

Abstract
Cannabinoid receptors (CBr) stimulation induces numerous central and peripheral effects. A growing interest in the beneficial properties of manipulating the endocannabinoid system has lead to the possible involvement of CBr in the control of brain inflammation. In the present study we examined the effect of the CBr agonist, (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)-pyrrolo[1,2,3-de]-1,4benzoxazin-6-yl]-1-naphthalenyl-methanone mesylate (WIN-55212-2), on microglial activation and spatial memory performance, using a well characterized animal model of chronic brain inflammation produced by the infusion of lipopolysaccharide (LPS, 250 ng/hr for 3 weeks) into the 4th ventricle of young rats. WIN-55212-2 (0.5 or 1.0 mg/kg/day, i.p.) was administered for three weeks. During the third week of treatment, spatial memory ability was examined using the Morris water-maze task. We found that 0.5 and 1 mg/kg WIN-55212-2 reduced the number of LPS-activated microglia, while 1 mg/kg WIN-55212-2 potentiated the LPS-induced impairment of performance in the watermaze task. CB1 receptors were not expressed by microglia and astrocytes, suggesting an indirect effect of WIN on microglia activation and memory impairment. Our results emphasize the potential use of CBr agonists in the regulation of inflammatory processes within the brain; this knowledge may lead to the use of CBr agonists in the treatment of neurodegenerative diseases associated with chronic neuroinflammation, such as Alzheimer disease.
Keywords: cannabinoid receptors, inflammation, activated microglia, Alzheimer’s disease, LPS, spatial memory
Microglial cells play a pivotal role as immune effectors in the central nervous system and may participate in the initiation and progression of neurological disorders, such as Alzheimer’s disease (AD), Parkinson’s disease and multiple sclerosis by releasing harmful molecules such as pro-inflammatory cytokines, reactive oxygen species or complement proteins (Akiyama et al., 2000; Kim and de Vellis, 2005). Many of the pathological, immunological, biochemical and behavioral changes seen in these and other neurodegenerative diseases can be reproduced in young rats by chronic infusion of lipopolysaccharide (LPS) into the 4th ventricle (Hauss-Wegrzyniak et al., 1998, Hauss-Wegrzyniak et al., 1999). Chronic infusion of LPS results in the activation of microglia within hippocampus and entorhinal cortex, brain regions involved in learning and memory formation (Hauss-Wegrzyniak et al., 1998; Rosi et al., 2004). Chronic brain inflammation is associated with impaired spatial memory, impaired induction of long-term potentiation, a loss of N-methyl-d-aspartate (NMDA) receptors, astrocytosis, elevated cytokines and related pro-inflammatory proteins and transcription factors (Hauss-Wegrzyniak et al., 1998, Hauss-Wegrzyniak et al., 1999; Rosi et al., 2004).
The endocannabinoid system may regulate many aspects of the brain’s inflammatory response, including the release of pro-inflammatory cytokines and modulation of microglial activation (Neumann, 2001; Klein, 2005). The endocannabinoid system is comprised of two G-protein-coupled receptors designated as CB1 and CB2, although not all endocannabinoid effects can be explained only by these two receptors (Begg et al., 2005). CB1 receptors are expressed in the brain and are responsible for most of the behavioral effects of the cannabinoids. CB2 receptors are expressed by immune and hematopoietic cells peripherally (Begg et al., 2005), and seem to be expressed on neurons in the brainstem and the brain (Van Sickle et al., 2005; Gong et al., 2006; Onaivi et al. 2006) although their presence in the brain is controversial (Munro et al., 1993). Two endogenous ligands for these receptors, arachydonylethanolamine and 2-arachidonoylglycerol (Stella, 2004), influence immune responses by inhibiting cytokine release and other anti-inflammatory actions (Klein et al., 2003; Klein, 2005). Microglia also express CBr and release cytokines in response to exposure to LPS or beta-amyloid protein; this effect can be inhibited by prior cannabinoid treatment (Facchinetti et al., 2003; Ramirez et al., 2005; Sheng et al., 2005). Astrocytes may also synthesize and release endocannabinoids (Walter et al., 2002). CB1 receptors have been widely studied because of their role in the psychoactive effects of the cannabis sativa plant (Δ9-etrahydrocannabinol or Δ9-THC). Δ9-THC can impair performance in rats, mice or monkeys under multiple experimental conditions (Castellano et al., 2003). Therefore, in the current study, we investigated the effect of a CBr agonist on microglial activation and spatial memory in a rodent model of chronic brain inflammation induced by LPS infusion into the 4th ventricle. We used the number of immunoreactive microglia (activated) as a biomarker of brain inflammation (Hauss-Wegrzyniak et al., 1998, Hauss-Wegrzyniak et al., 1999; Rosi et al., 2004, Rosi et al., 2005) to evaluate the potential anti-inflammatory properties of the WIN 55-212-2 compound.
