

Abstract
The human FMR1 gene contains a CGG repeat in its 5’ untranslated region. The repeat length in the normal population is polymorphic (5-55 CGG repeats). Lengths beyond 200 CGGs (full mutation) result in the absence of the FMR1 gene product, FMRP, through abnormal methylation and gene silencing. This causes Fragile X Syndrome, the most common inherited form of mental retardation. Elderly carriers of the premutation, defined as a repeat length between 55-200 CGGs can develop a progressive neurodegenerative syndrome: Fragile X-associated tremor/ataxia syndrome (FXTAS). In FXTAS, FMR1 mRNA levels are elevated and it has been hypothesised that FXTAS is caused by a pathogenic RNA gain-of-function mechanism. We have developed a knock-in mouse model carrying an expanded CGG-repeat (98 repeats), which shows repeat instability and displays biochemical, phenotypic and neuropathological characteristics of FXTAS. Here, we report further repeat instability, up to 230 CGGs. An expansion bias was observed, with the largest expansion being 43 CGG units and the largest contraction 80 CGG repeats. In humans, this length would be considered a full mutation and would be expected to result in gene silencing. Mice carrying long repeats (∼230 CGGs) display elevated mRNA levels and decreased FMRP levels, but absence of abnormal methylation, suggesting that modelling the Fragile X full mutation in mice require additional repeats, or other genetic manipulation.
Keywords: Fragile X Syndrome, CGG repeat, FXTAS, FMRP, FMR1, repeat instability, CpG methylation, mRNA
Introduction
The Fragile X mental retardation gene 1 (FMR1), involved in fragile X syndrome, contains a CGG repeat in its 5’-untranslated region. Depending on the length of this trinucleotide repeat, different clinical outcomes are possible. Repeats of normal individuals are within the range of 5 to 55 CGGs [1]. Repeat lengths greater than 200 CGGs (full mutation: FM) typically leads to methylation of both the CGG repeat and the FMR1 promoter resulting in transcriptional silencing of the gene. The consequent absence of FMRP in neurons is the cause of the mental retardation in Fragile X patients. Fragile X Syndrome is the most common genetic disorder associated with mental retardation [2].
The premutation (PM), comprising ∼55 to ∼200 unmethylated CGG repeats, was long thought to be associated only with a high risk of expansion to a full mutation upon maternal transmission. However, elevated FMR1 mRNA levels and normal or slightly reduced FMR1 protein (FMRP) were reported [3], [4]. Additionally, 20% of female PM carriers are at risk of developing premature ovarian failure [5]. In 2001 the first evidence of a new neurological syndrome (Fragile X Associated Tremor/Ataxia Syndrome: FXTAS) was published by Hagerman and colleagues, observed in five elderly male PM carriers. Patients presented with progressive intention tremor, leading to executive function deficits and generalised brain atrophy on MRI scans. Since this neurological syndrome is restricted to the PM range, reduced levels of FMRP are unlikely to be the underlying cause [6]. However, since elevation of FMR1 mRNA levels seems to be correlated to the length of the CGG repeat [4, 7, 8], and cognitive and functional impairment increases with the number of CGG repeats [9], an RNA-gain-of-function effect has been proposed, in which elevated levels of FMR1 mRNA containing an expanded CGG repeat lead to progressive neurodegeneration [6].
Little is known about when and how CGG repeat instability takes place. In order to be able to elucidate the timing and mechanism of CGG repeat instability and methylation of the FMR1 gene, a mouse model was generated by Bontekoe and colleagues [10]. In this model, the endogenous mouse CGG repeat (8 trinucleotides) of a wild type mouse was exchanged with a human CGG repeat of 98 trinucleotide units, which is in the PM range in humans. This ‘knock in’ CGG triplet mouse shows moderate repeat instability upon both maternal and paternal transmission and displays biochemical, phenotypic and neuropathological characteristics of FXTAS [10, 11].
In this paper, we report further repeat instability, up to lengths above 200 CGG units. In humans, this would implicate a full mutation, thus silencing of the gene. Mice carrying these long repeats (∼230 CGGs) display elevated mRNA levels and decreased FMRP levels, but absence of CpG methylation, which would mean that in mice a full mutation has not occurred as yet. However, additional mice with long CGG repeats will be necessary to fully evaluate this issue.
Materials and methods
Mice
Both the knock in CGG triplet mice and wild type mice (parent of the knock in mouse, with a mouse endogenous (CGG)8 repeat) were housed in standard conditions. All experiments were carried out with permission of the local ethical committee. Repeat lengths were determined for the whole mouse colony but only male mice were used for the experiments.
Isolation of DNA from mouse tails
DNA was extracted from mouse tails by incubating with 0.2 mg/ml Proteinase K (Roche Diagnostics) in 335 μl lysis buffer (50 mM Tris-HCl pH 7.5, 10 mM EDTA, 150 mM NaCl, 1% SDS) overnight at 55°C. After adding 100 μl saturated NaCl solution the next day, the suspension was centrifuged. Two volumes of 100% ethanol were added and gently mixed. The appearing DNA cloud was fished out with a plastic pipet tip and subsequently washed and centrifuged in 500 μl 70% ethanol. The DNA was then dissolved in 100 μl milliQ-H2O.
Repeat length determination
Following a method kindly provided by K. Usdin (personal communication), CGG repeat lengths were measured using the Expand High Fidelity PCR System (Roche Diagnostics). Approximately 300 ng of tail DNA was added to a PCR mixture with a total volume of 50 μl, containing 0.2 μM of each primer, 200 μM of each dNTP (Invitrogen), 2% DMSO (Sigma), 2.5 M Betaine (Sigma), 5U Expand High Fidelity Plus PCR System Enzyme and 10 μl of the 5x Expand HF buffer with Mg (7.5 μM). As forward primer 5’-CGGAGGCGCCGCTGCCAGG-3’ was used and 5’-TGCGGGCGCTCGAGGCCCAG-3’ as reverse. PCR conditions were 10 minutes initial denaturation at 95°C, followed by 35 cycles of denaturation for 1 minute at 95°C, annealing for 1 minute at 65°C and elongation for 5 minutes at 75°C, with a final elongation step of 10 minutes at 75°C. 25 μl of PCR product was mixed with an equal volume of loading dye (95% formamide, 0.1% brome phenol blue, 0.1% xylene cyanol) and loaded onto a denaturing 6% poly acrylamide (PA)(19:1) gel, which was run in 0.6x TBE at 18 Watts. Separated DNA was visualised on a UV-lamp after soaking the gel in 0.6x TBE with ethidium bromide. Approximate repeat lengths were determined using a standard curve, based on DNA samples of which repeat lengths were determined with an ABI-based Fragile X size polomorphism assay in the past [10].
As these primers are specific to the knock-in allele and do not detect wild type alleles, a separate PCR is necessary to distinguish females heterozygous or homozygous for the repeat. TaKaRa LA Taq was used with GC buffer II according to manufacturers instructions (Takara Bio Inc), with 5’-GCTCAGCTCCGTTTCGGTTTCACTTCCGGT-3’ as forward primer and 5’-AGCCCCGCACTTCCACCACCAGCTCCTCCA-3’ as reverse primer.
