

Abstract
The loss of mitochondrial function has been implicated in a number of maladies such as Huntington’s Disease, Parkinson’s Disease, cancer and cardiovascular disease. The objective of this research was to develop a radiolabeled mitochondrial probe. Two tracers, 7′-Z-iodorotenol and 7′-Z-iodorotenone, analogs of rotenone a natural product that inhibits Complex I of the mitochondrial electron transport chain, have been labeled with iodine-125 in 45-85% yield in a single step from the corresponding tribuytylstannyl precursor. In vivo distribution in adult male Sprague-Dawley rats for both compounds showed high accumulation in the heart (1.7-3.7 %ID/g at 1h), a tissue with high mitochondrial content. Z-Iodorotenol did not washout of most tissues between 1 and 2 h post injection whereas Z-iodorotenone showed moderate washout (7-26%) over the same period. By 24 h there was significant loss of both compounds from most tissues including the heart. Heart-to-blood, -lung and -liver ratios for Z-iodorotenone of 28.9, 10.7 and 2.4, respectively, were two- to four-fold higher than the Z-iodorotenol ratios. Compared to the current clinical perfusion tracers, 99mTc-sestamibi and 99mTc-tetrofosmin, Z-iodorotenone demonstrates similar 1h heart accumulation and significantly higher heart-to-lung ratio (P <0.001). Z-Iodorotenone heart-to-liver ratio is equivalent to 99mTc-sestamibi. 7′-Z-Iodorotenone possesses distribution characteristics of an improved tracer for SPECT perfusion studies.
Keywords: iodine-125, rotenone, mitochondria, complex I
1. Introduction
Mitochondrial dysfunction has been linked to normal aging processes as well as a variety of diseases including neurodegeneration (Alzheimer’s, Huntingon’s and Parkinson’s diseases) [1], cancer, osteoarthritis and cardiovascular disease. Wallace and colleagues have gathered evidence supporting a unified threshold hypothesis [2]. Wallace posits that each individual is born with a given mitochondrial mass. As one ages mutations to the mitochondrial DNA (mtDNA) diminishes the mitochondrial energy capacity. Organ failure or disease may ensue if the mitochondrial capacity falls below a certain threshold in a given organ system. Normal aging is defined under this hypothesis as a slow loss of energy capacity over ones lifetime ultimately leading to progressive system failure. Mitochondrial-related disease is characterized by either an inherited genetic defect that significantly lowers the initial amount of mitochondria or environmental factors such as oxidative damage to mtDNA that speeds up the energy capacity loss as postulated in the case of Parkinson’s Disease [3, 4].
Rotenone (1, Figure 1), a natural product found in the roots of the tropical Derris plant, has been shown to be a potent reversible inhibitor (IC50 = 0.25 nM) of Complex I of the mitochondrial electron transport chain [5]. Rotenone and preparation of crude root extracts have been widely used as “organic” insecticides and piscicides, due to their biological activity and rapid breakdown to environmentally benign byproducts [6, 7]. Several rotenonids have been isotopically labeled with 13C, 14C, 2H and 3H for structural studies [8] and to assess Complex I activity in vivo and in particular the loss of Complex I related to Parkinson’s Disease (PD) [9]. Tritiated (6′,7′)-dihydrorotenone [10] has been used to determine the regional distribution of Complex I in rodent brain [11, 12] as well as to assess binding to intact human platelets as a potential biomarker of PD [13].
Figure 1.
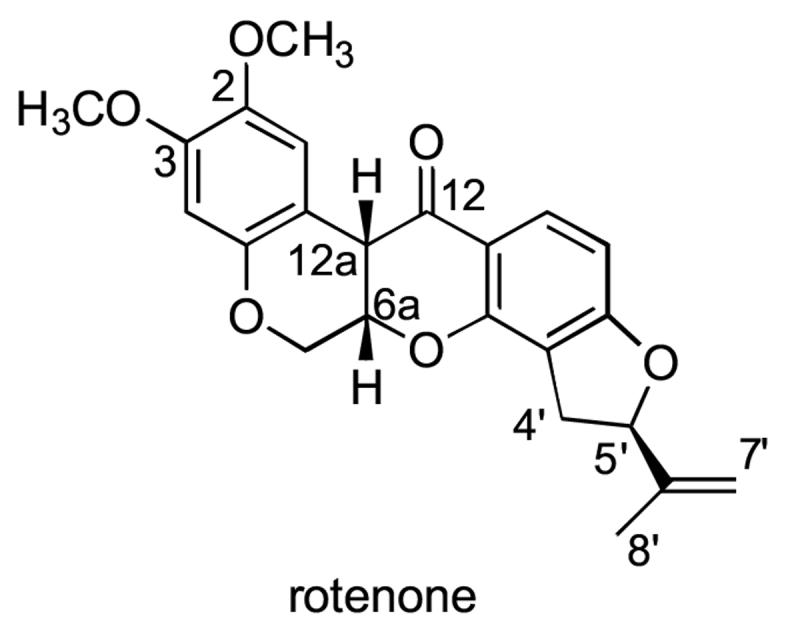
(5′R, 6aS, 12aS)-Rotenone structure shown with the conventional numbering system [8].
Positron labeled analogs have been developed and evaluated as prospective in vivo imaging probes of Complex I activity. The brain distribution of two carbon-11 labeled radiotracers, (2-[11C]methoxy)rotenone and (2-[11C]methoxy)-6′,7′-dihydroroten-12-ol, were evaluated in balb/c female mice [14, 15]. The regional distribution in the mouse brain was relatively equivalent with the overall brain uptake of the rotenol being higher than that of the rotenone. Further assessment of the [11C]dihydrorotenol in unilateral intrastriatal quinolinic acid and MPP+ lesioned rats demonstrated the in vivo sensitivity of the tracer to the loss of Complex I in the striatum [16, 17]. Fluorine-18 labeled rotenone analogs, 6′,7′-dihydro-7′-fluororotenone and 12-deoxy-6′,7′-dihydro-7′-fluororotenone, have been produced and evaluated in rats. Both compounds exhibited rapid clearance from the blood accompanied by early uptake in the brain and heart [18-20]. The 12-deoxy compound cleared from brain and heart indicative of its reduced Complex I affinity. While several of these tracers possess suitable imaging characteristics, the Complex I imaging capabilities of these tracers has yet to be fully exploited.
