
- 32.La Rosa C, Wang Z, Lacey SF, et al. In vitro expansion of polyclonal T-cell subsets for adoptive immunotherapy by recombinant modified vaccinia Ankara. Exp Hematol. 2006;34(4):497–507. doi: 10.1016/j.exphem.2005.12.018. [DOI] [PubMed] [Google Scholar]
- 33.La Rosa C, Krishnan R, Markel S, et al. Enhanced immune activity of cytotoxic T-lymphocyte epitope analogs derived from positional scanning synthetic combinatorial libraries. Blood. 2001;97(6):1776–1786. doi: 10.1182/blood.v97.6.1776. [DOI] [PubMed] [Google Scholar]
- 34.Lacey SF, Villacres MC, La Rosa C, et al. Relative dominance of HLA-B*07 restricted CD8+ T-lymphocyte immune responses to human cytomegalovirus pp65 in persons sharing HLA-A*02 and HLA-B*07 alleles. Hum Immunol. 2003;64(4):440–452. doi: 10.1016/s0198-8859(03)00028-4. [DOI] [PubMed] [Google Scholar]
- 35.Ourmanov I, Brown CR, Moss B, et al. Comparative efficacy of recombinant modified vaccinia virus Ankara expressing simian immunodeficiency virus (SIV) Gag-Pol and/or Env in macaques challenged with pathogenic SIV. J Virol. 2000;74(6):2740–2751. doi: 10.1128/jvi.74.6.2740-2751.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Drexler I, Staib C, Sutter G. Modified vaccinia virus Ankara as antigen delivery system: how can we best use its potential? Curr Opin Biotechnol. 2004;15(6):506–512. doi: 10.1016/j.copbio.2004.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carroll MW, Moss B. E. coli beta-glucuronidase (GUS) as a marker for recombinant vaccinia viruses. Biotechniques. 1995;19(3):352–354. 356. [PubMed] [Google Scholar]
- 38.Daftarian P, Ali S, Sharan R, et al. Immunization with Th-CTL fusion peptide and cytosine-phosphate-guanine DNA in transgenic HLA-A2 mice induces recognition of HIV-infected T cells and clears vaccinia virus challenge. J Immunol. 2003;171(8):4028–4039. doi: 10.4049/jimmunol.171.8.4028. [DOI] [PubMed] [Google Scholar]
- 39.Riddell SR, Watanabe KS, Goodrich JM, Li CR, Agha ME, Greenberg PD. Restoration of viral immunity in immunodeficient humans by the adoptive transfer of T cell clones. Science. 1992;257(5067):238–241. doi: 10.1126/science.1352912. [DOI] [PubMed] [Google Scholar]
- 40.Staib C, Drexler I, Sutter G. Construction and Isolation of Recombinant MVA. Methods Mol Biol. 2004;269:77–100. doi: 10.1385/1-59259-789-0:077. [DOI] [PubMed] [Google Scholar]
- 41.Meyer H, Sutter G, Mayr A. Mapping of deletions in the genome of the highly attenuated vaccinia virus MVA and their influence on virulence. J Gen Virol. 1991;72 ( Pt 5):1031–1038. doi: 10.1099/0022-1317-72-5-1031. [DOI] [PubMed] [Google Scholar]
- 42.Sutter G, Staib C. Vaccinia vectors as candidate vaccines: the development of modified vaccinia virus Ankara for antigen delivery. Curr Drug Targets Infect Disord. 2003;3(3):263–271. doi: 10.2174/1568005033481123. [DOI] [PubMed] [Google Scholar]
- 43.Earl PL, Americo JL, Wyatt LS, et al. Immunogenicity of a highly attenuated MVA smallpox vaccine and protection against monkeypox. Nature. 2004;428(6979):182–185. doi: 10.1038/nature02331. [DOI] [PubMed] [Google Scholar]