Transmission pattern of repeat alleles
The mode of transmission of the repeat allele was investigated, by comparison of the repeat lengths present in the tails of the breeding couple with those of their offspring. Since the ability of the PA gels to resolve lengths of PCR products in this range is limited, relatively large bin sizes were chosen to describe repeat length. Comparisons between parents and offspring were made using PCR products run on the same gel. Frequencies of contractions, stable transmissions and expansions were tested with a one-sample T test. Magnitude and direction (contraction, stable transmission or expansion) of the repeat instability, gender of the offspring and parental origin of the repeat allele were taken into account. Transmission groups were tested for differences in magnitude of contractions and expansion separately, using one-way ANOVA. Effect of gender of offspring, as well as effect of parental allele of origin was investigated with independent samples T-tests for both contractions and expansions. Because of small expected frequencies, Fisher’s Exact Test was performed to test for possible differences in frequencies of contractions, stable transmissions or expansions, among the different transmission groups. The same test was used to determine the significance of differences in frequencies of an allele fitting into the categories distinguishing magnitude of instability (categories shown in fig.2). A difference in absolute magnitude of contractions vs. expansions was investigated using an independent samples T-test.
Fig.2.
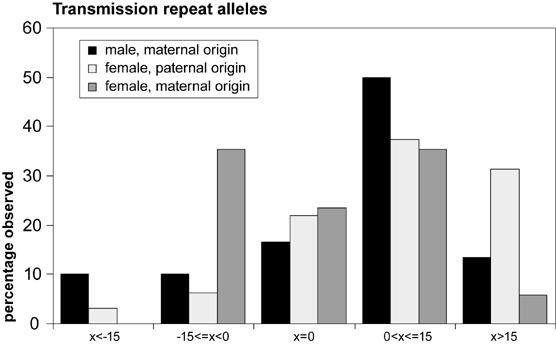
Histogram summarizing the observed instability of CGG repeat alleles upon transmission to the next generation. Bars represent the percentage of times a transmitted allele fits in an instability category (x=change in repeat length), within a transmission group (male offspring, maternal origin, or female offspring with either maternal or paternal origin). Fisher’s Exact Test revealed no statistically significant differences in frequencies of instability categories amongst the transmission groups (P=12.86, sd=0.09).
RNA isolation, RT and Q-PCR analysis
Mouse brains were homogenised on ice in buffer (PBS with 1% NP-40, Complete protease inhibitor cocktail tablets (Roche Diagnostics), 3 mM DTT (Invitrogen) and 20U RNAsin (Promega). 1 ml RNA bee (Tel-Test) was added to 100 μl brain homogenate. 200 μl chloroform was then added and the mixture was then spun at 4°C for phase separation. 1 Volume of isopropanol was added to the aqueous phase to precipitate the RNA. The pellet was washed with 80% ethanol and dissolved in DEPC-treated MilliQ-H2O. RNA concentration and purity was determined using a NanoDrop ND-1000 Spectrophotometer (NanoDrop Technologies).
RT-PCR was performed on 2 μg RNA using iScript cDNA Synthesis Kit (BioRad) according to manufacturer’s instructions.
For Q-PCR two primer sets for Fmr1 (transition exon 5/6 (5’-AGATCAAGCTGGAGGTGCCA-3’ as forward primer and 5’-CAGAGAAGGCACCAACTGCC-3’ as reverse) and transition exon 16/17 (5’-CCGAACAGATAATCGTCCACG-3’ as forward primer and 5’-ACGCTGTCTGGCTTTTCCTTC-3’ as reverse) and two primer sets for two distinct internal controls (Gapdh: forward: 5’-CCTGGAGAAACCTGCCAAGTAT-3’ and reverse: 5’-CCCTCAGATGCCTGCTTCA-3’) and Rpl13a: forward: 5’-TCTCAAGGTTGTTCGGCTGA-3’ and reverse: 5’-GTGGCTGTCACTGCCTGGTA-3’ were used. Since efficiencies of the different primer sets were not identical when tested, it was necessary to make a standard curve for each primer pair in every run. Input quantities were calculated by subjecting Ct values to the formula of the appropriate standard curves. A ratio of Fmr1 mRNA to the reference gene was then calculated for each mouse. This Fmr1/reference-ratio was standardised to 1 for wild type (wt) mice. Fmr1/reference -ratios were compared with the ratio in wt mice of the same age. 1/10 and 1/100 dilutions of RT product were used as input material. All dilutions were performed in duplicate.
Western blotting
Half brains (saggital) were homogenised in 500 μl HEPES-buffer (10 mM HEPES, 300 mM KCl, 3 mM MgCl2, 100 μM CaCl2, 0.45% Triton X-100 and 0.05% Tween-20, pH 7.6, with Complete protease inhibitor cocktail (Roche Diagnostics), while kept on ice. After incubating the homogenates on ice for 30 minutes, they were sonicated twice for 20 seconds. Cell debris was spun down and the supernatant was collected. Loading mix was added to 100 μg of protein, heated at 95°C for 5 minutes and loaded onto a 10% SDS-PAGE gel. After electroblotting the gel onto a nitrocellulose membrane, the me mbrane was incubated overnight at 4°C with the monoclonal 2F5-1 antibody specific for FMRP [12], in PBS-T with 5% milk powder. The lower part of the membrane was incubated, under the same conditions, with an antibody for synaptophysin. The next day the membrane was incubated with a horseradish peroxidase conjugated secondary antibody rabbit-α-mouse (DAKO), allowing chemiluminescence detection with an ECL KIT (Amersham).
Methylation status
DNA was checked for the presence of methylated CpGs in the promoter region in two ways. A quick screen consists of overnight digestion of 1 μg DNA with the methylation-sensitive enzyme BssHII (after pre-digestion with EcoRI to facilitate complete digestion with BssHII). The digested DNA is then subjected to PCR over the promoter region containing the BssHII-site (forward primer: 5’-AAATCTACCCAATGCCCCTCC-3’, reverser primer: 5’-CCTGGGTACCTGCCTCAGG-3’). The PCR mixture contained 10x PCR buffer, W-1, 1 unit Taq Polymerase (all Invitrogen) and 50 mM Spermidine (Fluka BioChemika). The PCR program was as follows: 5 minutes denaturation at 95°C, 35 cycles of 30 seconds at 95°C, 30 seconds annealing at 56°C and 1.5 minutes at 72°C, followed by a final elongation step of 10 minutes at 72°C. A non-methylated BssHII site can be cut by BssHII, and hence will not produce a PCR product. Methylated DNA prevents BssHII digestion, thus a PCR product can be produced.