Ongoing studies in our laboratory have corroborated the heart uptake seen in the Emory studies [18] and have demonstrated the avidity of the labeled rotenone analogs for the heart. Initial assessment of the 7′ (Z)-[125I]iodorotenone and 6′,7′-dihydro-7′-[18F]fluororotenone in the isovolumic, retrograde, red blood cell-perfused rabbit heart demonstrated the high extraction and retention of the rotenones as a function of blood flow [21, 22]. Both analogs possessed better linearity with flow than the current clinical tracer 99mTc-sestamibi, a tracer that accumulates in the heart tissue based on the mitochondrial membrane potential.[23, 24] It is not surprising that the rotenone analogs accumulate in the heart tissue given that mitochondria represent 20-30% of the myocardial mass.[25] Thus, given their distribution and biologic properties labeled rotenones represent a new class of potential cardiac perfusion tracers.
The detailed syntheses of two radioiodinated rotenone analogs, 7′-(Z)-iodorotenol (4, Scheme 1) and 7′-(Z)-iodorotenone (5, Scheme 1), are reported herein. The corresponding iodine-125 labeled derivatives were prepared by iododestannylation of the tributylstannane intermediates. Additionally, the biological distribution of both radiotracers was evaluated in normal Sprague-Dawley rats.
Scheme 1.

Preparation of unlabeled 7′(Z)-iodorotenol 4 and 7′(Z)-iodorotenone 5.
2. Materials and methods
2.1 General
All chemicals and solvents were obtained from Aldrich Chemical Co. (Milwaukee, WI) and were used without further purification. Solvents were dried by standard techniques. All reactions were performed under a nitrogen atmosphere. High-performance liquid chromatography (HPLC) was performed using a Waters 590 solvent pump with an in-line Linear™ Model UV-106 UV detector (254 nm). An in-line NaI(Tl) radioactivity detector was added for radioHPLC. Melting points were determined using a Mel-Temp apparatus and are reported uncorrected. The 1H NMR spectra were recorded with either a Bruker 300 or 400 MHz spectrometer with CDCl3 as the internal standard (δ 7.26 ppm). Elemental analyses were performed by the Microanalytical Laboratory, operated by the College of Chemistry, University of California, Berkeley. High resolution (HRMS) and low resolution (LRMS) mass spectral determinations (EI or FAB) were made at the Midwest Center for Mass Spectrometry, University of Nebraska-Lincoln. High specific activity (2175 Ci/mmol) Na[125I]iodide in pH 9-12 aqueous 0.1M NaOH was purchased from Perkin Elmer Life Sciences.
Rotenol (2) was prepared by previously reported methods [26].
2.2 6′-Oxo-7′-desmethylene-rotenol(3)
To a solution of 2 (4.7 g, 11.9 mmol) in THF (100 mL) and water (40 mL) was added a small crystal of OsO4. After stirring for 5 min, NaIO4 (7.6 g, 35.6 mmol) was added and stirring was continued for 8 h. The mixture was filtered through celite, and the filtrate was diluted with water and extracted with ethyl acetate. The extracts were washed with brine and dried (MgSO4). Concentration in vacuo afforded a residue that was purified by flash chromatography (3:2 EtOAc:hexane) providing oxo-rotenol 3 (3.8 g, 81%): m.p. 129-131 °C. 1H NMR δ 2.28 (s, 3 H, 8′-CH3), 3.22 (dd, J = 10.84 Hz, J = 16.08 Hz, 1 H, 4′-Hα), 3.38 (dd, J = 4.63 Hz, J = 5.10 Hz, 1 H, 12a-H), 3.42 (dd, J = 6.68 Hz, J = 16.08 Hz, 1 H, 4′-Hβ), 3.84 (s, 3 H, 2-OCH3), 3.85 (s, 3 H, 3-OCH3), 4.22 (dd, J = 5.18 Hz, J = 9.84 Hz, 1 H, 6-Hα), 4.58 (dd, J = 9.84 Hz, J = 11.07 Hz, 1 H, 6-Hβ), 4.82 (ddd, J = 5.10 Hz, J = 5.18 Hz, J = 11.07 Hz, 1 H, 6a-H), 4.92 (br s, 1 H, 12-H), 5.08 (dd, J = 6.68 Hz, J = 10.84 Hz, 1 H, 5′-H), 6.46 (s, 1 H, 4-H), 6.52 (d, J = 8.20 Hz, 1 H, 10-H), 6.70 (s, 1 H, 1-H), 7.09 (d, J = 8.20 Hz, 1 H, 11-H). Anal. calcd. for C22H22O7*0.5 H2O: C, 64.86; H, 5.41; found: C, 65.16; H, 5.94. HRMS: m/z 398.1363 (calcd. for C22H22O7, 398.1365). LRMS: m/z 398 (M), 380 (M-H2O, 32), 192 (base).
2.3 7′-(Z)-Iodorotenol(4)
To a suspension of Ph3PCH2I2 (6.0 g, 11.3 mmol) in THF (20 mL) was added NaN(TMS)2 (11.5 mL, 1.0 M solution in THF, 11.5 mmol). Upon dissolution, the flask was cooled to -78 °C and HMPA (3 mL) was added. Oxo-rotenol 3 (3.7 g, 9.3 mmol) in THF (10 mL) was added slowly, and the solution was allowed to warm to room temperature. The solution was diluted with water and extracted with CH2Cl2. The combined extracts were washed with aqueous NaHSO3, water, and then dried (MgSO4). Concentration in vacuo afforded a residue that was purified by flash chromatography on silica gel (1:9 EtOAc:CH2Cl2) providing Z-iodorotenol 4 (1.4 g, 30%) as a foam. A few milligrams of 4 were purified by HPLC (Whatman Partisil 10, 9.4 mm × 25 cm, 25% EtOAc/Hexane, tR = 23 min) for analysis. 1H NMR δ 1.89 (d, J = 1.42 Hz, 3 H, 8′-CH3), 2.83 (dd, J = 9.66 Hz, J = 15.98 Hz, 1 H, 4′-Hα), 3.40 (dd, J = 4.26 Hz, J = 4.98 Hz, 1 H, 12a-H), 3.48 (dd, J = 8.46 Hz, J = 15.98 Hz, 1 H, 4′-Hβ), 3.85 (s, 3 H, 2-OCH3), 3.86 (s, 3 H, 3-OCH3), 4.24 (dd, J = 5.13 Hz, J = 9.76 Hz, 1 H, 6-Hα), 4.61 (dd, J = 9.76 Hz, J = 11.34 Hz, 1 H, 6-Hβ), 4.84 (ddd, J = 4.98 Hz, J = 5.13 Hz, J = 11.34 Hz, 1 H, 6a-H), 4.93 (d, J = 4.26 Hz, 1 H, 12-H), 5.67 (dd, J = 8.46 Hz, J = 9.66 Hz, 1 H, 5′-H), 6.05 (d, J = 1.42 Hz, 1 H, 7′-H), 6.46 (d, J = 8.15 Hz, 1 H, 10-H), 6.48 (s, 1 H, 4-H), 6.71 (s, 1 H, 1-H), 7.07 (d, J = 8.15 Hz, 1 H, 11-H). Anal. calcd. for C23H23IO6: C, 52.87; H, 4.41; found: C, 53.42; H, 4.86. HRMS: m/z 522.0546 (calcd. for C23H23IO6 522.0539). LRMS(FAB): m/z 522 (M+, 26), 505 (m-OH, 30), 307 (6), 192 (base).