EXPERIMENTAL PROCEDURES
Subjects and surgical procedures
Fifty-four young (3 months old) male F-344 rats (Harlan Sprague-Dawley, Indianapolis, IN) were singly housed in Plexiglas cages with free access to food and water. The rats were maintained on a 12/12-h light-dark cycle in a temperature-controlled room (22°C) with lights off at 0800. All rats were given health checks, handled upon arrival and allowed at least one week to adapt to their new environment prior to surgery. Artificial cerebrospinal fluid (aCSF n=26) or LPS (Sigma, St-Louis, MO, E. coli, serotype 055:B5, TCA extraction, 1.0 mg/ml dissolved in aCSF, n=28) were chronically infused for 21 days through a cannula implanted into the 4th ventricle that was attached (via Tygon tubing, 0.06 O.D., and an osmotic pump connect, Model 3280P, Plastics One, Roanoke, VA) to an osmotic minipump (Alzet, Cupertino, CA, model #2004, to deliver 0.25 μl/h, Hauss-Wegrzyniak et al., 1998). The aCSF vehicle contained (in mM) 140 NaCl; 3.0 KCl; 2.5 CaCl2; 1.0 MgCl2; 1.2 Na2HPO4, adjusted to pH 7.4. Rats infused with either aCSF or LPS were also administered daily the synthetic cannabinoid CB1/CB2 receptor agonist (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)-pyrrolo[1,2,3-de]-1,4benzoxazin-6-yl]-1-naphthalenyl-methanone mesylate (WIN-55212-2, Sigma, St-Louis, MO, 0.5 or 1.0 mg/kg, i.p.) or vehicle (Dimethylsulfoxide (100%), DMSO, Sigma, St-Louis, MO). Rats were assigned to one of the following six groups: aCSF + vehicle, LPS+ vehicle, aCSF + WIN (0.5 mg/kg), LPS + WIN (0.5mg/kg), aCSF + WIN (1 mg/kg), LPS + WIN (1 mg/kg). Drug or vehicle administration began the first day after surgery and continued throughout the behavioral testing, which began on day 14 of the LPS or aCSF infusion.
Behavioral Testing
Spatial learning ability was assessed using a 185 cm diameter water maze with white walls. The water was maintained at 26-28°C and made opaque by adding white, non-toxic, paint. The pool was in the center of a 2.3 x 2.73 x 2.5m room with multiple visual stimuli on the wall as distal cues, and a chair and a metal board against the wall of the pool as proximal cues. The circular escape platform was 11.5 cm in diameter. For the spatial (hidden-platform) version of the water task, a circular escape platform was present in a constant location, submerged about 2.5 cm below the water surface. The rats were tracked by an overhead video camera connected to a VP114 tracking unit (HVS Image, England). Custom software was used to store and analyze each rat’s latency to find the submerged platform during each trial.
Each rat performed three training blocks per day (two training trials per block) for 4 days (24 trials total), with a 60-min inter-block interval. On each trial, the rat was released into the water from one of seven locations spaced evenly at the side of the pool, which varied randomly from trial to trial. After the rat found the escape platform or swam for a maximum of 60 sec, it was allowed to remain on the platform for 30 sec. To control for possible drug-induced deficits in visual acuity and swimming ability, the same rats were also tested on a second version of this task. In this version, a visible platform raised 2 cm above the surface of the water was moved randomly to one of four locations in the tank after each trial. A total of 4 visible-platform trials were performed. Drug administration was performed 20 minutes prior to the behavioral testing. The results, i.e. latency (sec) to find the hidden platform, were analyzed by ANOVA followed by post hoc comparisons according to the method of Bonferonni/Dunn.
Histological procedures
After behavioral testing was completed, each rat was deeply anesthetized with isoflurane and prepared for a transcardiac perfusion of the brain with cold saline containing 1 U/ml heparin, followed by 4% paraformaldehyde in 0.1 M phosphate buffer, pH 7.4. Brains were then removed and the placement of the cannula in the 4th ventricle was confirmed. The brains were then post-fixed one hour in the same fixative and then stored (4°C) in phosphate buffer saline (PBS), pH 7.4. Free-obtained using a vibratome from perfused tissues for staining with standard avidin/biotin peroxydase or fluorescence labeling methods. The monoclonal antibody OX-6 (final dilution 1:400, Pharmigen, San Diego, CA) was used to visualize activated microglia cells. This antibody is directed against Class II major histocompatibility complex (MHC II) antigen. After quenching endogenous peroxydase/activity and blocking nonspecific binding, the sections were incubated (4°C) overnight with primary antibodies directed against the specific epitope (MCH II). Thereafter, the sections were incubated for 2h (22°C) with the secondary monoclonal antibody, rat adsorbed biotinylated horse anti-mouse immunoglobulin G (final dilution 1:200, Vector, Burlingame, CA). Sections were than incubated for 1h (22°C) with avidin-biotinylated horseradish peroxydase (Vectastain, Elite ABC kit, Vector, Burlingame, CA). After washing again in PBS, the sections were incubated with 0.05% 3, 3’-diaminobenzidine tetrahydrochloride (Vector, Burlingame, CA) as chromogen. The reaction was stopped by washing the section with buffer. No staining was detected in the absence of the primary or secondary antibodies. Sections were mounted on slides, air-dried and coverslipped with cytoseal (Allan Scientific, Kalamazoo, MI) mounting medium. The location of immunohistochemically-defined cells was examined by light microscopy. Quantification of cell density in the reconstructed hippocampal coronal sections was assessed with MetaMorph imaging software (Universal Image Corporation, West Chester, PA). Briefly, areas of interest were determined as previously reported in detail (Rosi et al., 2005), their surface measured, and the immunoreactive cells numerated, allowing use to determine a number of immunoreactive cells by millimeter square in the areas of interest.