The other method consists of subjecting 1.5 μg of DNA to bisulphite conversion, using the Qiagen Epitect kit, according to manufacturers instructions. 1.5 μl of converted DNA was then used for a PCR using forward primers 5’-GAAGTGAAATCGAAACGTTTTTTAGCGTTT-3’ and 5’-GAAGTGAAATTGAAATGTTTTTTAGTGTTT-3’ and reverse primer 5’-ATTTATCACTTTTTTTTTACTTCCCTCC-3’. In order to be able to amplify a fragment with unknown methylation status, it is necessary to use a combination of primers, one of which matches the sequence after conversion in the case where the DNA was methylated and another primer in case it was unmethylated. The PCR mixture was prepared as described above for the PCR after methylation-sensitive digestion, except for that 50 mM MgCl2 was added. The PCR program was the same as described above with the exception of an annealing temperature of 63.5°C. 5 μl of PCR product was then used for a semi-nested re-PCR, since no PCR product was seen on a 2% agarose gel initially. Primers used in the second PCR reaction were forward: 5’-TTTTGTAAATAGTTAAATGATGTTATGTGATTTG-3’ and 5’-TTTTGTAAATAGTTAAACGATGTTACGTGATTCG-3’ and reverse: 5’-TTTTTTACTTCCCTCCCACCAC-3’ and 5’-TTTTTTACTTCCCTCCCGCCGC-3’. PCR products were visualised and cut out of a 2% agarose gel and the bisulphite converted PCR product was purified using an Illustra GFX PCR DNA and gel band purification kit (GE Healthcare). The resulting converted DNA was then sequenced as follows: amplification reactions were performed in 22 μl containing 1x Invitrogen PCR buffer, 1.5mM MgCl2, 0.01% W-1, 250 μM of each dNTP, 1 μM forward primer,1 μM reverse primer (primers were the same as the ones used in the second PCR reaction), 0.75 units of Platinum Taq DNA polymerase (Invitrogen) and 25 ng genomic DNA; DNA Cycle conditions: 7’30” 95°C; 10 cycles of 30” denaturation 94°C, annnealing 68°C minus 1°C per cycle, 1’ extension 72°C followed by 25 cycles of 30” denaturation 94°C, annealing 58°C, 1’ extension 72°C; final extension 5’ 72°C. Templates for the Direct Sequencing reactions were cleaned from dNTP’s and primers using 1 ul ExoSAP-IT (USB) during 30 minutes at 37°C followed by a 15 minutes inactivation step at 80°C. Direct sequencing of both strands was performed using Big Dye Terminator chemistry ver.3.1 (Applied Biosystems) as recommended by the manufacturer. Fragments were loaded on an ABI3100 automated sequencer and analyzed with DNA Sequencing Analysis (ver.3.7) and SeqScape (ver.2.1) software (Applied Biosystems).
Immunocytochemistry
Mice were sacrificed by cervical dislocation, brains were dissected immediately and fixed overnight in 3% paraformaldehyde. The brains were embedded in paraffin according to standard protocols. Sections (7 μm) were deparaffinized, followed by antigen retrieval using microwave treatment in 0.01 M sodium citrate solution. Endogenous peroxidase activity blocking and immunoincubation was performed as described before using monoclonal antibodies against FMRP (1C3) [13].
Results
The original knock in construct introduced 98 CGG repeats into the mouse Fmr1 locus and has been used as a model for the human premutation disorder, FXTAS [10, 11, 14]. This line was initially developed to study repeat instability of long CGG repeats in the mouse Fmr1 locus and has been maintained for several years now. In the course of breeding, animals with longer repeats were born and selected for breeding in an attempt to further increase the size of the CGG array, with the hypothesis that repeat instability will be greater as the repeat tract gets longer. We previously reported expansions of this tract to sizes of 110 CGG units [14]. CGG repeat lengths in the mouse model have now expanded to above the threshold of human FMR1 full mutations. This study focuses on the characteristics of these mice, which are summarised in table 1.
Table 1.
Main characteristics of the CGG repeat knock in mice described in this study. Noneof these mice were littermates.
Repeat instability
In order to investigate the mode of transmission of CGG repeats in the expanded CGG repeat mouse model, mice were bred and repeat lengths in the offspring were compared to repeat lengths in the breeding couple. The CGG repeat shows instability upon transmission to the next generation (figure 1). Both expansions and contractions occur in addition to stable transmissions. Predominantly small expansions are found along with stable transmissions. Both germ line and somatic mutations can be observed. In figure 1, for example, animal 2 is a mosaic for repeat length, showing two long repeat alleles (approx. 190 and 200 CGGs) as well as a shorter allele (approx. 60 CGGs), which must represent a mitotic contraction, which was transmitted to one of her male offspring (animal 3). Figure 1 also shows the mouse with the longest CGG repeat length (>230: animal d in figure 1) observed thus far, as well as the controls used for repeat length determination.
Fig. 1.
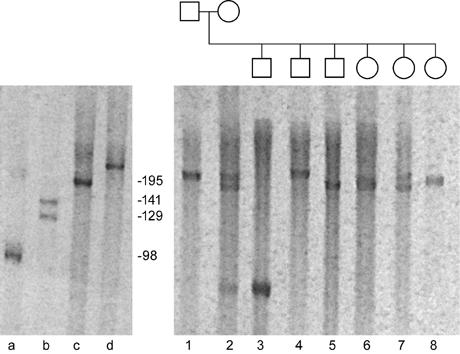
PA gel with PCR products of an expanded CGG repeat litter and the breeding couple. The CGG repeat shows both expansion and contraction upon transmission to the next generation.
Transmission pattern of repeat alleles
Mode of transmission of CGG repeat alleles was investigated by comparison of repeat alleles of breeding couples with those of their offspring and subjected to statistical analysis. Figure 2 shows instability of repeat alleles in three different transmission groups, based on gender of offspring and origin of the allele. Seventy-nine alleles, of which the parental allele of origin was known, were subjected to statistical analysis. Among all transmissions, 15 contractions (19%), 16 stable transmissions (20%) and 48 expansions (61%) were observed. A one-sample T-test revealed that these frequencies were statistically significantly different from expected frequencies (adjusted p-value for contractions=0.006, for stable transmissions: padj=0.006 and for expansions padj<0.001). The largest contraction was of 80 CGG repeats and the biggest expansion was 43 trinucleotide units. Comparing mean magnitude of instability between contractions (n=15, mean=15.93, sd=21.0) and expansions (n=48, mean=12.8, sd=10.5), among all alleles considered, with an independent samples T-test revealed no statistical significance (t=0.55, p=0.59). Table 2 summarises these data. One-way ANOVA revealed no statistically significant difference in magnitude of instability of contractions (F=3.28, p=0.07) between the different transmission groups, namely male offspring with a repeat allele of maternal origin (MM)(mean=-30.7, standard deviation (sd)= 11.3), female offspring with an allele of paternal origin (FP)(mean=-7.67, sd=4.7) and female offspring with a maternally derived repeat allele (FM)(mean=-5.33, sd=0.42). Neither was a difference found in magnitude of expansions (F=1.36, P=0.27) between MM (mean=11.32, sd=2.4), FP (mean=15.36, sd=2.5) and FM (mean=8.86, sd=1.9). When looking at the factors that determine the transmission groups, gender of offspring and parental allele of origin, separately, for both contractions and expansions, no specific transmission pattern is revealed with independent samples T-tests. Considering contractions, no effect (t=-2.15, p=0.08) for gender of offspring (male: n=6, mean=-30.7, sd=27.8, female: n=9, mean=-6.1, sd=4.3). It must be noted that numbers are small, so that the largest contraction, which occurred in a male mouse, has a relatively big influence on the outcome. There was no difference (t=-7.96, p=0.43) between magnitude of mean male expansion (11.32, sd=10.4, n=19) and mean female expansion (13.79, sd=10.6, n=29). Testing for an effect of parental allele of origin revealed no statistically significant outcome for contractions (t=0.75, p=0.47) nor expansions (t=1.6, p=0.12) (contractions: mean(paternal origin)=-7.7, sd=8.4, n=3, mean(maternal origin)=-18.0, n=12, sd=22.9; expansions: mean(paternal origin)=15.4, sd=11.5, n=22, mean(maternal origin)=10.7, sd=9.2, n=26). Fisher’s Exact Test revealed no statistically significant differences in observed frequencies of contractions, stable transmissions and expansions (P=5.70, p=0.33) between the transmission groups. Neither were there statistically significant differences in frequencies of instability categories amongst the transmission groups (P=12.86, sd=0.09). Means of the magnitude of repeat instability are shown in table 3 for the different transmission groups.