2.4 7′-(Z)-Iodorotenone (5)
To a solution of iodorotenol (100 mg, 0.192 mmol) in dry acetonitrile (5.0 mL) was added activated MnO2 (250 mg, 2.88 mmol) in one portion under argon atmosphere at room temperature. The mixture was vigorously stirred for 1 min and immediately filtered through a pad of celite. The filtrate was evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (1:3 EtOAc: Hexane) to give Z-iodorotenone 5 (57 mg, 58%) as a colorless solid and recovered iodorotenol 4 (22 mg, 22%). m.p. 201-203 °C. 1H NMR δ 1.87 (s, 3 H, 8′-CH3), 2.81 (dd, J = 9.63 Hz, J = 16.01 Hz, 1 H, 4′-Hα), 3.50 (dd, J = 8.88 Hz, J = 16.01 Hz, 1 H, 4′-Hβ), 3.76 (s, 3 H, 2-OCH3), 3.80 (s, 3 H, 3-OCH3), 3.84 (d, J = 4.26 Hz, 1 H, 12a-H), 4.17 (d, J = 12.03 Hz, 1 H, 6-Hα), 4.60 (dd, J = 3.24 Hz, J = 12.03 Hz, 1 H, 6-Hβ), 4.93 (m, 1 H, 6a-H), 5.68 (dd, J = 8.88 Hz, J = 9.63, 1 H, 5′-H), 6.07 (s, 1 H, 7′-H), 6.45 (s, 1 H, 4-H), 6.50 (d, J = 8.56 Hz, 1 H, 10-H), 6.74 (s, 1 H, 1-H), 7.83 (d, J = 8.56 Hz, 1 H, 11-H). Anal. calcd. for C23H21IO6: C, 53.08; H, 4.04; found: C, 52.93; H, 4.39. HRMS: m/z 520.0378 (calcd. for C23H21IO6, 520.0383). LRMS(FAB): m/z 520 (M+, 47), 307 (26), 192 (46), 154 (base).
2.5 7′-(Z)-Tributylstannylrotenol (6)
To a solution of iodorotenol 4 (1.44 g, 2.86 mmol) in toluene (22 mL) was added (Bu3Sn)2 (3.2 g, 5.5 mmol) and Pd(PPh3)4 (0.042 g, 0.036 mmol). The solution was refluxed under argon for 13 h and then allowed to cool to room temperature. The reaction mixture was passed through a short silica gel column with hexane, CH2Cl2, and 1:9 EtOAc:CH2Cl2 as successive eluents. Concentration of the appropriate fractions in vacuo afforded a residue that was further purified by HPLC (Whatman Partisil 10, 9.4 mm × 50 cm, 20% EtOAc/Hexane, 4 mL/min,) to give tributylstannylrotenol 6 (200 mg, 10%) as a colorless oil. 1H NMR δ 0.85-0.93 (m, 15 H, 3 CH3CH2), 1.26-1.36 (m, 6 H, 3 CH2), 1.44-1.50 (m, 6 H, 3 CH2), 1.89 (d, J = 1.22 Hz, 3 H, 8′-CH3), 2.95 (dd, J = 9.34 Hz, J = 15.75 Hz, 1 H, 4′-Hα), 3.25 (dd, J = 9.34 Hz, J = 15.75 Hz, 1 H, 4′-Hβ), 3.39 (dd, J = 4.29 Hz, J = 5.28 Hz, 1 H, 12a-H), 3.84 (s, 3 H, 2-OCH3), 3.85 (s, 3 H, 3-OCH3), 4.24 (dd, J = 4.72 Hz, J = 9.78 Hz, 1 H, 6-Hα), 4.62 (dd, J = 9.78 Hz, J = 11.34 Hz, 1 H, 6-Hβ), 4.83 (ddd, J = 4.72 Hz, J = 5.28 Hz, J = 11.34 Hz, 1 H, 6a-H), 4.92 (br s, 1 H, 12-H), 5.12 (t, J = 9.34 Hz, 1 H, 5′-H), 5.73 (d, J = 1.22 Hz, 1 H, 7′-H), 6.43 (d, J = 8.10 Hz, 1 H, 10-H), 6.46 (s, 1 H, 4-H), 6.70 (s, 1 H, 1-H), 7.04 (d, J = 8.10 Hz, 1 H, 11-H). Anal. calcd. for C35H50O6Sn: C, 61.40; H, 7.31; found: C, 61.56; H, 7.42.