A polyclonal antibody directed against the first ninety-nine amino-acid residue from the human CB1 (final dilution 1:500, Affinity Bioreagents, Golden, CO) was used to visualize CB1 receptors. After quenching endogenous peroxydase/activity and blocking nonspecific binding, the sections were rinsed in 0.1 M TRIS buffer (TB), pH 7.4, for 15min, in 0.1 M Tris buffered saline (TBS), pH 7.4, for 15 min, blocked using 2% avidin in TBS for 30 min, rinsed in TBS for 30 min, blocked using 2% biotin for 30min, and finally rinsed in TBS for 30 min. The tissue sections were then incubated in the anti-CB1 (diluted 1:500) overnight at 4°C. The antibody was diluted in a solution containing 0.1% Triton X-100 and 1% NGS in 0.1 M TBS. These sections were then rinsed in TBS for 60 min and incubated in biotinylated goat anti-rabbit IgG (diluted 1:200) for 90 min at room temperature. The sections were rinsed with TBS for 60 min and incubated for 1h (22°C) with avidin-biotinylated horseradish peroxydase (Vectastain, Elite ABC kit, Vector, Burlingame, CA). The sections were then rinsed with TBS for 30 min and then incubated with 0.05% 3,3’-diaminobenzidine tetrahydrochloride as chromogen. The reaction was stopped by washing the section with buffer. No staining was detected in the absence of the primary or secondary antibodies. Sections were mounted on slides, air-dried for 24 h.Counter staining (Cresyl Violet) was performed before slides were cover-slipped with cytoseal (Allan Scientific, Kalamazoo, MI) mounting medium.
Double immunofluorescence staining
Free floating sections were mounted on slides and air-dried under a hound. The tissues were than processed as described in details in Rosi et al., 2005. After washing in TBS solution the polyclonal rabbit anti-CB1 (Affinity Bioreagents, Golden, CO, dilution 1:500) was apply. After 48 h of incubation at 4°C, the sections were incubated for 2 h at room temperature with the secondary anti-rabbit biotinylated antibody (Vector, Burlingame, CA), followed by incubation with Avidin+Biotin amplification system (Vector, Burlingame, CA) for 45 minutes. The staining was visualized using the TSA fluorescence system CY3 (PerkinElmer Life Sciences, Emeryville, CA). After washing in TBS solution, the tissue was quenched and blocked again and incubated with the monoclonal antibody OX-6 (Pharmigen, San Diego, CA, final dilution 1:400) for 24 h or with the monoclonal antibody anti-GFAP (Chemicon, Temecula, CA, final dilution 1:500) for 24h. Before applying the biotinylated monoclonal secondary rat adsorbed antibody (Vector, Burlingame, CA) for 2 h, the tissue was incubated with Avidin Biotin Blocking Kit (Vector, Burlingame, CA) for 30 min to block cross reaction with the primary staining. Following treatment with an Avidin+Biotin amplification system (Vector, Burlingame, CA), the staining was visualized with TSA fluorescence system CY5 (PerkinElmer Life Sciences, Emeryville, CA) and the nuclei were counterstained with Sytox-Green (Molecular Probes, Eugene, OR). No staining was detected in the absence of the primary or secondary antibodies.
RESULTS
Chronic infusion of LPS and WIN-55212-2 injections were well tolerated by all rats: they gained weight normally for the duration of the study.
Behavior
No significant difference in locomotor activity (swim speed) was found across groups, p>0.1. An ANOVA performed on the latency results obtained in the water maze task revealed an overall main effect of testing day (F5, 218=16.057, p<0.0001) for all groups (See Figure 1) and an overall group effect (LPS versus aCSF) upon latency for each day of testing (F1, 328=11.367, p=0.0008 for day1; F1, 328=85.681, p<0.0001 for day2; F1,328=109.756, p<0.0001 for day3; F1, 328=96.25, p<0.0001 for day 4). There was no significant effect of treatment except for day 3 (F2, 328=5.788, p=0.0034). There was a significant interaction between group and drug on days 2 and 3 (F2, 328=3.982, p=0.0196 for day 2; F2, 328=12.641, p<0.0001 for day 3). Post-hoc analyses of each testing day revealed a significant treatment effect on day 1, where LPS+WIN 1 rats were significantly impaired compared to the aCSF+WIN 1 rats (p=0.0015). On days 2 and 3, all LPS-infused rats were significantly (p<0.0033) impaired, as compared to their respective control groups. On day 4, all LPS-infused rats were significantly (p<0.0033) impaired as compared to their respective aCSF controls. There was a significant interaction between treatment and LPS-infused animals (F2, 113=5.026, p=0.081). Post-hoc analyses of each testing day revealed a significant treatment effect on days 2 and 3; performance of LPS+WIN 1 rats was significantly (p<0.0033) worse as compared to the LPS+vehicle rats on those both days. Overall, the drug treatment (0.5 or 1 mg/kg) did not significantly impair performance of aCSF-infused rats and did not attenuate the impairment induced by the LPS infusion. The 1 mg/kg treatment, that did not cause any impairment in aCSF-infused rats, but did worsen the impairment observed in LPS-infused rats, demonstrating an interaction between inflammation and the highest dose of the drug used.