Table 2.
Summary of the observations on direction and magnitude of instability. SS: statistically significant, NSS: not statistically significant (see text for test statistics and pvalues).
| Type of instability | n | Largest instability | Mean Magnitude (sd) |
|---|---|---|---|
| Contractions | 15 | -80 | 16 (21.0) |
| Stable transmissions | 16 | - | - |
| Expansions | 48 | +43 | 13 (10.5) |
| Statistically significant? | SS | NSS | NSS |
Table 3.
Summary of the mean repeat instability observed in mice specified for the different transmission groups. Displayed are the mean magnitude of instability (sd), number of observations. See text for the outcome of statistical analyses.
| Transmission groups | Instability category | |||||
|---|---|---|---|---|---|---|
| Δrep < -15 | -15<Δrep <0 | Δrep=0 | 0<Δrep<15 | Δrep>15 | total | |
| Male, maternal | -51 (25.9), n=3 | -11 (7.5), n=3 | n=5 | 7 (3.9), n=15 | 28 (10.5), n=4 | 1 (21.9), n=30 |
| Female, paternal | -17, n=1 | -3 (1.4), n=2 | n=7 | 7 (4.0), n=12 | 26 (8.5), n=10 | 10 (12.9), n=32 |
| Female, maternal | n=0 | -5 (1.0), n=6 | n=4 | 8 (4.4), n=6 | 16, n=1 | 2 (7.2), n=17 |
| total | -42 (27.0), n=4 | -6 (4.5), n=11 | n=16 | 7 (3.9), n=33 | 26 (8.9), n=15 | 5 (16.5), n=79 |
Q-PCR analysis
Since human carriers of the premutation have elevated Fmr1 mRNA levels [3, 4], we investigated mRNA levels of mice with different repeat lengths, to determine whether repeat length has an influence on mRNA abundance. mRNA ratios proved to be independent of the amount of input RNA, tested by using two dilutions of input material (data not shown). First we investigated wt mice of different ages and found a two-fold increase of Fmr1 mRNA in 100-week old wt mice, as compared with 25-week old wt mice. Next we tested Fmr1 mRNA levels in CGG repeat mice of different repeat lengths, with reference to age-matched wt mice. 100-week old mice with a CGG repeat of 110 and 130 trinucleotide units showed a two-fold increase of Fmr1 mRNA, with reference to age-matched wt mice (data not shown). Figure 3 shows Fmr1 mRNA levels for mice around 25 weeks of age, with reference to 25-week old wt mice. Bars represent two mice in each category, with an exception of category >230, which represents a single mouse. Four combinations of two primer pairs within the Fmr1 gene and two primer pairs for two distinct reference genes, Gapdh and Rpl13a, were measured and calculated. Figure 3 shows comparison of Fmr1 mRNA levels as measured with a primer pair amplifying the transition of exon 16 to exon 17 and Gapdh as a reference gene (the ratio in wt mice is standardised to 1). This graph is representative for all combinations of primer pairs. Both Gapdh and Rpl13a showed stable Ct values throughout all CGG length categories, thus proved to be suitable reference genes in this assay. The range of Ct values obtained for the different genes were 26 to 28 for Fmr1, 20 to 22 for Gapdh and 19 to 21 for Rpl13a. Throughout all repeat length categories an approximately two-fold increase of the Fmr1 mRNA is observed with reference to the age-matched wt, with exception of the single observation of the mouse in category >230, which shows a five-fold increase.
Fig. 3.
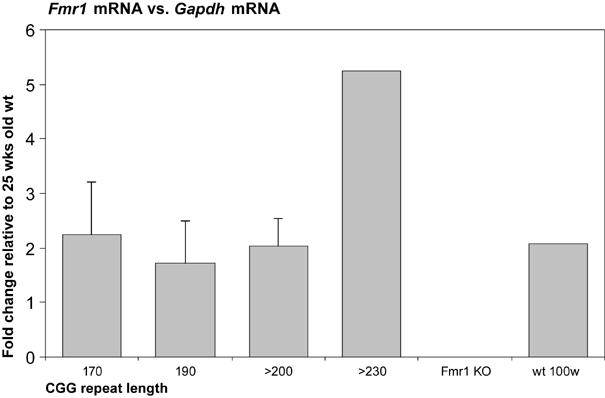
Brain Fmr1 mRNA levels are shown relative to 25-week old male mice. Bars represent two male mice of roughly the same repeat length (SEM were 0.9, 0.8 and 0.5 respectively for 170, 190 and >200 CGGs), with exception of >230, which only represents one male mouse.
Protein levels
After finding elevated Fmr1 mRNA levels in a mouse with over 230 CGG repeats, we were interested in Fmr1 protein levels in these mice. We performed Western blotting on whole brain lysates and found significantly reduced Fmrp levels (21%, 5% and 51% of wt levels respectively (corrected for input material)) in three mice with approximately 230 CGG repeats, one of which (C), with the longest repeat tract, is the mouse that showed five-fold Fmr1 mRNA levels (figure 4).
Fig.4.
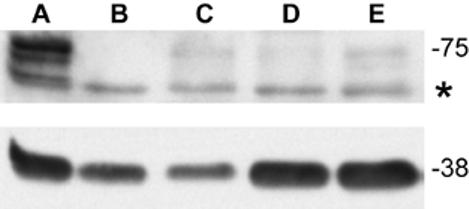
Western blot of brain tissue showing Fmrp isoforms (at 70-80kD) of wt (A), Fmr1 KO male mouse (B) and male mice with approximately 230 CGGs (C, D and E). Mouse C,D and E show clearly reduced Fmrp levels: 21%, 5% and 51% of wt levels respectively (corrected for input material). The asterisk marks a background band. Synapthophysin (at 38 kD) was used as a loading control.