2.6 7′-(Z)-Tributylstannylrotenone (7)
To a solution of tributylstannylrotenol 6 (75 mg, 0.11 mmol) in CH3CN (10 mL) was added MnO2 (250 mg, 2.8 mmol). The mixture was stirred 10 min at room temperature and then filtered through celite. Concentration of the filtrate in vacuo afforded a residue which was purified by HPLC (Whatman Partisil 10, 9.4 mm × 25 cm, 20% EtOAc/Hexane, tR = 15 min) to give the tributylstannylrotenone 7 (29 mg, 39%) as an oily foam. 1H NMR δ 0.84-0.91 (m, 15 H, 3 CH3CH2), 1.22-1.34 (m, 6 H, 3 CH2), 1.42-1.52 (m, 6 H, 3 CH2), 1.87 (d, J = 1.27 Hz, 3 H, 8′-CH3), 2.96 (dd, J = 9.33 Hz, J = 15.88 Hz, 1 H, 4′-Hα), 3.29 (dd, J = 9.33 Hz, J = 15.88 Hz, 1 H, 4′-Hβ), 3.76 (s, 3 H, 2-OCH3), 3.81 (s, 3 H, 3-OCH3), 3.84 (d, J = 3.48 Hz, 1 H, 12a-H), 4.18 (d, J = 12.06 Hz, 1 H, 6-Hα), 4.62 (dd, J = 3.03 Hz, J = 12.06 Hz, 1 H, 6-Hβ), 4.93 (dd, J = 3.03 Hz, J = 3.48 Hz, 1 H, 6a-H), 5.16 (t, J = 9.33 Hz, 1 H, 5′-H), 5.77 (d, J = 1.27 Hz, 1 H, 7′-H), 6.46 (s, 1 H, 4-H), 6.49 (d, J = 8.56 Hz, 1 H, 10-H), 6.78 (s, 1 H, 1-H), 7.84 (d, J = 8.56 Hz, 1 H, 11-H). Anal. calcd. for C35H48O6Sn*1/2 H2O: C, 60.78; H, 6.95; found: C, 60.50; H, 6.92; LRMS(FAB): m/z 683 (M+, 19), 627 (M-C4H9, base), 513 (M-3xC4H9, 11), 192 (37), 179 (65).
2.7 7′-(Z)-[125I]Iodorotenol (8)
To a solution of tributylstannylrotenol 6 (1 mg) in THF (200 μL) were successively added Na[125I]I (1.5 mCi), 0.61 M NaOAc (pH 4.5, 50 μL), and 2:1 30% H2O2:AcOH (25 μL). The reaction mixture was stirred for 1 min at room temperature and then quenched with 50 μL 10% Na2S2O3(aq). The mixture was diluted with 350 μL of water and was injected onto a Whatman Partisil 10 ODS-3 HPLC column (9.4 mm × 50 cm, 60/40 MeOH/H2O, 4 mL/min). Product fractions were pooled and diluted with an equal volume of water and passed through a Waters C18 Sep-Pak® Classic cartridge. The trapped product was eluted from the Sep-Pak with 3 mL ethanol providing 1.3 mCi (81%) of Z-[125I]iodorotenol 8. Radiochemical purity of >95% was determined by HPLC on a Waters μ-Bondapack column (3.9 mm × 30 cm, 70/30 MeOH/H2O -20 mM triethylammonium phosphate pH 7.4, 2 mL/min, tR = 3.4 min). The product was stored in ethanol at -80 °C in a sealed vial.
2.8 7′-(Z)-[125I]Iodorotenone (9)
To a solution of tributylstannylrotenone 7 (0.75 mg, 1.1 mmol) in THF (200 μL) were successively added Na[125I]I (5 mCi), 0.61 M NaOAc (pH 4.5, 50 μL), and 2:1 30% H2O2:AcOH (25 μL). The reaction mixture was stirred for 1 min at room temperature and then quenched with 40 μL 10% Na2S2O3(aq). The mixture was diluted with 350 μL of water and was injected onto a Whatman Partisil 10 ODS-3 HPLC column (9.4 mm × 50 cm, 60/40 MeOH/H2O, 6 mL/min). Product fractions were pooled and diluted with an equal volume of water and passed through a Waters C18 Sep-Pak® cartridge. The trapped product was eluted from the Sep-Pak with 3 mL ethanol providing 2.4 mCi (48%) of Z-[125I]iodorotenone 9. Radiochemical purity of >95% was determined by HPLC on a Waters μ-Bondapack column (3.9 mm × 30 cm, 70/30 MeOH/H2O-triethylammonium phosphate pH 7.4, 2 mL/min, tR = 6.0 min). The product was stored in ethanol at -80 °C in a sealed vial.
2.5 In Vivo Biodistribution Studies
All animal experiments were carried out in compliance with the Guidelines for Care and Use of Research Animals established by the Lawrence Berkeley National Laboratory Animal Welfare and Research Committee.
The ethanol solution of either Z-[125I]iodorotenol 8 or Z-[125I]iodorotenone 9 was diluted with isotonic saline to a final concentration of 5% ethanol. Isoflurane anesthetized Sprague-Dawley male rats (∼200 g) were injected in the tail vein with the 15-20 μCi of the either 8 or 9. At 1h, 2h and 24h post injection, rats (n=3-7 per time point) were euthanized by decapitation under general anesthesia. Blood and organs were removed, weighed and assayed for radioactivity using a Packard Gamma Counter. The percent injected dose per gram of tissue was determined.
3. Results
3.1 Synthesis of Iodorotenol, Iodorotenone and the corresponding tributylstannyl precursors
The synthetic pathway for the preparation of iodorotenol 4 and iodorotenone 5 is shown in Scheme 1. Commercially available rotenone 1 was quantitatively converted to a single isomer of rotenol 2 by selective sodium borohydride reduction of the 12-carbonyl group [26]. Oxidative cleavage of the 6′-7′ olefin with osmium tetroxide gave the 6′ ketone intermediate 3 in 61% yield. The subsequent Wittig reaction with (iodomethyl)-triphenylphosphonium iodide ylide gave exclusively the 7′-(Z)-iodorotenol 4 in 25-30% yield. Based on NMR and HPLC examination a single isomer was predominantly formed (>99%). The Z stereoisomer configuration was independently confirmed by NOE NMR studies (unpublished results). The overall yield of iodorotenol from rotenone was 18%.
Oxidation of the 12 hydroxy group of Z-iodorotenol 4 gave Z-iodorotenone 5 in one minute using activated manganese dioxide. Typical yields for this reaction ranged from 25-58% with recovery of a significant amount (∼25%) of starting iodorotenol and a by-product. When the oxidation time was increased to five minutes the starting material was completely consumed, however, the Z-iodorotenone yield decreased significantly with a concomitant increase in the by-product. The overall yield of Z-iodorotenone for the four-step conversion from rotenone was 5-11%.
The synthesis of the vinyl stannane labeling precursors from the common intermediate iodorotenol 4 is shown in Scheme 2. Reaction of Z-iodorotenol 4 with hexabutylditin using catalytic tetrakis(triphenylphosphine)-palladium (0) gave the 7′-(Z)-tributylstannylrotenol 6 in low yield (10-15%). Subsequent oxidation of 6 to the corresponding 7′-(Z)-tributylstannylrotenone 7 was accomplished with manganese dioxide. The constraints of the oxidation reaction are the same as previously described for the conversion of 4 to 5 and account for the moderate oxidation yield of 40%. The overall yield of the stannylrotenol 6 and the stannylrotenone 7 from starting rotenone is 1.8% and 0.7%, respectively.