- 44.Falkner FG, Moss B. Escherichia coli gpt gene provides dominant selection for vaccinia virus open reading frame expression vectors. J Virol. 1988;62(6):1849–1854. doi: 10.1128/jvi.62.6.1849-1854.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Staib C, Drexler I, Ohlmann M, Wintersperger S, Erfle V, Sutter G. Transient host range selection for genetic engineering of modified vaccinia virus Ankara. Biotechniques. 2000;28(6):1137–1142. doi: 10.2144/00286st04. 1144–1136, 1148. [DOI] [PubMed] [Google Scholar]
- 46.Brubaker JO, Thompson CM, Morrison LA, Knipe DM, Siber GR, Finberg RW. Th1-associated immune responses to beta-galactosidase expressed by a replication-defective herpes simplex virus. J Immunol. 1996;157(4):1598–1604. [PubMed] [Google Scholar]
- 47.Marktel S, Magnani Z, Ciceri F, et al. Immunologic potential of donor lymphocytes expressing a suicide gene for early immune reconstitution after hematopoietic T-cell-depleted stem cell transplantation. Blood. 2003;101(4):1290–1298. doi: 10.1182/blood-2002-08-2351. [DOI] [PubMed] [Google Scholar]
- 48.Bonini C, Grez M, Traversari C, et al. Safety of retroviral gene marking with a truncated NGF receptor. Nat Med. 2003;9(4):367–369. doi: 10.1038/nm0403-367. [DOI] [PubMed] [Google Scholar]
- 49.Scheiflinger F, Dorner F, Falkner FG. Transient marker stabilisation: a general procedure to construct marker-free recombinant vaccinia virus. Arch Virol. 1998;143(3):467–474. doi: 10.1007/s007050050303. [DOI] [PubMed] [Google Scholar]
- 50.Hill AV. Pre-erythrocytic malaria vaccines: towards greater efficacy. Nat Rev Immunol. 2006;6(1):21–32. doi: 10.1038/nri1746. [DOI] [PubMed] [Google Scholar]
- 51.Morse MA, Clay TM, Hobeika AC, et al. Phase I study of immunization with dendritic cells modified with fowlpox encoding carcinoembryonic antigen and costimulatory molecules. Clin Cancer Res. 2005;11(8):3017–3024. doi: 10.1158/1078-0432.CCR-04-2172. [DOI] [PubMed] [Google Scholar]
- 52.Goonetilleke N, Moore S, Dally L, et al. Induction of Multifunctional Human Immunodeficiency Virus Type 1 (HIV-1)-Specific T Cells Capable of Proliferation in Healthy Subjects by Using a Prime-Boost Regimen of DNA- and Modified Vaccinia Virus Ankara-Vectored Vaccines Expressing HIV-1 Gag Coupled to CD8+ T-Cell Epitopes. J Virol. 2006;80(10):4717–4728. doi: 10.1128/JVI.80.10.4717-4728.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stittelaar KJ, Kuiken T, de Swart RL, et al. Safety of modified vaccinia virus Ankara (MVA) in immune-suppressed macaques. Vaccine. 2001;19(27):3700–3709. doi: 10.1016/s0264-410x(01)00075-5. [DOI] [PubMed] [Google Scholar]
- 54.Cosma A, Nagaraj R, Buhler S, et al. Therapeutic vaccination with MVA-HIV-1 nef elicits Nef-specific T-helper cell responses in chronically HIV-1 infected individuals. Vaccine. 2003;22(1):21–29. doi: 10.1016/s0264-410x(03)00538-3. [DOI] [PubMed] [Google Scholar]
- 55.Hanke T, McMichael AJ, Dennis MJ, et al. Biodistribution and persistence of an MVA-vectored candidate HIV vaccine in SIV-infected rhesus macaques and SCID mice. Vaccine. 2005;23(12):1507–1514. doi: 10.1016/j.vaccine.2004.08.050. [DOI] [PubMed] [Google Scholar]