Immunocytochemistry
It was of interest to investigate the decrease in Fmrp expression in vivo, using immunohistochemistry on brains of two of the three mice tested by Western blotting. Mice with approximately 230 CGG repeats show significantly diminished Fmrp expression throughout the brain, although detectable Fmrp remained (figure 5). Strikingly, the CA2 and CA3 region of the hippocampus showed the highest levels of FMRP expression in the ∼230 CGG mice. This region, however, is also highly immunoreactive for Fmrp in wt mice.
Fig.5.
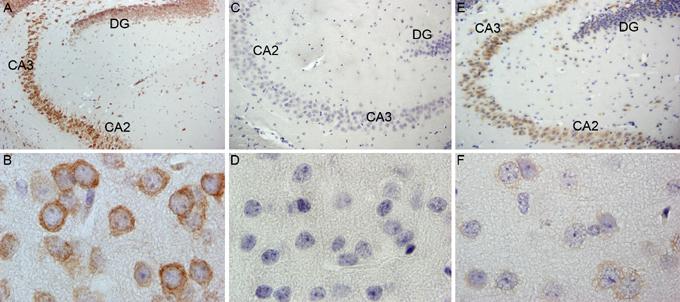
Mouse brains from wt (A and B), Fmr1 KO (C and D) and the mouse with >230 CGG repeats (E and F) were stained with antibodies against Fmrp. Photos A,C and E show hippocampus and B,D, and F show cortex tissue. DG: dentate gyrus
Methylation status
The decrease in Fmrp expression led to the question of whether the Fmr1 promoter and CGG repeats are methylated in mice carrying ∼230 repeats. To obtain a general idea thereof, a methylation sensitive digestion was performed, followed by PCR over the promoter region. No PCR products were seen after methylation-sensitive digestion of tail DNA of all male mice tested thus far, suggesting that the restriction site (BssHII) was not methylated (data not shown). We confirmed the unmethylated status of the Fmr1 promoter by applying bisulphite conversion followed by sequencing of PCR products of the converted DNA. We have successfully applied the bisulphite-conversion method in the past [15],[16] Sequencing revealed complete bisulphite conversion of cytosines to uracil indicating that no methylated cytosines were present in the Fmr1 promoter region of the starting material (data not shown).
Discussion
We have managed to obtain mice carrying full mutation length CGG repeats in the Fmr1 locus through selective breeding of a line of mice that were created by knocking in 98 CGGs. In this study, we have characterised these animals for repeat instability during transmission and for expression of the Fmr1 mRNA and protein. We find that mice carrying large CGG repeat alleles express increased amounts of mRNA, but significantly reduced levels of Fmrp. These findings are consistent with the observation that the large CGG alleles, which would be considered full mutations in humans, remain unmethylated in male mice.
CGG repeat instability
In humans, instability of a PM to a FM occurs exclusively upon maternal transmission [17]. Also general instability seems to be greater when the repeat allele is of maternal origin. No effect on instability was observed for gender of offspring in humans [18]. In our mouse model, we find a significant excess of repeat expansions over both contractions and stable transmissions, which is consistent with observations of human premutations. Statistical analysis revealed no significant differences in magnitude or direction of instability for the factors that defined transmissions groups, namely gender of offspring or parental allele of origin.
Different mouse models with random (autosomal) insertion of unstable human PM alleles (maximum uninterrupted CGG stretch of 97 repeats) showed a very high incidence of small instabilities and occasional large deletions, but no large expansions [19, 20]. Random integration on an autosome, rather than on the X chromosome and the absence of necessary cis-acting factors might explain the different findings. Peier and Nelson described the generation of transgenic mice carrying PM-sized CGG repeats on a YAC, which enables them to study instability of these sequences within their chromosomal context. When looking at different constructs with varying (uninterrupted) CGG repeat lengths, they observed length-dependent instability. More expansions, however of smaller magnitude, were found than contractions. Instability of the CGG repeat was seen in about 17% of transmissions [21]. In humans, AGG interruptions in the CGG repeat have a stabilising role. Loss of AGG interruptions typically occurs often before a big jump towards the full mutation range, thus reflecting greater instability [22]. The expanded CGG repeat allele present in the knock-in mouse does not contain AGG interruptions [10]. Thus, it could be expected that greater instability already occurred right after the generation of the mouse model. Our initial report on the instability of an expanded CGG repeat in the Fmr1 promoter described a much lower frequency of instability than currently seen [10]. However, in earlier studies a different method was applied to determine CGG repeat length, so no accurate comparison can be made with the methodology employed in this study. However, no obvious change in magnitude of repeat instability is observed over time. It seems that the risk of becoming unstable upon transmission becomes greater as the CGG repeat gets longer, rather than the magnitude of the instability.
Many questions remain unanswered as to how, why and when trinucleotide repeat instability occurs. However, some mechanisms have been proposed and some evidence has been given for the underlying mechanism of repeat instability. The observation in human fragile X syndrome that expansions to a full mutation (FM) only occur upon maternal transmission of a premutation allele, means that repeat expansion must occur during meiosis in the oocyte. Alternatively, it could be due to mitotic events in the embryonic development of the germline [23, 24]. The mosaic pattern, as often seen in Fragile X patients, must result from mitotic contraction or expansion events in the developing embryo, leading to the presence of alleles with repeats in both the FM and PM range [10]. In male Fragile X patients, only PM alleles are present in sperm and testes [25, 26], despite the presence of FM in peripheral lymphocytes and other tissues [27]. In a 13-week old FM fetus, no FMRP expression could be demonstrated in primordial germ cells (PGCs), while in a 17-week old FM fetus some primordial germ cells expressed FMRP. It was then concluded by the authors that a PM allele must have been present in the PGCs to support protein production. This suggests that contraction from FM to PM may occur during the maturation of the PGCs, after the initial replication event [24]. In both mouse and human zygotes, the paternal chromatin undergoes active demethylation soon after fertilisation [28]. This process, confined to the paternal pronucleus, occurs prior to DNA replication [29, 30]. In mouse, the demethylation process continues up to the morula stage [28]. After completion of the first cell cycle passive demethylation occurs, due to the absence of a maintenance methylase [31]. Soon after implantation, DNA methylation is restored and maintained thereafter [32]. CpG methylation appears to stabilise the CGG repeat instability [33]. Hence, paternally derived alleles can be prone to deletions upon demethylation. Cells in the testis that have undergone contraction to PM lengths, thus producing FMRP, might be selectively maintained, as FMRP is thought to play a role in gonadal development [34].
Despite the abundance of repetitive DNA in the genome, no general microsatellite instability is found in fragile X patients, thus repeat instability as seen in the FMR1 gene is limited to the disease locus only. This implies that expansions do not result from aberrant trans-acting factors involved in DNA replication, repair and recombination [35]..