Scheme 2.

Synthesis of iodine-125 labeled 7′(Z)-iodorotenol 8 and 7′(Z)-iodorotenone 9 from the corresponding tributylstannane precursors.
3.2 Synthesis of radioiodinated iodorotenone and iodorotenol
The radioiodination reactions are shown in Scheme 2. The formation of both radioiodinated compounds occured rapidly by iododestannylation using sodium [125I]iodide in the presence of in situ derived peracetic acid. The isolated yield of Z-[125I]iodorotenol (75-80%) was typically greater than that of the [125I]iodorotenone (∼50%). The Z-[125I]iodoroteno1 8 and Z-[125I]iodorotenone 9 were very stable when stored in ethanol at -80°C. The degradation of the stored material, verified by HPLC, was less than 3% per month.
3.3 In Vivo Biodistribution Studies
The distribution of the radioiodinated rotenone analogs at 1h, 2h and 24h was evaluated in ∼200 gram male Sprague-Dawley rats. The tissue biodistribution of 7′-(Z)-[125I]iodorotenol 8 is presented in Table 1 and that of 7′-(Z)-[125I]iodorotenone 9 in Table 2. Both compounds were rapidly taken up in the heart (1.7 %ID/g for 8 and 3.7%ID/g for 9). The initial amount of tracer that remained in the blood was inversely proportional to the heart uptake with the Z-iodorotenol 8 blood values nearly double that of the Z-iodorotenone 9. The 1h uptake in the rest of the tissues was comparable for both compounds. Interestingly, there is considerable uptake of the tracer in the brain for both compounds during the first hour.
Table 1.
Biodistribution of 7’(Z)-[125I]iodorotenol (8) in Sprague-Dawley male rats
| Tissue | 1 h (n=3) | 2 h (n=4) | 24 h (n=4) |
|---|---|---|---|
| blood | 0.259 ± 0.027 | 0.297 ± 0.031 | 0.080 ± 0.013 |
| heart | 1.698 ± 0.074 | 1.586 ± 0.240 | 0.241 ± 0.042 |
| lung | 0.548 ± 0.035 | 0.521 ± 0.050 | 0.078 ± 0.017 |
| liver | 1.497 ± 0.104 | 1.376 ± 0.151 | 0.138 ± 0.018 |
| kidney | 0.934 ± 0.009 | 0.822 ± 0.062 | 0.109 ± 0.009 |
| spleen | 0.364 ± 0.024 | 0.335 ± 0.026 | 0.047 ± 0.005 |
| muscle | 0.518 ± 0.093 | 0.491 ± 0.046 | 0.086 ± 0.012 |
| fat | 1.066 ± 0.269 | 1.469 ± 0.385 | 0.213 ± 0.038 |
| brain | 0.427 ± 0.030 | 0.297 ± 0.023 | 0.015 ± 0.002 |
| thyroida | 0.139 ± 0.047 | 0.241 ± 0.072 | 5.037 ± 2.376 |
| heart:blood | 6.600 ± 0.645 | 5.316 ± 0.363 | 3.016 ± 0.303 |
| heart:lung | 3.107 ± 0.203 | 3.081 ± 0.629 | 3.199 ± 0.892 |
| heart:liver | 1.136 ± 0.048 | 1.151 ± 0.109 | 1.755 ± 0.207 |
Male Sprague-Dawley rats (∼200 grams) were injected iv with 15-20 μCi of Z-[125I]iodorotenol in 10% ethanol saline.
Tissue distribution values are given as percent injected dose / gram and presented as mean ± standard deviation.
Thyroid values are given as percent injected dose/ organ.
Table 2.
Biodistribution of 7’ (Z)-[125I]iodorotenone (9) in Sprague-Dawley male rats
| Tissue | 1 h (n=7) | 2 h (n=6) | 24 h (n=4) |
|---|---|---|---|
| blood | 0.130 ± 0.020 | 0.135 ± 0.019 | 0.063 ± 0.010 |
| heart | 3.707 ± 0.203 | 2.959 ± 0.244 | 0.607 ± 0.109 |
| lung | 0.352 ± 0.055 | 0.326 ± 0.050 | 0.102 ± 0.017 |
| liver | 1.557 ± 0.201 | 1.169 ± 0.188 | 0.267 ± 0.052 |
| kidney | 1.257 ± 0.118 | 0.933 ± 0.092 | 0.212 ± 0.037 |
| spleen | 0.275 ± 0.036 | 0.229 ± 0.013 | 0.062 ± 0.012 |
| muscle | 0.507 ± 0.077 | 0.536 ± 0.074 | 0.199 ± 0.050 |
| fat | 0.723 ± 0.193 | 1.018 ± 0.284 | 1.034 ± 0.144 |
| brain | 0.436 ± 0.038 | 0.341 ± 0.035 | 0.040 ± 0.004 |
| thyroida | 0.154 ± 0.057 | 0.339 ± 0.096 | 3.784 ± 0.647 |
| heart:blood | 28.94 ± 3.61 | 22.35 ± 3.83 | 9.737 ± 2.239 |
| heart:lung | 10.68 ± 1.34 | 9.316 ± 1.978 | 6.118 ± 1.716 |
| heart:liver | 2.402 ± 0.209 | 2.570 ± 0.334 | 2.284 ± 0.310 |
Male Sprague-Dawley rats (∼200 grams) were injected iv with 15-20 μCi of Z-[125I]iodorotenone in 10% ethanol saline.
Tissue distribution values are given as percent injected dose / gram and presented as mean ± standard deviation.
Thyroid values are given as percent injected dose/ organ.
The distribution of Z-iodorotenol 8 does not significantly change in most tissues between 1 h and 2 h post injection, with the exception of the kidneys and brain (P < 0.02), yet by 24h there is significant washout (43-92%) from all tissues with the exception of the thyroid. Z-Iodorotenone 9 shows significant moderate (7-26%; P < 0.03) washout by 2 h from all tissues except for blood, lung, muscle, thyroid and fat. By 24 h there is significant loss of tracer (53-89%) from all tissues with the exception of thyroid and fat.