Figure 1.
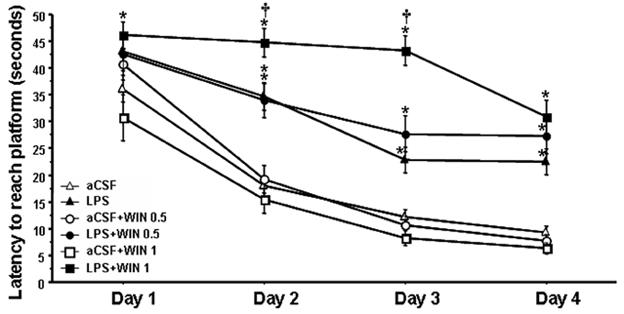
Water maze performance. On days 2 and 3, all LPS-infused animals (closed triangles, squares and circles) were significantly impaired (*p<0.0033, Bonferroni/Dunn post hoc test) compared to their control groups. On days 2 and 3, LPS+WIN 1 rats were significantly impaired († p<0.0033, Bonferroni/Dunn post hoc test) compared to the LPS+vehicle rats. WIN 55212-2 did not impair performance (open squares and circles) of aCSF-infused rats and did not attenuate the impairment due to the LPS infusion (closed squares and circles)
Histology
Immunostaining for OX-6 (Figure 2A) revealed numerous highly activated microglia cells distributed throughout the hippocampus of LPS-infused rats (Figure 2A d). The activated microglia had a characteristic bushy morphology with increased cell body size and contracted and ramified processes (Figure 2A d). Rats infused with aCSF had very few mildly activated microglia evenly scattered throughout the brain, consistent with results from previous studies (Hauss-Wegrzyniak et al., 1998; Rosi et al., 2005). No difference in staining was evident between the aCSF group and the aCSF rats injected with either dose of WIN (Figure 2A a-c). In LPS-infused rats that were also treated with WIN (Figure 2A e-f) the OX-6 antibody stained fewer activated microglial cells.
Figure 2.
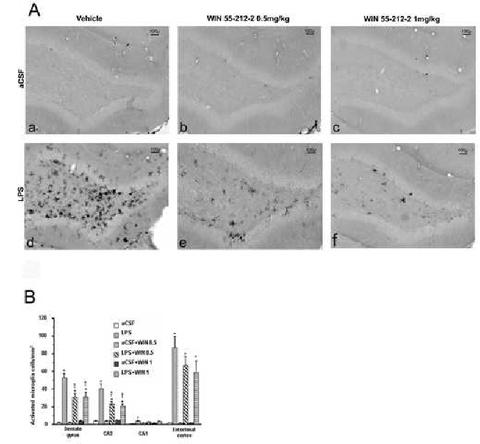
(A.) Activated microglia in the dentate gyrus. Note the diminution of activated microglia cells in the dentate gyrus of animals treated with either doses of WIN 55212-2 (e,f) compare to the LPS+vehicle group (d) (B.) Density of activated microglial cells in different areas of interest. Similar results were obtained for the two doses of WIN-55212-2 used in the dentate gyrus and CA3 areas. For both of them, LPS infused animal were significantly different from their control group (*p<0.0033, Bonferroni/Dunn post hoc test). The injection of both doses (0.5 and 1 mg/kg) reversed partially the LPS induction of activated microglia († p<0.0033, Bonferroni/Dunn post hoc test). No significant effect of the treatment was found for either CA1 or the entorhinal cortex.
Activated microglia cell counts
The number of activated microglia per millimeter square was determined in 4 different brain regions: dentate gyrus (DG), CA3 and CA1 regions of the hippocampus and the entorhinal cortex (EC) (Figure 2B). These brain regions were examined for their known implication in spatial learning (Nadel and Land, 2000). An ANOVA of the data revealed an overall main region effect (F5,210=16.057, p<0.0001) for all groups and an overall main effect of the infusion of LPS in each region examined (F5,48=32.557, p<0.0001 for DG; F5,48=23.552, p<0.0001 for CA3; F5,48=3.828, p=0.0053 for CA1;F5,48=19.308, p<0.0001 for EC).
In the DG, CA3 and EC, all LPS infused rats had a significantly (p<0.0033) higher density of activated microglia compared to their respective control groups, consistent with our previous results (Rosi et al., 2005). In the DG and CA3 the number of activated microglia was significantly (p<0.0033) reduced in LPS-infused rats given either dose of WIN, as compared to rats in the LPS+vehicle group (DG, 41.4% reduction for 0.5mg/kg and 40.6% reduction for 1mg/kg; CA3, 43.7% reduction for WIN 0.5mg/kg and 49.4% reduction for WIN 1mg/kg). In the EC, despite a 24.2% reduction for WIN 0.5mg/kg and 32.4% reduction for WIN 1mg/kg, no significant difference was found between the LPS-infused groups. In the CA1 region of the hippocampus, there was a significant (p<0.0033) difference only between the aCSF and LPS+vehicle groups. Overall, the effects of WIN were not dose-dependent in the DG and CA3 and not significant within the CA1 or EC brain regions.