- 56.Wang Z, La Rosa C, Lacey SF, et al. Attenuated poxvirus expressing three immunodominant CMV antigens as a vaccine strategy for CMV infection. J Clin Virol. 2006;35(3):324–331. doi: 10.1016/j.jcv.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 57.Johnson PR, Schnepp BC, Connell MJ, et al. Novel adeno-associated virus vector vaccine restricts replication of simian immunodeficiency virus in macaques. J Virol. 2005;79(2):955–965. doi: 10.1128/JVI.79.2.955-965.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shiver JW, Fu TM, Chen L, et al. Replication-incompetent adenoviral vaccine vector elicits effective anti-immunodeficiency-virus immunity. Nature. 2002;415(6869):331–335. doi: 10.1038/415331a. [DOI] [PubMed] [Google Scholar]
- 59.Xiang Z, Gao G, Reyes-Sandoval A, et al. Novel, chimpanzee serotype 68-based adenoviral vaccine carrier for induction of antibodies to a transgene product. J Virol. 2002;76(6):2667–2675. doi: 10.1128/JVI.76.6.2667-2675.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shiver JW, Emini EA. Recent advances in the development of HIV-1 vaccines using replication-incompetent adenovirus vectors. Annu Rev Med. 2004;55:355–372. doi: 10.1146/annurev.med.55.091902.104344. [DOI] [PubMed] [Google Scholar]
- 61.Publicover J, Ramsburg E, Rose JK. Characterization of nonpathogenic, live, viral vaccine vectors inducing potent cellular immune responses. J Virol. 2004;78(17):9317–9324. doi: 10.1128/JVI.78.17.9317-9324.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Strauss JH, Strauss EG. The alphaviruses: gene expression, replication, and evolution. Microbiol Rev. 1994;58(3):491–562. doi: 10.1128/mr.58.3.491-562.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schlesinger S, Dubensky TW. Alphavirus vectors for gene expression and vaccines. Curr Opin Biotechnol. 1999;10(5):434–439. doi: 10.1016/s0958-1669(99)00006-3. [DOI] [PubMed] [Google Scholar]
- 64.Endresz V, Burian K, Berencsi K, et al. Optimization of DNA immunization against human cytomegalovirus. Vaccine. 2001;19(28–29):3972–3980. doi: 10.1016/s0264-410x(01)00116-5. [DOI] [PubMed] [Google Scholar]
- 65.Shimamura M, Mach M, Britt WJ. Human cytomegalovirus infection elicits a glycoprotein M (gM)/gN-specific virus-neutralizing antibody response. J Virol. 2006;80(9):4591–4600. doi: 10.1128/JVI.80.9.4591-4600.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mach M, Kropff B, Dal Monte P, Britt W. Complex formation by human cytomegalovirus glycoproteins M (gpUL100) and N (gpUL73) J Virol. 2000;74(24):11881–11892. doi: 10.1128/jvi.74.24.11881-11892.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rasmussen L, Geissler A, Cowan C, Chase A, Winters M. The genes encoding the gCIII complex of human cytomegalovirus exist in highly diverse combinations in clinical isolates. J Virol. 2002;76(21):10841–10848. doi: 10.1128/JVI.76.21.10841-10848.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schleiss MR, Stroup G, Pogorzelski K, McGregor A. Protection against congenital cytomegalovirus (CMV) disease, conferred by a replication-disabled, bacterial artificial chromosome (BAC)-based DNA vaccine. Vaccine. 2006 doi: 10.1016/j.vaccine.2006.06.077. [DOI] [PubMed] [Google Scholar]
- 69.Barry PA, Lockridge KM, Salamat S, et al. Nonhuman primate models of intrauterine cytomegalovirus infection. Ilar J. 2006;47(1):49–64. doi: 10.1093/ilar.47.1.49. [DOI] [PubMed] [Google Scholar]