The rate of in vivo deiodination as evidenced by the percent injected dose in the thyroid is comparable for both compounds. The calculated linear thyroid accumulation rate appears to be about 0.15-0.2% per hour over the first 24 h. The rats were not pretreated with potassium chloride to block radioiodide uptake in the thyroid tissue.
Heart-to-tissue ratios were calculated for the blood, lung and liver. These values are given for Z-iodorotenol 8 and Z-iodorotenone 9 in Tables 1 and 2, respectively. The heart-to-blood ratio was the highest at the 1 h time point for each compound with the Z-iodorotenone ratio being four-fold higher than the Z-iodorotenol (28.9 compared to 6.6). The heart-to-blood ratios for both compounds decreased significantly (P < 0.03) at each time point up to the end of the study at 24 h post injection. The heart-to-lung ratio was again highest at 1 h post injection for both tracers (3.1 for 8 and 10.7 for 9). There was no significant change in the ratio for either tracer between 1 and 2 hours. By 24 h the heart-to-lung ratio for Z-iodorotenone dropped by nearly 43% to 6.1 while there was no change in the ratio (3.1-3.2) for the Z-iodorotenol over the entire study period. The 1 h heart-to-liver ratio for Z-iodorotenol was just over 1. It remained unchanged at 2h but increased to 1.8 between 2h and 24h. Z-Iodorotenone heart-to-liver ratios remained around 2.4 throughout the course of the study.
4.0 Discussion
The two iodinated rotenoids were synthesized in good yield from commercially available rotenone in three to four steps. All of the reactions gave predominantly the desired products with the exception of the manganese dioxide mediated oxidation of the rotenols to rotenones. Based on our work with the fluororotenones and literature precedence [27, 28] the major by-product of the oxidation was identified as the 6a-12a unsaturated compound. The oxidation of the iodorotenone to the 6a-12a dehydro-iodorotenone competes with the conversion of iodorotenol to iodorotenone. Thus, optimizing the reaction time and amount of manganese dioxide is critical for suitable yields of the iodorotenone.
As with the manganese dioxide oxidations, unavoidable oxidation by the peracetic acid during the radiochemical reactions may be impacting the Z-[125I]iodorotenone yield. It does not appear that the Z-[125I]iodorotenol yield is affected by the peracetic acid as the yields are consistently high, 75-80%. On the other hand the Z-[125I]iodorotenone or the tributylstannylrotenone precursor appear to be sensitive to peracetic acid oxidation as evidenced by the significantly lower product yield, ∼50%, and increased by-product formation observed on the semi-preparative HPLC chromatogram. While a thorough investigation of oxidant concentrations or alternative oxidants was not undertaken for the iodorotenone reaction, increasing the reaction time prior to quenching decreased the iodorotenone to by-product ratio. It is known from our work with the rotenoids (unpublished data) that a variety of other oxidizing agents did not improve the overall yield nor the desired rotenone-to-byproduct ratio.
An optimal cardiac flow tracer possesses the following characteristics, rapid and high extraction from the blood into the heart tissue, good retention in the cardiac tissue, linear uptake with flow at all flow rates, high heart-to-blood, high heart-to-lung and high heart-to-liver ratios as well as suitable clearance from liver and kidneys. There has been considerable effort to produce an ideal technetium-99m labeled tracer for SPECT perfusion imaging that surpass the less than ideal characteristics of the current clinical tracers, [99mTc]sestamibi and [99mTc]tetrofosmin.
In the present study, Z-[125I]iodorotenol and Z-[125I]iodorotenone demonstrate good accumulation in the heart tissue at the 1 h time point. The uptake of Z-iodorotenone is comparable to the rodent heart uptake of [99mTc]sestamibi and [99mTc]tetrofosmin, 3.7 versus 3.2 and 2.8 %ID/g respectively [29]. While the rate of accumulation, initial extraction and overall retention of the tracers is not captured in the present biodistribution study, Z-iodorotenone has been shown to have high first pass extraction (∼84%) and better retention versus sestamibi (∼48% first pass extraction) in isolated perfused rabbit heart studies [21]. The rodent biodistribution model does show tracer accumulation in the heart but is unable to discriminate the differences in extraction and retention that are important for selecting improved perfusion tracers. More sophisticated models of perfusion, such as the isolated perfused rabbit heart model, are necessary to examine the flow characteristics.
Both Z-iodorotenol and Z-iodorotenone are retained in the heart tissue from 1-2 h but ∼85% of the activity washes out of the heart tissue from 2 to 24 h. Without a gold standard it is difficult to evaluate the relative retention of these tracers in the intact rodent model. However, in the isolated perfused rabbit heart studies iodorotenone is retained in the heart to a greater extent than [99mTc]sestamibi. [21]. The mechanism of accumulation and retention of the labeled rotenone analogs is based on the known inhibitory properties of rotenoids for NADH2 oxidase (Complex I of the electron transport chain) found on the inner membrane of mitochondria. This is strikingly similar to the known mechanism of cationic [99mTc]sestamibi and [99mTc]tetrofosmin accumulation and retention based on the potential gradient sustained in viable mitochondria.[30-32] The high concentration of mitochondria in the heart tissue provides a reservoir for the flow tracers.
The avidity of these tracers for Complex I may also enable the evaluation of mitochondrial density in normal and diseased tissues. The degree of tracer uptake may correlate with the mass of mitochondria or with mitochondrial activity. This would include the original purpose for designing these tracers that was to measure the known loss of Complex I associated with neurodegenerative diseases.
The uptake in the tissues surrounding the heart, most notably the lungs, define the contrast value of the tracer as a cardiac perfusion imaging agent. The crown of the liver also appears in the images near the heart so it is desirable to have a high heart-to-liver ratio in addition to a high heart-to lung ratio. The 1 h heart-to-lung and heart-to-liver ratios for Z-[125I]iodorotenol 8 and Z-[125I]iodorotenone 9 are compared to the two clinical cationic SPECT tracers [99mTc]sestamibi and [99mTc]tetrofosmin in Figure 2. While the absolute heart uptake of iodorotenone was similar to sestamibi and tetrofosmin, Z-iodorotenone demonstrated significantly superior (P < 0.001) heart-to-lung ratios in the rodent at 1 h post injection. This contrast was sustained out to 2 h post injection. The heart-to-liver ratio for iodorotenone was equivalent to that of [99mTc]sestamibi (2.4 vs. 2.6:1) yet 2.5-fold less than [99mTc]tetrofosmin (5.8:1). Taken together the distribution characteristics of Z-[125I]iodorotenone are favorable compared to the current clinical technetium-99m tracers.