CB1 receptors
The distribution of the CB1 receptors observed following our staining is in accordance with previous studies (Tsou et al., 1998) (Figure 3a). Neuronal CB1 immunoreactivity was found in the hippocampus, striatum, amygdala as well as in the somatosensory, cingulate and entorhinal cortices. The apparent density of immunoreactive cells in the areas of interest (DG, CA3, CA1, or EC) did not vary across groups.
Figure 3.
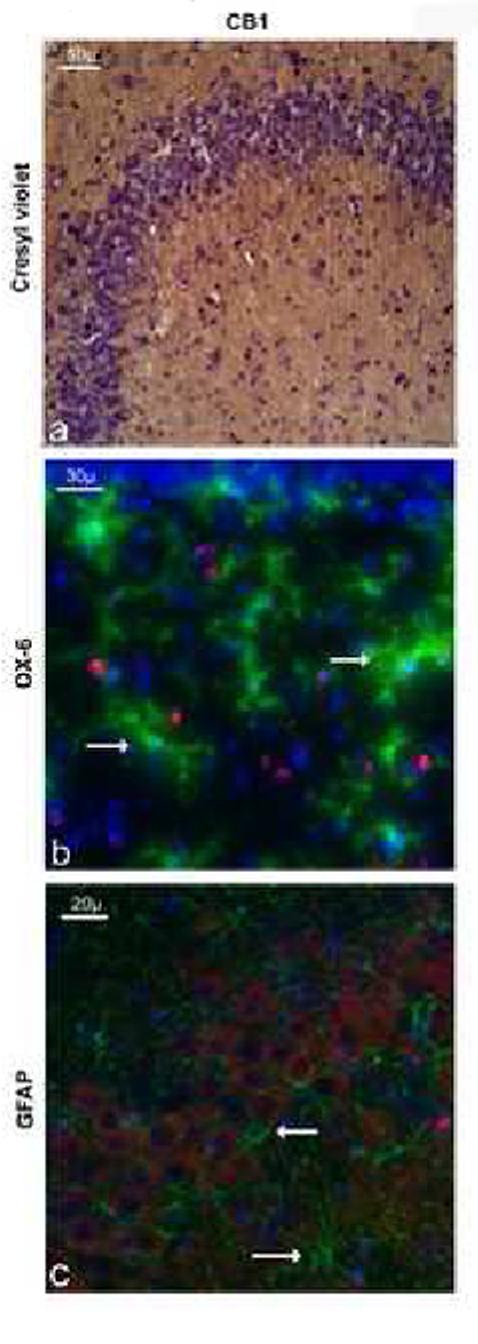
Immunoreactivity (IR) of CBr in the CA3 region of the hippocampus. (a) CB1-IR (DAB stain) is seen in the cytoplasm of all CA3 neuronal cell bodies (counterstaining with cresyl violet). (b) CB1-IR (red) and MCH-II-IR (green) do no co-localize, as indicated by white arrows. (c) CB1-IR (red) and GFAP-IR (green) do no co-localize as indicated by white arrows. CB1-IR thus seem to be located only within neurons in the hippocampus. These photomicrographs were obtained from only LPS-infused rats that are representative of the staining observed in aroups.
No co-localization between CB1 receptors and activated microglial cells
Double-immunofluorescence staining for CB1 and activated microglial cells performed on the brains of all LPS-infused groups did not show any co-localization (Figure 3b). These results indicate that CB1 receptors are not present on activated microglial cells in response to LPS infusion or treatment with a CBr agonist.
No co-localization between CB1 and astrocytes
Double-immunofluorescence staining for CB1 and astrocytes performed on the brains of all LPS-infused groups did not show any co-localization (Figure 3c). These results indicate that CB1 are not present on astrocytes in response to LPS infusion or treatment with a CBr agonist.
DISCUSSION
The results demonstrate that a CB1/CB2 agonist, WIN-55212-2, prevents microglial cell activation during LPS-induced chronic neuroinflammation in young rats. The effects of this agonist were not dependent on direct CB1 receptors stimulation of microglia or astrocytes, were region dependant and did not reverse the LPS-induced impairment in a spatial memory task. The neurodegeneration associated with AD may result from prolonged activation of microglia and a chronic elevation of cytokines and nitric oxide (Akiyama et al., 2000; Streit, 2004) leading to a cascade of self-propagating cellular events that alters the expression of cannabinoid receptors (Minghetti and Levi, 1998; Bernardino et al., 2005; Klein, 2005). Endocannabinoids are implicated in the modulation of the central inflammatory response by neurons and glia (Klein et al., 2003; Klein, 2005) and may have a neuroprotective role in several neuroinflammation related diseases (Klein, 2005). Cannabinoid agonists can prevent the activation of microglia by β-amyloid, reduce the subsequent release of TNF-α(Ramirez et al., 2005), and attenuate the induction and release of nitric oxide by cultured microglia (Waksman et al., 1999). An inhibition of glutamate release by cannabinoids and the subsequent reduction of the calcium influx via NMDA channels (Piomelli, 2003; Takahashi and Castillo, 2006) have also been demonstrated. Additionally, cannabinoids can attenuate oxidative stress and subsequent toxicity (Hampson and Grimaldi, 2001) and induce the expression of brain-derived neurotrophic factor following the infusion of kainic acid (Marsicano et al., 2003).