Repeat instability is dependent on both the repeat length and the purity of the CGG-tract, i.e. the number and locations of AGG interruptions, which in turn determine the likelihood and stability of alternate DNA structures such as hairpins [36]. Also, the location of transcription start sites has been shown to be affected. It was demonstrated that a shift to more upstream transcription initiation sites takes place as the CGG repeat expands to the premutation range. This was interpreted as that the CGG repeat might act as a downstream enhancer or modulator of transcription. This phenomenon was observed both in neuronal and non-neuronal cell lines [37]. The expanded CGG repeat mouse model as described here might provide insights about repeat instability in vivo, especially with the expanded range of repeat lengths now available.
Transcription and translation
Q-PCR analysis on RNA isolated from mouse brains revealed that Fmr1 mRNA levels are two-fold higher in 100-week old wt animals, as opposed to 25-week old wt mice, illustrating that Fmr1 mRNA is more abundant in old wt mice. On average a two-fold increase of Fmr1 mRNA levels has been reported at different ages (1-72 weeks) in expanded CGG repeat mice, when compared with age-matched wt mice [14]. Here we report two-fold elevated Fmr1 mRNA levels for mice with 110-200 CGG repeats, in comparison with age-matched wt mice, which is in line with earlier findings in the expanded CGG repeat mouse [14]. So, based on the current data, in contrast to observations in humans [4, 7, 8], in mice Fmr1 mRNA levels do not seem to elevate further with added CGG repeat length. Interestingly, one exception to this observation was seen in the mouse with over 230 CGG repeats, which showed a five-fold increase. More mice with repeats of this length should be investigated in the future, in order to be able to assess to what extent mice resemble humans with regard to Fmr1 mRNA levels. Also, it should be taken into account that in mice brain mRNA was measured, whereas findings in humans represent blood mRNA. Tassone and co-workers have shown that Fmr1 mRNA levels, relative to a reference gene, are higher in brain than in blood in humans. However, the ratio of Fmr1 mRNA levels in PM carriers is higher in blood (3.8x) than in brain (1.5x) [38]. Thus our observations of a two-fold elevation of Fmr1 mRNA levels in CGG repeat mice seems to mimic the human situation.
The underlying molecular mechanism of the elevated FMR1 transcript levels has not been defined yet. Tassone and colleagues were the first to report elevated FMR1 mRNA levels (at least five-fold) in leukocytes of carriers of the high PM range (100-200 CGGs), in combination with a reduced percentage of FMRP-immunoreactive cells [3]. They proposed a model wherein an increase of transcription compensates for a diminished translational efficiency that might occur due to an expanded CGG repeat, in an attempt to overcome an arising protein deficit. A (linear) correlation between FMR1 mRNA levels and repeat size was recently confirmed in a large cohort of both males and females, among whom were carriers of common, intermediate and PM alleles [7]. Similar correlations between FMR1 transcript levels and repeat length were observed in human lymphoblastoid cell lines of PM carriers. Also, an inverse correlation was observed in these cell lines between FMRP levels and repeat length [4, 8]. Primerano and co-workers found an up to five-fold increase of Fmr1 mRNA in a cell line of a carrier with 195 repeats [8], while in the study by Kenneson and colleagues transcription levels did not exceed a three-fold increase when repeat length approached 200 units [4]. Both authors explained their observations by the occurrence of a translational defect that arises as the mRNA carries a longer CGG tract. Primerano et al revealed a dramatic change in translational efficiency of Fmr1 mRNA specifically; they demonstrated a reduced percentage of Fmr1 mRNA associated with polysomes in cell lines expressing a longer CGG repeat tract. However, they showed that a CGG repeat does not impair overall translational activity [8].
When comparing our own observations in mice with what has been described for human PM carriers, it appears that the positive linear relation between CGG repeat length and Fmr1 transcript levels is absent. Furthermore, in humans the increase in FMR1 mRNA levels as compared to controls was greater than in mice, although in the study performed by Kenneson and colleagues this increase was less pronounced [4]. However, the mouse carrying the largest CGG expansion detected in our lab thus far, did exhibit a five-fold increase in Fmr1 mRNA levels, when compared to age-matched wild type control mice. It must be stressed that this was only observed in a single mouse. This mouse also showed a striking difference in protein expression level in brain, as visualised with immunohistochemistry. Brain tissue of a second mouse with approximately 230 CGGs showed the same decreased Fmrp expression, despite the two-fold increase of Fmr1 mRNA. Throughout the brain a remarkable decrease in Fmrp expression could be detected, which was confirmed with Western blot. Interestingly, the CA2 and CA3 region of the hippocampus showed the strongest Fmrp expression. This is a region with the highest Fmrp expression in wt mice.
It is tempting to regard this particular expanded CGG repeat mouse, with the longest repeat tract as yet and the highest Fmr1 mRNA levels, as a parallel to the human situation where at the end of the PM range mRNA levels increase significantly and the number of FMRP-immunoreactive lymphocytes drops slightly, reflecting a decrease in translation [3]. However, it is too early to draw conclusions, since, due to limited availability of mice with repeat tracts this long; thus far this has only been a single observation. Hence, in the near future more mice with repeat lengths over 230 CGGs should be investigated in order to be able to distinguish a parallel to the human situation from an outlier.
To explain the impaired translation, many authors refer to a study performed by Feng et al. in 1995, in which they showed that the 40S ribosomal subunit is hampered in scanning for initiation codons when the FMR1 mRNA contains over 200 CGGs [39].However, individuals carrying an unmethylated full mutation have been reported, who showed normal FMRP expression in their EBV-transformed ly mphoblasts and show normal cognitive functioning [40]. The observation that mice with repeat lengths of over 200 trinucleotide units, which in humans would constitute a full mutation, still express Fmrp confirms that in murine cells ribosomes are still capable of translating mRNAs carrying CGG repeats of this size.
When considering that a FM in humans is defined as a CGG repeat length greater than 200 CGG triplets, and that these alleles are typically hypermethylated [2, 41], it can be concluded that a different situation occurs in mice carrying Fmr1 CGG repeats of this length. It is as yet unknown whether a mechanism similar to that seen in humans might happen at a greater repeat length in mice, or whether a transition to a silenced Fmr1 gene might never take place in mice. It is possible that certain methyltransferases that are responsible for CpG methylation of the FMR1 promoter and the CGG repeat in humans are not present in mice. In embryonic development, differences exist in (de)methylation mechanisms between man and mouse. Active demethylation occurs up to a later stage in mouse, as compared with in humans [28]. After the first cell cycle, passive demethylation occurs, as a result of the absence of a maintenance methylase [31]. It could be that similar mechanisms play a role in mice with respect to the Fmr1 gene, preventing this region from becoming methylated once the CGG tract exceeds 200 CGG repeats.
This mouse model provides an excellent tool to investigate whether methylation and silencing of the Fmr1 gene takes place in mice. In case a mechanism similar to that in humans does occur, the events that trigger methylation, the timing and sequence of methylation and gene silencing events can be studied in detail with special emphasis to the gonads. Furthermore, it will be interesting to perform behavioural tests in mice with lowered protein expression and possibly in mice with silenced genes to investigate learning and memory processes.