Figure 2.
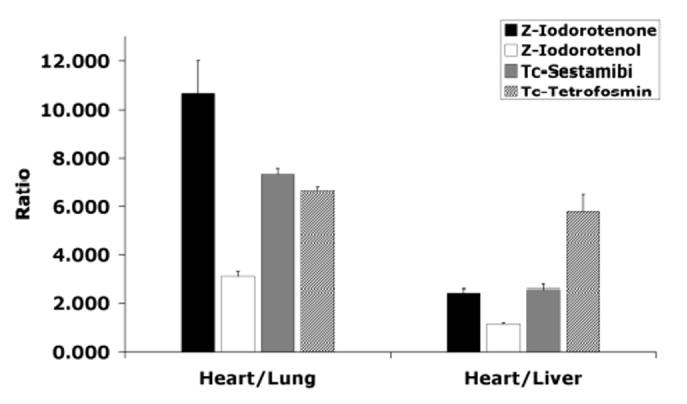
Heart-to-Liver and Heart-to-Lung uptake ratios for 8, 9, 99mTc-sestamibi and 99mTc-tetrofosmin. Data for the technetium compounds were taken from reference [33]. Heart-to-Lung: Z-iodorotenone P < 0.001 vs. Z-iodorotenol; P < 0.001 vs 99mTc-sestamibi; and P < 0.001 vs 99mTc-tetrofosmin. Heart-to-Liver: Z-iodorotenone P< 0.001 vs. Z-iodorotenol; P > 0.1 vs 99mTc-sestamibi; and P < 0.001 vs 99mTc-tetrofosmin.
5.0 Conclusion
A new class of mitochondrial avid perfusion tracers have been identified. Z-[125I]Iodorotenone and Z-[125I]iodorotenol were produced in moderate to high yield and in a single step from their respective tributylstannyl precursors. The good heart uptake combined with the high heart-to-blood and heart-to-lung ratios and good clearance from the kidneys demonstrates the favorable distribution characteristics of iodorotenone. Combined with the studies conducted in the isolated perfused rabbit heart, iodorotenone possesses desirable properties of an improved cardiac flow tracer that may prove valuable as a more sensitive identifier of significant coronary lesions.
Acknowledgements
This work was supported by Director, Office of Science, Office of Biological and Environmental Research, Medical Science Division of the U.S. Department of Energy under contract no. DE-AC03-76SF00098 and NIH grants AG10129, HL25840. The authors would like to thank Heidi Mauer, Katie Brennan, Michelle Heusman, Pattie Powers-Risius and Dr. Scott Taylor for their assistance with the in vivo biodistribution studies, Dr. David Glover for detailed data on the rodent distribution of sestamibi and tetrofosmin and Dr. Mustafa Janabi for assistance with manuscript review.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Albers DS, Beal MF. Mitochondrial dysfunction and oxidative stress in aging and neurodegenerative disease. J Neural Transm Suppl. 2000;59:133–54. doi: 10.1007/978-3-7091-6781-6_16. [DOI] [PubMed] [Google Scholar]
- [2].Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A Dawn for Evolutionary Medicine. Ann Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Beal MF. Mitochondrial dysfunction and oxidative damage in Alzheimer’s and Parkinson’s diseases and coenzyme Q10 as a potential treatment. J Bioenerg Biomembr. 2004;36:381–6. doi: 10.1023/B:JOBB.0000041772.74810.92. [DOI] [PubMed] [Google Scholar]
- [4].Sherer TB, Betarbet R, Greenamyre JT. Environment, mitochondria, and Parkinson’s disease. Neuroscientist. 2002;8:192–7. doi: 10.1177/1073858402008003004. [DOI] [PubMed] [Google Scholar]
- [5].Haley TJ. A review of the literature of rotenone 1,2,12,12a-tetrahydro-8,9-dimethoxy-2-(1-methylethenyl)-1-benzopyrano[3,5-b]furo[2,3-h][1]benzopyran-6(6h)-one. J Env Path Tox. 1978;1:315–37. [PubMed] [Google Scholar]
- [6].Fukami H, Nakajima M. In: Naturally occurring insecticides. Jacobson M, Crosby DG, editors. Marcel Dekker, Inc.; New York: 1971. pp. 71–75. [Google Scholar]
- [7].Rach JJ, Gingerich WH. Distribution and accumulation of rotenone in tissues of warmwater fishes. Trans Amer Fisheries Soc. 1986;115:214–19. [Google Scholar]
- [8].Crombie L, Whiting DA. Review article number 135. Biosynthesis in the rotenoid group of natural products: applications of isotope methodology. Phytochemistry. 1998;49:1479–507. doi: 10.1016/s0031-9422(98)00178-2. [DOI] [PubMed] [Google Scholar]
- [9].Greenamyre JT, Higgins DS, Eller RV. Quantitative autoradiography of dihydrorotenone binding to complex I of the electron transport chain. J Neurochem. 1992;59:746–9. doi: 10.1111/j.1471-4159.1992.tb09431.x. [DOI] [PubMed] [Google Scholar]
- [10].O’Neil JP, VanBrocklin HF, Morimoto H, Williams PG. Synthesis of 3H Labeled Dihydrorotenone. J Labelled Comp Radiopharm. 1997;39:215–21. [Google Scholar]
- [11].Talpade DJ, Greene JG, Higgins DS, Jr., Greenamyre JT. In vivo labeling of mitochondrial complex I (NADH:ubiquinone oxidoreductase) in rat brain using [(3)H]dihydrorotenone. J Neurochem. 2000;75:2611–21. doi: 10.1046/j.1471-4159.2000.0752611.x. [DOI] [PubMed] [Google Scholar]
- [12].Higgins DS, Jr., Greenamyre JT. [3H]dihydrorotenone binding to NADH: ubiquinone reductase (complex I) of the electron transport chain: an autoradiographic study. J Neurosci. 1996;16:3807–16. doi: 10.1523/JNEUROSCI.16-12-03807.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Blandini F, Greenamyre JT. Assay of [3H]dihydrorotenone binding to complex I in intact human platelets. Anal Biochem. 1995;230:16–9. doi: 10.1006/abio.1995.1430. [DOI] [PubMed] [Google Scholar]
- [14].Charalambous A, Tluczek L, Frey KA, Higgins DS, Jr., Greenamyre TJ, Kilbourn MR. Synthesis and biological evaluation in mice of (2-[11C]methoxy)-6′,7′-dihydrorotenol, a second generation rotenoid for marking mitochondrial complex I activity. Nucl Med Biol. 1995;22:491–6. doi: 10.1016/0969-8051(94)00129-8. [DOI] [PubMed] [Google Scholar]