CBr are expressed in senile plaques (Benito et al., 2003; Ramirez et al., 2005) and the number of CB1 receptor-immunoreactive neurons is greatly reduced in areas of microglial activation in the AD brain (Ramirez et al., 2005). In contrast, we did not find evidence for changes in CB1 receptor expression on hippocampal neurons in response to brain inflammation or cannabinoid receptor stimulation. This absence of changes in CB1 receptors on neurons in our model may be related to the fact that the chronic infusion of LPS into young rats does not induce senile plaque formation (Hauss-Wegrzyniak et al., 1998) or neuronal loss in the hippocampus (Rosi et al., 2005), important factors that likely influence the expression of CBr-positive neuron in post-mortem studies on AD brains from aged humans. The LPS infusion into the 4th ventricle produces widespread activation of glia and impaired performance in the water maze task (Hauss-Wegrzyniak et al., 1998). In contrast, an infusion of amyloid into a lateral ventricle (Ramirez et al., 2005) produced a more localized and limited glial activation (frontal cortex) and performance impairment that responded positively to treatment with WIN 55-212-2 (Ramirez et al., 2005). This difference may explain why WIN 55-212-2 in our model did not reverse the impairment induced by LPS in the 4th ventricle.
In the current study, CB1 receptors were not co-localized with MCH II-positive microglia or GFAP-positive astrocytes within the hippocampus; the presence of these receptors appeared to be solely neuronal (Figure 3). This suggests an indirect role of CBr upon microglia that is linked to the modulation of neuronal activity by stimulation of the endocannabinoid system. Our results are consistent with previous in vivo findings (Tsou et al., 1998) that describe CBr receptors located on hippocampal neurons, and their modulator role upon glutamatergic and GABAergic function (Takahashi and Castillo, 2006; Katona et al., 1999). CBr are present on astrocytes and microglial cells taken from humans, monkeys, rats and mice when studied in vitro (Stella 2004). The contradictory findings we report may be related to many factors, such as the different species that have been studied, the microenvironment of the cells (in vivo vs in vitro), antibody sensitivity and selectivity, and the age or pathological condition under examination. Further experiments must be performed to determine clearly the characteristic and changes of the endocannabinoid system in our model and thus explain our present findings.
We have previously speculated that LPS induces a cascade of inflammatory processes that leads to an elevation in extracellular glutamate and activation of NMDA receptors (Wenk et al., 2006). The selective antagonism of NMDA receptors reduced microglia activation (Rosi et al., 2006) similar to that reported in the current study using WIN-55212-2. Taken together these findings are consistent with the hypothesis that the ability of endocannabinoids to reduce the release of glutamate within the hippocampus underlies the reduction of microglia activation by WIN-55212-2.
Interestingly, our results demonstrated an interaction between the presence of brain inflammation and the actions of the cannabinoid agonist at a dose (1 mg/kg/day i.p. of WIN 55-212-2) that did not impair performance in the control rats. Surprisingly, those doses of agonist (0.5 and 1 mg/kg/day i.p. of WIN 55-212-2) also did not improve performance, as might be expected given the results of our previous investigations using more typical anti-inflammatory drugs (Hauss-Wegrzyniak et al., 1999). We speculate that this was because the WIN 55-212-2 treatment was not able to reverse totally the activation of microglia that had been induced by the chronic LPS infusion; in the current study, the reduction in the number of activated microglia was only about 40-50%, in contrast to the ability of an non-steroidal anti-inflammatory drug’s ability to reduce the number of activated microglia almost completely. This partial reduction in microglia activation might thus not be sufficient to reverse the spatial memory impairment produced by the LPS infusion. This result is of particular importance with regard to patients suffering with a disease associated with brain inflammation, e.g. AD, Parkinson’s disease or multiple sclerosis, who are also using marijuana.
The current report is the first to our knowledge to demonstrate the modulatory role of cannabinoids in an animal model of chronic neuroinflammation, pointing out the effectiveness of a CBr agonist on the consequences of LPS mediated neuroinflammation at a dose (0.5 mg/kg/day i.p. of WIN-55212-2) that does not impair performance in a patial memory task. These results further advocate for the manipulation of the endocannabinoid system to diminish the consequences of neuroinflammation in progression of AD and others inflammation-related diseases (Klein, 2005).
Acknowledgments
We thank A. Sy, P. Raniero, F. Cerbai for technical assistance and Dr V. Ramirez-Amaya for critical experimental procedure assistance.