Acknowledgments
We are thankful to Dr. K. Usdin for providing the PCR protocol for amplification of the CGG repeat. Also, we would like to thank Prof. J. Fallon for kindly providing us with the 2F5 antibody against FMRP. Furthermore, we are grateful to Elisabeth Lodder for useful discussion and to Ruud Koppenol and Tom de Vries Lentsch for support with graphics. This study was financially supported by the Prinses Beatrix Fonds (JB), NFXF (RW) and NIH ROI HD38038 (B.A.O and D.L.N).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fu YH, Kuhl DP, Pizzuti A, Pieretti M, Sutcliffe JS, Richards S, Verkerk AJ, Holden JJ, Fenwick R, Jr., Warren ST, Oostra BA, Nelson DL, Caskey CT. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell. 1991;67:1047–1058. doi: 10.1016/0092-8674(91)90283-5. [DOI] [PubMed] [Google Scholar]
- 2.Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP, Eussen BE, Van Ommen GJ, Blonden B, Riggins LAJ, Chastain GJ, Kunst JL, Galjaard CB, Caskey H, Nelson CT, Oostra DL, and BA, Warren ST. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- 3.Tassone F, Hagerman RJ, Taylor AK, Gane LW, Godfrey TE, Hagerman PJ. Elevated levels of FMR1 mRNA in carrier males: A new mechanism of involvement in the Fragile-X syndrome. Am J Hum Genet. 2000;66:6–15. doi: 10.1086/302720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kenneson A, Zhang F, Hagedorn CH, Warren ST. Reduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediate-length and premutation carriers. Hum Mol Genet. 2001;10:1449–1454. doi: 10.1093/hmg/10.14.1449. [DOI] [PubMed] [Google Scholar]
- 5.Sherman SL. Premature Ovarian Failure among Fragile X Premutation Carriers: Parent-of-Origin Effect? Am J Hum Genet. 2000;67:11–13. doi: 10.1086/302985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hagerman RJ, Leehey M, Heinrichs W, Tassone F, Wilson R, Hills J, Grigsby J, Gage B, Hagerman PJ. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology. 2001;57:127–30. doi: 10.1212/wnl.57.1.127. [DOI] [PubMed] [Google Scholar]
- 7.Allen EG, He W, Yadav-Shah M, Sherman SL. A study of the distributional characteristics of FMR1 transcript levels in 238 individuals. Hum Genet. 2004;114:439–447. doi: 10.1007/s00439-004-1086-x. [DOI] [PubMed] [Google Scholar]
- 8.Primerano B, Tassone F, Hagerman RJ, Hagerman P, Ama ldi F, Bagni C. Reduced FMR1 mRNA translation efficiency in Fragile X patients with premutations. RNA. 2002;8:1–7. [PMC free article] [PubMed] [Google Scholar]
- 9.Grigsby J, Brega AG, Jacquemont S, Loesch DZ, Leehey MA, Goodrich GK, Hagerman RJ, Epstein J, Wilson R, Cogswell JB, Jardini T, Tassone F, Hagerman PJ. Impairment in the cognitive functioning of men with fragile X-associated tremor/ataxia syndrome (FXTAS) J Neurol Sci. 2006 doi: 10.1016/j.jns.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 10.Bontekoe CJ, Bakker CE, Nieuwenhuizen IM, van Der Linde H, Lans H, de Lange D, Hirst MC, Oostra BA. Instability of a (CGG)(98) repeat in the Fmr1 promoter. Hum Mol Genet. 2001;10:1693–9. doi: 10.1093/hmg/10.16.1693. [DOI] [PubMed] [Google Scholar]
- 11.Van Dam D, Errijgers V, Kooy RF, Willemsen R, Mientjes E, Oostra BA, De Deyn PP. Cognitive decline, neuromotor and behavioural disturbances in a mouse model for Fragile-X-associated tremor/ataxia syndrome (FXTAS) Behavioural Brain Research. 2005;162:233–239. doi: 10.1016/j.bbr.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 12.Gabel LA, Won S, Kawai H, McKinney M, Tartakoff AM, Fallon JR. Visual Experience Regulates Transient Expression and Dendritic Localization of Fragile X Mental Retardation Protein. J Neurosci. 2004;24:10579–10583. doi: 10.1523/JNEUROSCI.2185-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bakker CE, de Diego Otero Y, Bontekoe C, Raghoe P, Luteijn T, Hoogeveen AT, Oostra BA, Willemsen R. Immunocytochemical and biochemical characterization of FMRP, FXR1P, and FXR2P in the mouse. Exp Cell Res. 2000;258:162–70. doi: 10.1006/excr.2000.4932. [DOI] [PubMed] [Google Scholar]
- 14.Willemsen R, Hoogeveen-Westerveld M, Reis S, Holstege J, Severijnen L, Nieuwenhuizen I, Schrier M, VanUnen L, Tassone F, Hoogeveen A, Hagerman P, Mientjes E, Oostra BA. The FMR1 CGG repeat mouse displays ubiquitin-positive intranuclear neuronal inclusions; implications for the cerebellar tremor/ataxia syndrome. Hum Mol Genet. 2003;12:949–59. doi: 10.1093/hmg/ddg114. [DOI] [PubMed] [Google Scholar]
- 15.Pietrobono R, Pomponi MG, Tabolacci E, Oostra B, Chiurazzi P, Neri G. Quantitative analysis of DNA demethylation and transcriptional reactivation of the FMR1 gene in fragile X cells treated with 5-azadeoxycytidine. Nucleic Acids Res. 2002;30:3278–85. doi: 10.1093/nar/gkf434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stoyanova V, Oostra BA. The CGG Repeat and the FMR1 Gene. Methods Mol Biol. 2004;277:173–84. doi: 10.1385/1-59259-804-8:173. [DOI] [PubMed] [Google Scholar]
- 17.Rousseau F, Heitz D, Biancalana V, Blumenfeld S, Kretz C, Boue J, Tommerup N, Van Der Hagen C, DeLozier-Blanchet C, Croquette MF, Gilgenkranz S, Jalbert P, Voelckel MA, Oberlé I, Mandel JL. Direct diagnosis by DNA analysis of the fragile X syndrome of mental retardation. N Engl J Med. 1991;325:1673–1681. doi: 10.1056/NEJM199112123252401. [DOI] [PubMed] [Google Scholar]
- 18.Nolin SL, Brown WT, Glicksman A, Houck GE, Jr, Gargano AD, Sullivan A, Biancalana V, Brondum-Nielsen K, Hjalgrim H, HolinskiFeder E, Kooy F, Longshore J, Macpherson J, Mandel JL. Expansion of the Fragile X CGG Repeat in Females with Premutation or Intermediate Alleles. Am J Hum Genet. 2003;72:454–464. doi: 10.1086/367713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lavedan C, Grabczyk E, Usdin K, Nussbaum RL. Long uninterrupted CGG repeats within the first exon of the human FMR1 gene are not intrinsically unstable in transgenic mice. Genomics. 1998;50:229–240. doi: 10.1006/geno.1998.5299. [DOI] [PubMed] [Google Scholar]