- [15].Charalambous A, Mangner TJ, Kilbourn MR. Synthesis of (2-[11C]methoxy)rotenone, a marker of mitochondrial complex I activity. Nucl Med Biol. 1995;22:65–9. doi: 10.1016/0969-8051(94)00075-u. [DOI] [PubMed] [Google Scholar]
- [16].Snyder SE, Sherman PS, Desmond TJ, Frey KA, Kilbourn MR. (-)-6′7′-[11C]Dihydroroten-12a-ol ((-)-[11C]DHROL) for In Vivo Measurement of Mitochondrial Complex I. J Labelled Comp Radiopharm. 1999;47:641–52. [Google Scholar]
- [17].Kilbourn MR, Charalambous A, Frey KA, Sherman P, Higgins DS, Jr., Greenamyre JT. Intrastriatal neurotoxin injections reduce in vitro and in vivo binding of radiolabeled rotenoids to mitochondrial complex I. J Cereb Blood Flow Metab. 1997;17:265–72. doi: 10.1097/00004647-199703000-00003. [DOI] [PubMed] [Google Scholar]
- [18].Martarello L, Greenamyre JT, Goodman MM. Synthesis and evaluation of a new fluorine-18 labeled rotenoid as a potential pet probe of mitochondrial complex I activity. J Label Compound Radiopharm. 1999;42:1039–51. [Google Scholar]
- [19].VanBrocklin H, Enas J, Hanrahan S, O’Neil J. Fluorine-18 Labeled Dihydrorotenone Analogs: Preparation and evaluation of PET mitochondrial probes. J Lab Comp Radiopharm. 1995;37:217–19. [Google Scholar]
- [20].Kenski DM, VanBrocklin HF, O’Neil JP. Fluorine-18 Labeled Rotenone Analogs: Preparation and Evaluation of PET Mitochondrial Probes. J Labelled Comp Radiopharm. 1999;42(suppl 1):S333–35. [Google Scholar]
- [21].Marshall RC, Powers-Risius P, Reutter BW, Taylor SE, VanBrocklin HF, Huesman RH, Budinger TF. Kinetic Analysis of [125I]iodorotenone as a deposited myocardial flow tracer: comparison to [99mTc]sestamibi. J Nucl Med. 2001;42:272–81. [PubMed] [Google Scholar]
- [22].Marshall R, Powers-Risius P, Reutter B, O’Neil J, La Belle M, Huesman R, VanBrocklin H. Kinetic analysis of 18F-fluorodihydrorotenone as a deposited myocardial flow tracer: comparison to 201Tl. J Nucl Med. 2004;45:1950–9. [PubMed] [Google Scholar]
- [23].Crane P, Laliberte R, Heminway S, Thoolen M, Orlandi C. Effect of mitochondrial viability and metabolism on technetium-99m-sestamibi myocardial retention. Eur J Nucl Med. 1993:20. doi: 10.1007/BF02261241. [DOI] [PubMed] [Google Scholar]
- [24].Carvalho P, Chiu M, Kronauge J, Kawamura M, Jones A, Holman B, Piwnica-Worms D. Subcellular distribution and analysis of technetium-99m-MIBI in isolated perfused rat hearts. J Nucl Med. 1992;33:1516–22. [PubMed] [Google Scholar]
- [25].Schaper J, Meiser E, Stammler G. Ultrastructural morphometric analysis of myocardium from dogs, rats, hamsters, mice, and from human hearts. Circ Res. 1985;56:377–91. doi: 10.1161/01.res.56.3.377. [DOI] [PubMed] [Google Scholar]
- [26].Buchi G, Crombie L, Godin P, Kaltenbronn J, Siddalingaiah K, Whiting D. The absolute configuration of rotenone. J Chem Soc. 1961:2843–60. [Google Scholar]
- [27].Begley M, Crombie L, bin A, Hadi A, Josephs J. Synthesis of novel labile rotenoids with unnatural trans-B/C Ring Systems. J Chem Soc Perkin Trans I. 1989:204–05. [Google Scholar]
- [28].Crombie L, Godin P. The structure and stereochemistry of the rotenolones, rotenolols, isorotenolones, and isorotenolols. JCS. 1961:2861–75. [Google Scholar]
- [29].Boschi A, Uccelli L, Bolzati C, Duatti A, Sabba N, Moretti E, Di Domenico G, Zavattini G, Refosco F, Giganti M. Synthesis and biologic evaluation of monocationic asymmetric 99mTc-nitride heterocomplexes showing high heart uptake and improved imaging properties. J Nucl Med. 2003;44:806–14. [PubMed] [Google Scholar]
- [30].Younes A, Songadele JA, Maublant J, Platts E, Pickett R, Veyre A. Mechanism of uptake of technetium-tetrofosmin. II: Uptake into isolated adult rat heart mitochondria. J Nucl Cardiol. 1995;2:327–33. doi: 10.1016/s1071-3581(05)80077-7. [DOI] [PubMed] [Google Scholar]
- [31].Platts EA, North TL, Pickett RD, Kelly JD. Mechanism of uptake of technetiumtetrofosmin. I: Uptake into isolated adult rat ventricular myocytes and subcellular localization. J Nucl Cardiol. 1995;2:317–26. doi: 10.1016/s1071-3581(05)80076-5. [DOI] [PubMed] [Google Scholar]
- [32].Chiu M, Kronauge J, Piwnica-Worms D. Effect of mitochondrial and plasma membrane potentials on accumulation of hexakis (2-methoxyisobutylisonitrile) technetium(I) in cultured mouse fibroblasts. J Nucl Med. 1990;31:1646–53. [PubMed] [Google Scholar]
- [33].Hatada K, Riou LM, Ruiz M, Yamamichi Y, Duatti A, Lima RL, Goode AR, Watson DD, Beller GA, Glover DK. 99mTc-N-DBODC5, a new myocardial perfusion imaging agent with rapid liver clearance: comparison with 99mTc-sestamibi and 99mTc-tetrofosmin in rats. J Nucl Med. 2004;45:2095–101. [PubMed] [Google Scholar]