List of abbreviations
- Δ9-THC
Δ9-tetrahydrocannabinol
- aCSF
artificial cerebral spinal fluid
- AD
Alzheimer’s disease
- CB1
cannabinoid receptor 1
- CB2
cannabinoid receptor 2
- CBr
cannabinoid receptors
- DG
Dentate gyrus
- EC
Entorhinal cortex
- LPS
Lipopolysaccharide
- NMDA
N-methyl-d-aspartate
- PBS
phosphate buffer saline
- TBS
Tris buffer saline
- WIN-55212-2
(R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)-pyrrolo[1,2,3-de]-1,4benzoxazin-6-yl]-1-naphthalenyl-methanone mesylate
Footnotes
Financial support:This study has been supported by AG10546 (GLW).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Section editor: (j) Systems Neuroscience
REFERENCES
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cooper NR, Eikelenboom P, Emmerling M, Fiebich B, Finch CE, Frautschy S, Griffin WST, Hampel H, Landreth G, McGeer PL, Mrak R, MacKenzie I, O’Banion K, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray A. Inflammation in Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begg M, Pacher P, Batkai S, Osei-Hyiaman D, Offertaler L, Mo FM, Liu J, Kunos G. Evidence for novel cannabinoid receptors. Pharmacol Ther. 2005;2:133–145. doi: 10.1016/j.pharmthera.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Benito C, Nunez E, Tolon RM, Carrier EJ, Rabano A, Hillard CJ, Romero J. Cannabinoid CB2 receptors and fatty acid amide hydrolase are selectively overexpressed in neuritic plaque-associated glia in Alzheimer′s disease brains. J Neurosci. 2003;23:11136–11141. doi: 10.1523/JNEUROSCI.23-35-11136.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardino L, Xapelli S, Silva AP, Jakobsen B, Poulsen FR, Oliveira CR, Vezzani A, Malva JO, Zimmer J. Modulator effects of interleukin-1beta and tumor necrosis factor-alpha on AMPA-induced excitotoxicity in mouse organotypic hippocampal slice cultures. J Neurosci. 2005;25:6734–6744. doi: 10.1523/JNEUROSCI.1510-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano C, Rossi-Arnaud C, Cestari V, Costanzi M. Cannabinoids and memory: animal studies. Curr Drug Targets CNS Neurol Disord. 2003;2:389–402. doi: 10.2174/1568007033482670. [DOI] [PubMed] [Google Scholar]
- Facchinetti F, Del Giudice E, Furegato S, Passarotto M, Leon A. Cannabinoids ablate release of TNFalpha in rat microglial cells stimulated with lypopolysaccharide. Glia. 2003;41:161–168. doi: 10.1002/glia.10177. [DOI] [PubMed] [Google Scholar]
- Gong JP, Onaivi ES, Ishiguro H, Liu QR, Tagliaferro PA, Brusco A, Uhl GR. Cannabinoid CB2 receptors: immunohistochemical localization in rat brain. Brain Res. 2006;1071:10–23. doi: 10.1016/j.brainres.2005.11.035. [DOI] [PubMed] [Google Scholar]
- Hampson AJ, Grimaldi M. Cannabinoid receptor activation and elevated cyclic AMP reduce glutamate neurotoxicity. Eur J Neurosci. 2001;13:1529–1536. doi: 10.1046/j.0953-816x.2001.01536.x. [DOI] [PubMed] [Google Scholar]
- Hauss-Wegrzyniak B, Dobrzanski P, Stoehr JD, Wenk GL. Chronic neuroinflammation in rats reproduces components of the neurobiology of Alzheimer’s disease. Brain Res. 1998;780:294–303. doi: 10.1016/s0006-8993(97)01215-8. [DOI] [PubMed] [Google Scholar]
- Hauss-Wegrzyniak B, Vraniak P, Wenk GL. The effects of a novel NSAID upon chronic neuroinflammation are age dependent. Neurobiol Aging. 1999;20:305–313. doi: 10.1016/s0197-4580(99)00028-7. [DOI] [PubMed] [Google Scholar]
- Katona I, Sperlagh B, Sik A, Kafalvi A, Vizi ES, Mackie K, Freund TF. Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci. 1999;19(11):4544–58. doi: 10.1523/JNEUROSCI.19-11-04544.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SU, de Vellis J. Microglia in health and disease. J Neurosci Res. 2005;81:302–313. doi: 10.1002/jnr.20562. [DOI] [PubMed] [Google Scholar]
- Klein TW, Newton C, Larsen K, Lu L, Perkins I, Nong L, Friedman H. The cannabinoid system and immune modulation. J Leukoc Biol. 2003;74:486–496. doi: 10.1189/jlb.0303101. [DOI] [PubMed] [Google Scholar]
- Klein TW. Cannabinoid-based drugs as anti-inflammatory therapeutics. Nat Rev Immunol. 2005;5:400–411. doi: 10.1038/nri1602. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Cannich A, Azad SC, Cascio MG, Gutiérrez SO, van de Stelt M, López-Rodriguez ML, Casanova E, Schütz G, Zieglgänsberger W, Di Marzo V, Behl C, Lutz B. [Google Scholar]