- 20.Fleming K, Riser DK, Kumari D, Usdin K. Instability of the fragile X syndrome repeat in mice: the effect of age, diet and mutations in genes that affect DNA replication, recombination and repair proficiency. Cytogenet Genome Res. 2003;100:140–6. doi: 10.1159/000072848. [DOI] [PubMed] [Google Scholar]
- 21.Peier A, Nelson D. Instability of a premutation-sized CGG repeat in FMR1 YAC transgenic mice. Genomics. 2002;80:423–432. doi: 10.1006/geno.2002.6849. [DOI] [PubMed] [Google Scholar]
- 22.Poon PM, Chen QL, Zhong N, Lam ST, Lai KY, Wong CK, Pang CP. AGG interspersion analysis of the FMR1 CGG repeats in mental retardation of unspecific cause. Clin Biochem. 2006;39:244–248. doi: 10.1016/j.clinbiochem.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 23.Moutou C, Vincent MC, Biancalana V, Mandel JL. Transition from premutation to full mutation in fragile X syndrome is likely to be prezygotic. Hum Mol Genet. 1997;6:971–979. doi: 10.1093/hmg/6.7.971. [DOI] [PubMed] [Google Scholar]
- 24.Malter HE, Iber JC, Willemsen R, De Graaff E, Tarleton JC, Leisti J, Warren ST, Oostra BA. Characterization of the full fragile X syndrome mutation in fetal gametes. Nature Genet. 1997;15:165–169. doi: 10.1038/ng0297-165. [DOI] [PubMed] [Google Scholar]
- 25.Tassone F, Hagerman RJ, Gane LW, Taylor AK. Strong similarities of the FMR1 mutation in multiple tissues: postmortem studies of a male with a full mutation and a male carrier of a premutation. Am J Med Genet. 1999;84:240–4. [PubMed] [Google Scholar]
- 26.Reyniers E, Martin JJ, Cras P, Van Marck E, Handig I, Jorens HZ, Oostra BA, Kooy RF, Willems PJ. Postmortem examination of two fragile X brothers with an FMR1 full mutation. Am J Med Genet. 1999;84:245–9. doi: 10.1002/(sici)1096-8628(19990528)84:3<245::aid-ajmg16>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 27.Reyniers E, Vits L, De Boulle K, Van Roy B, Van Velzen D, de Graaff E, Verkerk AJMH, Jorens HZ, Darby JK, Oostra BA, Willems PJ. The full mutation in the FMR-1 gene of male fragile X patients is absent in their sperm. Nature Genet. 1993;4:143–146. doi: 10.1038/ng0693-143. [DOI] [PubMed] [Google Scholar]
- 28.Fulka H, Mrazek M, Tepla O, Fulka J., Jr. DNA methylation pattern in human zygotes and developing embryos. Reproduction. 2004;128:703–8. doi: 10.1530/rep.1.00217. [DOI] [PubMed] [Google Scholar]
- 29.Mayer W, Niveleau A, Walter J, Fundele R, Haaf T. Demethylation of the zygotic paternal genome. Nature. 2000;403:501–2. doi: 10.1038/35000656. [DOI] [PubMed] [Google Scholar]
- 30.Oswald J, Engemann S, Lane N, Mayer W, Olek A, Fundele R, Dean W, Reik W, Walter J. Active demethylation of the paternal genome in the mouse zygote. Curr Biol. 2000;10:475–8. doi: 10.1016/s0960-9822(00)00448-6. [DOI] [PubMed] [Google Scholar]
- 31.Monk M, Adams RL, Rinaldi A. Decrease in DNA methylase activity during preimplantation development in the mouse. Development. 1991;112:189–92. doi: 10.1242/dev.112.1.189. [DOI] [PubMed] [Google Scholar]
- 32.Monk M, Boubelik M, Lehnert S. Temporal and regional changes in DNA methylation in the embryonic, extraembryonic and germ cell lineages during mouse embryo development. Development. 1987;99:371–82. doi: 10.1242/dev.99.3.371. [DOI] [PubMed] [Google Scholar]
- 33.Nichol Edamura K, Pearson CE. DNA Methylation and Replication: Implications for the “Deletion Hotspot” Region of FMR1. Hum Genet. 2005;118:301–304. doi: 10.1007/s00439-005-0037-5. [DOI] [PubMed] [Google Scholar]
- 34.Tamanini F, Willemsen R, van Unen L, Bontekoe C, Galjaard H, Oostra BA, Hoogeveen AT. Differential expression of FMR1, FXR1 and FXR2 proteins in human brain and testis. Hum Mol Genet. 1997;6:1315–1322. doi: 10.1093/hmg/6.8.1315. [DOI] [PubMed] [Google Scholar]
- 35.Cleary JD, Pearson CE. Replication fork dynamics and dynamic mutations: the fork-shift model of repeat instability. Trends Genet. 2005;21:272–80. doi: 10.1016/j.tig.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 36.Pearson CE, Eichler EE, Lorenzetti D, Kramer SF, Zoghbi HY, Nelson DL, Sinden RR. Interruptions in the triplet repeats of SCA1 and FRAXA reduce the propensity and complexity of slipped strand DNA (S-DNA) formation. Biochemistry. 1998;37:2701–8. doi: 10.1021/bi972546c. [DOI] [PubMed] [Google Scholar]
- 37.Beilina A, Tassone F, Schwartz PH, Sahota P, Hagerman PJ. Redistribution of transcription start sites within the FMR1 promoter region with expansion of the downstream CGG-repeat element. Hum Mol Genet. 2004;13:543–549. doi: 10.1093/hmg/ddh053. [DOI] [PubMed] [Google Scholar]
- 38.Tassone F, Hagerman RJ, Garcia-Arocena D, Khandjian EW, Greco CM, Hagerman PJ. Intranuclear inclusions in neural cells with premutation alleles in fragile X associated tremor/ataxia syndrome. J Med Genet. 2004;41:E43. doi: 10.1136/jmg.2003.012518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feng Y, Zhang FP, Lokey LK, Chastain JL, Lakkis L, Eberhart D, Warren ST. Translational suppression by trinucleotide repeat expansion at FMR1. Science. 1995;268:731–734. doi: 10.1126/science.7732383. [DOI] [PubMed] [Google Scholar]
- 40.Smeets H, Smits A, Verheij CE, Theelen J, Willemsen R, Losekoot M, Van de Burgt I, Hoogeveen AT, Oosterwijk J, Oostra BA. Normal phenotype in two brothers with a full FMR1 mutation. Hum Mol Genet. 1995;4:2103–2108. doi: 10.1093/hmg/4.11.2103. [DOI] [PubMed] [Google Scholar]
- 41.Oberle I, Rousseau F, Heitz D, Kretz C, Devys D, Hanauer A, Boue J, Bertheas MF, Mandel JL. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science. 1991;252:1097–102. doi: 10.1126/science.252.5009.1097. [DOI] [PubMed] [Google Scholar]