- Minghetti L, Levi G. Microglia as effector cells in brain damage and repair: focus on prostanoids and nitric oxide. Prog Neurobiol. 1998;54:99–125. doi: 10.1016/s0301-0082(97)00052-x. [DOI] [PubMed] [Google Scholar]
- Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- Nadel L, Land C. Memory traces revisited. Nat Rev Neurosci. 2000;1:209–212. doi: 10.1038/35044572. [DOI] [PubMed] [Google Scholar]
- Neumann H. Control of glial immune function by neurons. Glia. 2001;36:191–199. doi: 10.1002/glia.1108. [DOI] [PubMed] [Google Scholar]
- Onaivi ES, Ishigurao H, Gong J, Patel S, Perchuk A, Meozzi P, Myers L, Mora Z, Tagliaferro P, Gardner E, Brusco A, Akinshola BE, Liu Q, Hope B, Iwasaki S, Arinami T, Teasenfitz L, Uhl GR. Discovery of the Presence and Functional Expression of Cannabinoid CB2 Receptors in Brain. Annals of the New York Academy of Sciences. 2006;1074:514–536. doi: 10.1196/annals.1369.052. [DOI] [PubMed] [Google Scholar]
- Piomelli D. The molecular logic of endocannabinoid signalling. Nat Rev Neurosci. 2003;4:873–884. doi: 10.1038/nrn1247. [DOI] [PubMed] [Google Scholar]
- Ramirez BG, Blazquez C, Gomez del Pulgar T, Guzman M, de Ceballos ML. Prevention of Alzheimer’s disease pathology by cannabinoids: neuroprotection mediated by blockade of microglial activation. J Neurosci. 2005;25:1904–1913. doi: 10.1523/JNEUROSCI.4540-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosi S, Ramirez-Amaya V, Hauss-Wegrzyniak B, Wenk GL. Chronic brain inflammation leads to a decline in hippocampal NMDA-R1 receptors. J Neuroinflammation. 2004;1:12. doi: 10.1186/1742-2094-1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosi S, Ramirez-Amaya V, Vazdarjanova A, Worley PF, Barnes CA, Wenk GL. Neuroinflammation alters the hippocampal pattern of behaviorally induced Arc expression. J Neurosci. 2005;25:723–731. doi: 10.1523/JNEUROSCI.4469-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosi S, Vazdarjanova A, Ramirez-Amaya V, Worley PF, Barnes CA, Wenk GL. Memantine reduces the consequences of chronic neuroinflammation and restores the hippocampal pattern of behaviorally-induced Arc expression. Neuroscience. 2006 doi: 10.1523/JNEUROSCI.4469-04.2005. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng WS, Hu S, Min X, Cabral GA, Lokensgard JR, Peterson PK. Synthetic cannabinoid WIN55,212-2 inhibits generation of inflammatory mediators by IL-1β -stimulated human Astrocytes. Glia. 2005;49:211–219. doi: 10.1002/glia.20108. [DOI] [PubMed] [Google Scholar]
- Stella N. Cannabinoid signaling in glial cells. Glia. 2004;48:267–277. doi: 10.1002/glia.20084. [DOI] [PubMed] [Google Scholar]
- Streit WJ. Microglia and Alzheimer′s disease pathogenesis. J Neurosci Res. 2004;77:1–8. doi: 10.1002/jnr.20093. [DOI] [PubMed] [Google Scholar]
- Takahashi KA, Castillo PE. The CB1 cannabinoid receptor mediates glutamatergic synaptic suppression in the hippocampus. Neuroscience. 2006;139:795–802. doi: 10.1016/j.neuroscience.2006.01.024. [DOI] [PubMed] [Google Scholar]
- Tsou K, Brown S, Sanudo-Pena MC, Mackie K, Walker JM. Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience. 1998;83:393–411. doi: 10.1016/s0306-4522(97)00436-3. [DOI] [PubMed] [Google Scholar]
- Van Sickle MD, Duncan M, Kingsley PJ, Mouihate A, Urbani P, Mackie K, Stella N, Makriyannis A, Piomelli D, Davison JS, Marnett LJ, Di Marzo V, Pittman QJ, Patel D, Sharkey KA. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science. 2005;310:329–32. doi: 10.1126/science.1115740. [DOI] [PubMed] [Google Scholar]
- Waksman Y, Olson J, Carlisle SJ, Cabral GA. The central cannabinoid receptor (CB1) mediates inhibition of nitric oxide production by rat microglial cells. J Pharmacol Exp Ther. 1999;288:1357–1366. [PubMed] [Google Scholar]
- Walter L, Franklin A, Witting A, Moller T, Stella N. Astrocytes in culture produce anandamide and other acylethanolamides. J Biol Chem. 2002;277:20869–20876. doi: 10.1074/jbc.M110813200. [DOI] [PubMed] [Google Scholar]
- Wenk GL, Parson C, Danysz W. Potential role of NMDA receptors as executors of neurodegeneration resulting from diverse insults -focus on memantine. Behavioral Pharmacology. 2006 doi: 10.1097/00008877-200609000-00007. in press. [DOI] [PubMed] [Google Scholar]